
Abstract
Introduction
The receptor for advanced glycation end products (RAGE) is a member of the immunoglobulin superfamily of cell surface receptor molecules. High concentrations of three of its putative proinflammatory ligands, S100A8/A9 complex (calprotectin), S100A8, and S100A12, are found in rheumatoid arthritis (RA) serum and synovial fluid. In contrast, soluble RAGE (sRAGE) may prevent proinflammatory effects by acting as a decoy. This study evaluated the serum levels of S100A9, S100A8, S100A12 and sRAGE in RA patients, to determine their relationship to inflammation and joint and vascular damage.
Methods
Serum sRAGE, S100A9, S100A8 and S100A12 levels from 138 patients with established RA and 44 healthy controls were measured by ELISA and compared by unpaired t test. In RA patients, associations with disease activity and severity variables were analyzed by simple and multiple linear regressions.
Results
Serum S100A9, S100A8 and S100A12 levels were correlated in RA patients. S100A9 levels were associated with body mass index (BMI), and with serum levels of S100A8 and S100A12. S100A8 levels were associated with serum levels of S100A9, presence of anti-citrullinated peptide antibodies (ACPA), and rheumatoid factor (RF). S100A12 levels were associated with presence of ACPA, history of diabetes, and serum S100A9 levels. sRAGE levels were negatively associated with serum levels of C-reactive protein (CRP) and high-density lipoprotein (HDL), history of vasculitis, and the presence of the RAGE 82Ser polymorphism.
Conclusions
sRAGE and S100 proteins were associated not just with RA inflammation and autoantibody production, but also with classical vascular risk factors for end-organ damage. Consistent with its role as a RAGE decoy molecule, sRAGE had the opposite effects to S100 proteins in that S100 proteins were associated with autoantibodies and vascular risk, whereas sRAGE was associated with protection against joint and vascular damage. These data suggest that RAGE activity influences co-development of joint and vascular disease in rheumatoid arthritis patients.
Similar content being viewed by others

Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease that leads to bone and cartilage destruction and extra-articular complications, including atherosclerotic vascular disease and premature mortality [1]. The receptor for advanced glycation end products (RAGE) has been implicated in the pathogenesis of RA through its ability to amplify inflammatory pathways [2, 3]. A member of the immunoglobulin superfamily of cell surface receptors, RAGE binds advanced glycation end products (AGEs), which are non-enzymatically glycated or oxidized proteins, lipids and nucleic acids formed under conditions of oxidative stress and hyperglycemia (reviewed in [4]). In addition to these, RAGE binds some proinflammatory ligands, including members of the S100/calgranulin family, and high mobility group box chromosomal protein 1 (HMGB-1), which is implicated in cell signaling by synergizing with DNA CpG motifs [5, 6]. Several RAGE ligands are characteristically overexpressed in RA and psoriatic arthritis, compared to healthy controls [7–9]. S100A8/A9 (calprotectin) and S100A12 (calgranulin C, EN-RAGE) levels are significantly elevated in serum and synovial fluid from RA patients compared to healthy normal donors [3, 10]. S100A8/A9 levels are also higher in supernatants of cultured RA synoviocytes than of osteoarthritis synoviocytes [11].
Soluble C-truncated RAGE (sRAGE) lacks the transmembrane and cytosolic domains of the full-length receptor and can prevent proinflammatory effects of RAGE signaling by acting as a decoy [12–14]. For example, in a collagen-induced arthritis (CIA) murine model, treatment with murine sRAGE significantly reduced joint inflammation and destruction [15]. Serum or plasma levels of sRAGE from patients with RA, hypertension or metabolic syndrome were lower than those in healthy subjects [16–18], suggesting that sRAGE levels may identify those RA patients exposed to high levels of RAGE ligands. A gain-of-function Gly82Ser polymorphism in the RAGE gene (RAGE 82Ser) occurs more frequently in RA patients than in healthy controls [19]. Monocytes expressing the RAGE 82Ser allele activated a stronger inflammatory response to S100A12 in vitro [15]. Although this might be predicted to contribute to enhanced proinflammatory mechanisms in RA, we found no evidence that patients with the RAGE 82S allele had higher levels of inflammation, or a greater likelihood of complicating cardiovascular (CV) events [19].
Most S100 proteins have a mass between 9 and 14 kDa, and are characterized by two calcium binding sites of the EF-hand type (helix-loop-helix) [20]. S100A8 and S100A9, generally functioning as the S100A8/A9 heterocomplex, and S100A12 are implicated in non-infectious chronic inflammatory diseases such as RA, psoriasis and inflammatory bowel disease [21–25]. Longitudinal and cross-sectional studies suggest a relationship between S100A12 and RA disease activity [26–28]. The S100A12 gene is rapidly upregulated in human monocytoid cells and blood monocytes by tumor necrosis factor (TNF) and lipopolysaccharide (LPS), suggesting its production in response to proinflammatory signals in RA [10, 25]. S100A12 is a potent monocyte chemoattractant and activates mast cells, which are important effector cells in RA and atherosclerosis [25, 29, 30]. S100A12 is also proposed to promote proinflammatory activities by binding and activating RAGE [31]. However, these studies were established using a murine model, and since it was later shown that mice have no S100A12 in their genome [20], alternate receptors are implicated [25]. In addition, recombinant S100 ligands may contain contaminating endotoxin, and their effects may not always be fully RAGE dependent [32].
S100A8 and S100A9 regulate leukocyte migration and adhesion [33]. The S100A8/A9 complex has antimicrobial effects, transports arachidonic acid to endothelial cells, and activates expression of endothelial cell adhesion molecules [11, 34, 35]. Although the receptor for S100A8/A9 complex is still unknown, RAGE has been implicated in some circumstances [36]. Murine S100A8 stimulates proatherogenic activity, such as uptake of low-density lipoprotein (LDL), in macrophages. S100A8 is a key target of oxidation by peroxide, hypochlorite and nitric oxide [37, 38]. Furthermore, S100A9 and S100A12 are implicated in vascular damage, whereas sRAGE is associated with vascular protection in atherosclerosis [30, 39–41].
The relationship between S100 protein levels and vascular disease or risk factors in RA patients has not been examined to date. We measured serum levels of S100A8, S100A9 heterocomplexes, S100A12 and sRAGE in a previously characterized cohort of established RA patients to identify their possible relationship to joint and vascular damage and risk factors in RA patients [19]. We report associations of each protein with both joint and vascular disease and their risk factors.
Materials and methods
Subjects
The cohort of RA patients met the American College of Rheumatology (ACR) 1987 revised criteria for the classification of RA, and has been previously described [42]. These patients presented for a scheduled appointment over a 5-month period (July to November 2003) at our tertiary hospital rheumatology clinic, as described previously [19]. Patients completed a questionnaire detailing CV history, risk factors, treatment, and details of RA. Each patient was clinically evaluated, with chart review to confirm history, at least once. The study protocol was approved by the Princess Alexandra Hospital Research Ethics Committee. Healthy controls (n = 44) without RA or CV disease were recruited by advertisement. All patients and controls signed informed consent to participate. No prospective follow-up was carried out in this study.
Measurement of S100 proteins
The serum levels of S100A8, S100A9 and S100A12 levels were measured using in-house affinity-purified rabbit polyclonal sandwich ELISAs exactly as described for S100A12 [25]. Antibodies to S100A8 did not cross-react with S100A9 (and did not recognize S100A8/A9 complexes) or S100A12, anti-S100A9 detected free S100A9 and S100A9 as an S100A8/A9 complex; anti-S100A12 was immunoadsorbed with S100A8 and S100A9 [25] and did not cross-react with these when tested by ELISA or immunoblotting. Standard curves were constructed with the relevant recombinant S100 proteins.
Measurement of sRAGE
sRAGE levels in sera were determined by RAGE Immunoassay (R&D Systems, Minneapolis, MN, USA) in an ELISA format, with wells coated with murine anti-human RAGE mAb in which serum samples (50 μl/well, normally 1:2 v/v dilution) were incubated. A polyclonal capture antibody against the extracellular domain of RAGE was used for detection. The minimum detectable sRAGE concentration is 4.12 pg/ml according to the manufacturer, and the interassay coefficient of variation is < 8% [41].
Ascertainment of CV events and risk factors, and features of RA
To ascertain CV events, patients were asked for a history, dates and treatments of myocardial infarction, angina, stroke, transient ischemic attack or peripheral vascular disease, and these events were verified by medical record review. Although a number of patients had events prior to the diagnosis of RA, only those CV events that occurred after RA diagnosis were included in the current analysis. Patients with multiple events had only one event counted per person. Myocardial infarction was identified if subjects developed either of; (1) typical rise and fall of biochemical markers (troponin or creatine kinase-MB (CK-MB)) consistent with myocardial necrosis with at least one of the following (a) ischemic symptoms, (b) development of pathological Q waves on the electrocardiogram (ECG), (c) ECG changes indicative of ischemia (ST segment elevation or depression); (2) either new pathological Q waves on serial ECGs or pathological changes of healed or healing infarction [43]. Stroke or transient ischemic attack were identified if subjects had been admitted to the hospital with CT evidence of ischemic occlusion or with carotid endarterectomy, or the subject presented with stroke/transient ischemic attack (TIA) symptoms with significant plaque on the carotid ultrasound and neurological sequelae, with exclusion of subarachnoid hemorrhage and space occupying lesions. Peripheral vascular disease was confirmed if Doppler ultrasonography showed significant large vessel disease.
Cigarette smoking was assessed by questionnaire, which included details about past and present smoking habits, number of cigarettes smoked per day and smoking duration. History of diabetes mellitus was identified if subjects had been diagnosed by a physician, were taking anti-diabetic medications, or had an elevated fasting glucose at the time of the assessment. Family history of CV disease or cerebrovascular attack before age of 65 in first-degree relatives was determined by questionnaire. History was not included if a stroke was deemed hemorrhagic. Body mass index (BMI) was calculated as weight in kilograms divided by the square of the height in meters. Blood pressure was measured at the time of evaluation. History of hypercholesterolemia and hypertension were identified if the diagnoses were recorded in medical records by a physician, if patients were taking lipid-lowering or antihypertensive drugs, or if elevated blood pressure or fasting cholesterol levels were found at the time of the evaluation. The percentage risk of coronary heart disease over the next 10 years was estimated using the 'CVD Risk Calculator' based on the Framingham Study [44] for patients between 30 and 74 years of age and without a history of coronary heart disease. Metabolic syndrome (modified American Heart Association (AHA) standard [45]) was identified by the presence of three or more of these components: (1) BMI > 30; (2) fasting blood triglycerides ≥ 150 mg/dl; (3) blood high-density lipoprotein (HDL) cholesterol (men: < 40 mg/dl (1.03 mmol/l), women: < 50 mg/dl (1.3mmol/l)); (4) blood pressure ≥ 130/85 mmHg; and (5) fasting glucose ≥ 100 mg/dl.
Laboratory data collected at the time of clinical evaluation included fasting total cholesterol, LDL, HDL, very low-density lipoprotein (VLDL), triglycerides, LDL/HDL cholesterol ratio, glucose, creatinine, C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), anti-citrullinated peptide antibodies (ACPA) and rheumatoid factor (RF). A 12-lead ECG carried out within the previous 12 months was scored for evidence of Q waves to ascertain possible silent coronary disease. Creatinine clearance (CrCl) was estimated for each patient on the basis of serum creatinine (SCr), age (years), and ideal body weight (kg) using the Cockcroft and Gault method as follows: CrCl (ml/min) = [(140 - age)(ideal wt)]/833 × SCr (mmol/l) × 0.85 for females [46]. Hand radiographs carried out at the time of evaluation were scored for erosions and joint space narrowing using the modified Sharp score [47].
Genotyping
High resolution human leukocyte antigen (HLA)-DRB1 genotyping was carried out on buffy coat DNA using PCR and sequence-specific oligonucleotide probes. PCR-based restriction fragment length polymorphism (RFLP) analysis was used to delineate the RAGE Gly82Ser and protein tyrosine phosphatase, non-receptor type 22 (PTPN22) Cys1858Thr polymorphisms as described [15, 48]. Shared epitope was considered positive when at least one DRB1 allele was one of the RA susceptibility alleles, as previously described [49].
Statistical analysis
Data were analyzed using STATA 9.1 (StataCorp, College Station, TX, USA). The variables included age, sex, BMI, current and previous smoking status, RF, ACPA, history of CV events, fasting glucose, homocysteine, cholesterol and triglyceride, ESR, CRP, HDL, LDL, creatinine, CrCl, systolic and diastolic blood pressure, history of diabetes or elevated blood sugar level, history of hyperlipidemia or elevated cholesterol, HLA-DRB1 genotype, Sharp erosion score, Sharp joint space narrowing score, RAGE Gly82Ser polymorphism, history of hypertension or elevated blood pressure, metabolic syndrome (modified AHA standard), serum S100A9, S100A8, S100A12 and sRAGE. Before further analysis, each variable was examined for normal distribution by histogram and box plot. If a variable was not normally distributed, it was transformed (either logarithmic base e or square root transformation) before further analysis. Results are reported as mean ± standard deviation (SD).
Unpaired t tests compared the serum levels of S100A9, S100A8, S100A12 and sRAGE between RA patients and healthy controls. Simple linear regression analysis was used to evaluate the relationship between a variable and the serum concentration of sRAGE or S100 proteins. Variables with P < 0.1 using this method were then subjected to multiple linear regression (MLR) analysis. An interaction and residual analysis was also performed on the MLR data. P values < 0.05 (two-tailed) were considered statistically significant.
Results
Clinical features of the RA cohort
We studied 138 patients with RA (mean age 64.0 years, range 17 to 87 years) and 44 healthy controls (mean age 62 years, range 44 to 80 years) with neither RA nor CV disease. The RA patients were characterized for RA clinical variables, CV risk factors, and RA complications such as vasculitis, radiographic changes, and CV events (Table 1).
Increased serum concentrations of the S100 proteins, but not sRAGE, in patients with established RA
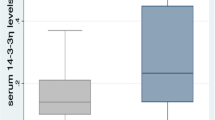
Serum levels of S100A9, S100A8 and S100A12 in patients with RA (n = 138) were increased relative to serum levels in healthy controls (n = 44, P < 0.001). The S100A9 levels detected in patient sera with an anti-S100A9 antibody that detected S100A9, and S100A9 complexed with S100A8, were some 100-fold lower than those reported in other studies [26, 27]. This could reflect differences in the specificity of the anti-calprotectin (an antibody generated against the S100A8/A9 complex) used by others; the anti-S100A9 used by us was generated against pure S100A9. In contrast to the S100 proteins, serum levels of sRAGE were not different (Figure 1a–d).

Serum sRAGE, S100A9, S100A8 and S100A12 levels in rheumatoid arthritis (RA) patients and healthy controls. Levels of S100A12 (a), S100A9 (b), S100A8 (c), and soluble receptor for advanced glycation end products (sRAGE) (d) were measured in serum of 138 patients with established RA and 44 healthy controls by ELISA. The horizontal line represents the mean value. *** P < 0.001, * P < 0.05.
Factors associated with serum levels of S100A9, S100A8 and S100A12 in patients with RA
We analyzed the cohort of 138 RA patients for associations between serum levels of S100A9, S100A8, S100A12 and sRAGE with RA clinical variables, CV risk factors, and with complications such as vasculitis, radiographic changes, and CV events. In simple linear regression analysis, we found that serum levels of S100A9 in RA patients were positively associated with the presence of the PTPN22 Cys1858Thr genetic polymorphism, serum levels of S100A12, and serum levels of S100A8 (Table 2, P < 0.05). Serum levels of S100A9 in MLR model analysis were positively associated with body mass index, and with serum levels of S100A8 and S100A12 (Table 2, P < 0.05).
In simple linear regression analysis, serum levels of S100A8 in RA patients were positively associated with ACPA, RF, carriage of HLA-DRB1*0401, RAGE 82S and of the RA-associated HLA-DR shared epitope [50], serum levels of S100A9, S100A12 and radiographic joint space narrowing. Conversely, serum levels of S100A8 were negatively associated with age and serum levels of sRAGE (Table 3, P < 0.05). The serum levels of S100A8 in MLR analysis were positively associated with RF, ACPA and serum levels of S100A9 (Table 3, P < 0.05).
In simple linear regression analysis, the serum levels of S100A12 in RA patients were positively associated with ACPA, RF, history of diabetes, and serum levels of S100A8 and S100A9 (Table 4, P < 0.05). The serum levels of S100A12 in RA patients in MLR analysis were positively associated with ACPA, a history of diabetes, and serum levels of S100A9 (Table 4, P < 0.05).
Factors associated with serum levels of sRAGE in patients with established RA
Simple linear regression analysis showed that serum sRAGE levels in RA patients were negatively associated with current smoking, family history of CV disease, history of vasculitis, diastolic blood pressure, RF, carriage of RAGE 82Ser, and serum levels of CRP and S100A8 (Table 5, P < 0.05). MLR analysis of sRAGE levels confirmed the negative associations with RAGE 82Ser, history of vasculitis, and with serum levels of CRP and HDL (Table 5, P < 0.05).
Discussion
We found associations of sRAGE and S100 proteins with clinical inflammatory factors, complications, and CV risk factors in established RA patients. S100 A8, A9 and A12 were all elevated in serum from patients with established RA relative to healthy controls, and their levels were correlated in RA patients. By contrast, serum sRAGE levels did not differ in healthy controls and patients with established RA on treatment. Although a previous study reported reduced levels of sRAGE in RA compared to healthy control sera [17], it seems likely that we observed similar levels because of good control of inflammation in the RA group. In support of this, elevated serum sRAGE levels were generally associated with a more favorable vascular risk profile in our RA cohort, and potentially associated with concomitant reduction in proinflammatory and/or pro-atherogenic RAGE ligand binding.
Despite the generally negative association of sRAGE with vascular risk factors, the single factor that showed a reverse trend in multivariate models was serum HDL. Serum HDL levels were also negatively associated with serum sRAGE among diabetic subjects with CV disease [51]. In contrast, no association with serum HDL and sRAGE was found in patients with essential hypertension [16]. In spite of its known protective role, HDL can become proinflammatory [52–54], and inflammatory HDL may increase the risk of atherosclerosis in SLE and RA patients [55, 56]. Moreover, HDL function, which is partly independent of HDL concentration, may be a more critical determinant of the atheroprotective capacity of HDL [57].
The positive association of S100A9 with S100A8 and S100A12 suggests that these proteins may be co-regulated in RA. This is supported by a previous study of S100 proteins in RA patients [58], and is in keeping with the high S100 gene expression profiles reported in RA [59]. Despite the positive associations between of S100A8, A9 and A12 levels, only S100A12 and S100A8 were associated with RA autoantibodies including ACPA and RF. Presence of ACPA or RF predicts a more aggressive RA disease course, including joint erosion and destruction [60]. Although S100A8/A9 from macrophages in RA patients amplified proinflammatory cytokine production in one study [11], the properties of serum S100A8/A9 are still debated. S100A8/A9 expression is seen in macrophages at the cartilage-pannus junction in RA, and expression of S100A8 significantly increased in macrophages in RA patients treated with high dose glucocorticoids compared to pre-treatment samples [61, 62]. Interestingly, glucocorticoids amplify LPS-induced S100A8 transcription in macrophages in an interleukin 10-dependent manner. Since we found S100A9 levels were associated with body mass index, it will be of interest to explore the relationship of this protein with endogenous or exogenous glucocorticoids. Recently, S100A9 or S100A8/A9 were reported to promote de-differentiation of dendritic cells and macrophages to myeloid suppressor cells in a tumor-bearing mouse model, suggesting anti-inflammatory effects of S100A9 which may reduce antigen-specific priming, for example, of cytotoxic T cell responses [63]. In support of an anti-inflammatory role for S100A9, S100A8 induced TNF in murine bone marrow cells through TLR4 signaling, and S100A9 negated this activity [64]. We found S100A9 to be associated with dystrophic calcification [39], which may be of relevance to atherosclerotic disease, and warrants future investigation in RA. Thus, the S100A8/A9 complex might have anti-inflammatory properties, or may be related to repair function in damaged or inflamed joints and vessels. It is also plausible that S100A8/A9 has variable effects depending on the presence of other disease factors or treatments. Finally, our assay measured S100A9, whether monomeric or heterocomplexed with S100A8. The ratio of S100A8:A9 may also play a role, given that the heterocomplex can have functions distinct from each protein alone.
In patients with Kawasaki disease, or with chronic hyperglycemia, serum levels of S100A12 were inversely associated with serum levels of sRAGE [65, 66]. Although the inverse correlation between sRAGE and S100A12 did not achieve statistical significance in the current study, the associations we found suggest opposing effects on RA severity. S100A12 has potent inflammatory effects. In chronic inflammatory arthritis, S100A12 is expressed by infiltrating granulocytes and by synovial macrophages, is a potent monocyte chemoattractant and activates mast cells to sequester them in inflammatory lesions [25, 29].
Our analysis indicates that sRAGE and S100 proteins are associated not only with RA inflammation and autoantibody production, but also with the recruitment of classical vascular risk factors to end-organ damage. This association with vascular risk supports previous reports of low sRAGE and high S100A8/A9 and S100A12 levels in patients with type 1 and type 2 diabetes, and essential hypertension [16, 66–68]. These data support evidence from clinical studies of atherosclerosis, suggesting that the roles of classical risk factors and inflammation are difficult to separate in RA [69]. As we observed here, increasing sRAGE levels are associated with a favorable vascular risk profile, potentially associated with concomitant reduction in proinflammatory and/or pro-atherogenic RAGE ligand binding [70, 71].
Finally, we observed a novel association of low sRAGE levels with presence of the RAGE 82Ser polymorphism, which is found more frequently in RA patients [15, 19]. It is conceivable that this, or other linked polymorphisms in the RAGE gene affect splicing of the C-truncated, endogenously secreted form of the receptor, or susceptibility to cell surface RAGE cleavage by matrix metalloproteinases [72], thus altering the ratio of soluble to membrane RAGE.
Several studies have been published which demonstrate the role of sRAGE, S100A8/A9 and S100A12 in the long-term development of vascular disease. These include a negative association between sRAGE levels and coronary artery disease in non-diabetic men [41], prediction of unstable plaque by S100A8/A9 levels in acute coronary syndromes [73], and of accelerated atherosclerosis by high levels of S100A12 in hemodialysis patients [74]. However, this is the first time such an association has been shown with CVD and RA.
Conclusions
sRAGE and S100 proteins were associated with RA inflammatory factors and autoantibody production, and with the recruitment of classical vascular risk factors to end-organ damage. Consistent with its role as a RAGE decoy molecule, sRAGE had opposing effects to S100A12 and S100A8 in RA. Our data suggest that RAGE may mediate a key pathway coordinating conventional risk factors in the inflammatory RA setting for co-development of joint and vascular disease. Prospective studies will be of interest to determine the utility of these proteins as prognostic biomarkers of joint and vascular damage.
Abbreviations
- ACPA:
-
anti-citrullinated peptide antibodies
- ACR:
-
American College of Rheumatology
- AGE:
-
advanced glycation end product
- BMI:
-
body-mass index
- CrCl:
-
creatinine clearance
- CRP:
-
C-reactive protein
- CT:
-
computed tomography
- CV:
-
cardiovascular
- ECG:
-
electrocardiogram
- ESR:
-
erythrocyte sedimentation rate
- HDL:
-
high-density lipoprotein
- HMGB1:
-
high mobility group box chromosomal protein
- HR:
-
hazard ratio
- LDL:
-
low density lipoprotein
- MI:
-
myocardial infarction
- PCR:
-
polymerase chain reaction
- RA:
-
rheumatoid arthritis
- RAGE:
-
receptor for advanced glycation end products
- RF:
-
rheumatoid factor
- SCr:
-
serum creatinine
- TG:
-
triglyceride
- TIA:
-
transient ischemic attack
- TNF:
-
tumor necrosis factor
- VLDL:
-
very-low-density lipoprotein.
References
Carroll L, Hannawi S, Marwick T, Thomas R: Rheumatoid arthritis: links with cardiovascular disease and the receptor for advanced glycation end products. Wien Med Wochenschr. 2006, 156: 42-52. 10.1007/s10354-005-0242-9.
van Beijnum JR, Buurman WA, Griffioen AW: Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis. 2008, 11: 91-99. 10.1007/s10456-008-9093-5.
Sunahori K, Yamamura M, Yamana J, Takasugi K, Kawashima M, Makino H: Increased expression of receptor for advanced glycation end products by synovial tissue macrophages in rheumatoid arthritis. Arthritis Rheum. 2006, 54: 97-104. 10.1002/art.21524.
Mohamed AK, Bierhaus A, Schiekofer S, Tritschler H, Ziegler R, Nawroth PP: The role of oxidative stress and NF-kappaB activation in late diabetic complications. Biofactors. 1999, 10: 157-167. 10.1002/biof.5520100211.
Bucciarelli LG, Wendt T, Rong L, Lalla E, Hofmann MA, Goova MT, Taguchi A, Yan SF, Yan SD, Stern DM, Schmidt AM: RAGE is a multiligand receptor of the immunoglobulin superfamily: implications for homeostasis and chronic disease. Cell Mol Life Sci. 2002, 59: 1117-1128. 10.1007/s00018-002-8491-x.
Taniguchi N, Kawahara K, Yone K, Hashiguchi T, Yamakuchi M, Goto M, Inoue K, Yamada S, Ijiri K, Matsunaga S, Nakajima T, Komiya S, Maruyama I: High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003, 48: 971-981. 10.1002/art.10859.
Batliwalla FM, Baechler EC, Xiao X, Li W, Balasubramanian S, Khalili H, Damle A, Ortmann WA, Perrone A, Kantor AB, Gulko PS, Kern M, Furie R, Behrens TW, Gregersen PK: Peripheral blood gene expression profiling in rheumatoid arthritis. Genes Immun. 2005, 6: 388-397. 10.1038/sj.gene.6364209.
Liao H, Wu J, Kuhn E, Chin W, Chang B, Jones MD, O'Neil S, Clauser KR, Karl J, Hasler F, Roubenoff R, Zolg W, Guild BC: Use of mass spectrometry to identify protein biomarkers of disease severity in the synovial fluid and serum of patients with rheumatoid arthritis. Arthritis Rheum. 2004, 50: 3792-3803. 10.1002/art.20720.
Edwards CJ, Feldman JL, Beech J, Shields KM, Stover JA, Trepicchio WL, Larsen G, Foxwell BM, Brennan FM, Feldmann M, Pittman DD: Molecular profile of peripheral blood mononuclear cells from patients with rheumatoid arthritis. Mol Med. 2007, 13: 40-58. 10.2119/2006-00056.Edwards.
Foell D, Kane D, Bresnihan B, Vogl T, Nacken W, Sorg C, Fitzgerald O, Roth J: Expression of the pro-inflammatory protein S100A12 (EN-RAGE) in rheumatoid and psoriatic arthritis. Rheumatology (Oxford). 2003, 42: 1383-1389. 10.1093/rheumatology/keg385.
Sunahori K, Yamamura M, Yamana J, Takasugi K, Kawashima M, Yamamoto H, Chazin WJ, Nakatani Y, Yui S, Makino H: The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res Ther. 2006, 8: R69-10.1186/ar1939.
Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, Yasui K, Takeuchi M, Makita Z, Takasawa S, Okamoto H, Watanabe T, Yamamoto H: Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J. 2003, 370: 1097-1109. 10.1042/BJ20021371.
Pullerits R, Brisslert M, Jonsson IM, Tarkowski A: Soluble receptor for advanced glycation end products triggers a proinflammatory cytokine cascade via beta2 integrin Mac-1. Arthritis Rheum. 2006, 54: 3898-3907. 10.1002/art.22217.
Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Grone HJ, Kurschus FC, Schmidt AM, Yan SD, Martin E, Schleicher E, Stern DM, Hammerling GG, Nawroth PP, Arnold B: Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004, 113: 1641-1650.
Hofmann MA, Drury S, Hudson BI, Gleason MR, Qu W, Lu Y, Lalla E, Chitnis S, Monteiro J, Stickland MH, Bucciarelli LG, Moser B, Moxley G, Itescu S, Grant PJ, Gregersen PK, Stern DM, Schmidt AM: RAGE and arthritis: the G82S polymorphism amplifies the inflammatory response. Genes Immun. 2002, 3: 123-135. 10.1038/sj.gene.6363861.
Geroldi D, Falcone C, Emanuele E, D'Angelo A, Calcagnino M, Buzzi MP, Scioli GA, Fogari R: Decreased plasma levels of soluble receptor for advanced glycation end-products in patients with essential hypertension. J Hypertens. 2005, 23: 1725-1729. 10.1097/01.hjh.0000177535.45785.64.
Pullerits R, Bokarewa M, Dahlberg L, Tarkowski A: Decreased levels of soluble receptor for advanced glycation end products in patients with rheumatoid arthritis indicating deficient inflammatory control. Arthritis Res Ther. 2005, 7: R817-824. 10.1186/ar1749.
Koyama H, Shoji T, Yokoyama H, Motoyama K, Mori K, Fukumoto S, Emoto M, Shoji T, Tamei H, Matsuki H, Sakurai S, Yamamoto Y, Yonekura H, Watanabe T, Yamamoto H, Nishizawa Y: Plasma level of endogenous secretory RAGE is associated with components of the metabolic syndrome and atherosclerosis. Arterioscler Thromb Vasc Biol. 2005, 25: 2587-2593. 10.1161/01.ATV.0000190660.32863.cd.
Carroll L, Frazer IH, Turner M, Marwick TH, Thomas R: Receptor for advanced glycation end products glycine 82 serine polymorphism and risk of cardiovascular events in rheumatoid arthritis. Arthritis Res Ther. 2007, 9: R39-10.1186/ar2175.
Ravasi T, Hsu K, Goyette J, Schroder K, Yang Z, Rahimi F, Miranda LP, Alewood PF, Hume DA, Geczy C: Probing the S100 protein family through genomic and functional analysis. Genomics. 2004, 84: 10-22. 10.1016/j.ygeno.2004.02.002.
Foell D, Frosch M, Sorg C, Roth J: Phagocyte-specific calcium-binding S100 proteins as clinical laboratory markers of inflammation. Clin Chim Acta. 2004, 344: 37-51. 10.1016/j.cccn.2004.02.023.
Renaud W, Merten M, Figarella C: Increased coexpression of CFTR and S100 calcium binding proteins MRP8 and MRP14 mRNAs in cystic fibrosis human tracheal gland cells. Biochem Biophys Res Commun. 1994, 201: 1518-1525. 10.1006/bbrc.1994.1876.
Zwadlo G, Bruggen J, Gerhards G, Schlegel R, Sorg C: Two calcium-binding proteins associated with specific stages of myeloid cell differentiation are expressed by subsets of macrophages in inflammatory tissues. Clin Exp Immunol. 1988, 72: 510-515.
Yui S, Nakatani Y, Mikami M: Calprotectin (S100A8/S100A9), an inflammatory protein complex from neutrophils with a broad apoptosis-inducing activity. Biol Pharm Bull. 2003, 26: 753-760. 10.1248/bpb.26.753.
Yang Z, Tao T, Raftery MJ, Youssef P, Di Girolamo N, Geczy CL: Proinflammatory properties of the human S100 protein S100A12. J Leukoc Biol. 2001, 69: 986-994.
Hammer HB, Odegard S, Fagerhol MK, Landewe R, Heijde van der D, Uhlig T, Mowinckel P, Kvien TK: Calprotectin (a major leucocyte protein) is strongly and independently correlated with joint inflammation and damage in rheumatoid arthritis. Ann Rheum Dis. 2007, 66: 1093-1097. 10.1136/ard.2006.064741.
Madland TM, Hordvik M, Haga HJ, Jonsson R, Brun JG: Leukocyte protein calprotectin and outcome in rheumatoid arthritis. A longitudinal study. Scand J Rheumatol. 2002, 31: 351-354. 10.1080/030097402320817077.
Brun JG, Haga HJ, Boe E, Kallay I, Lekven C, Berntzen HB, Fagerhol MK: Calprotectin in patients with rheumatoid arthritis: relation to clinical and laboratory variables of disease activity. J Rheumatol. 1992, 19: 859-862.
Yang Z, Yan WX, Cai H, Tedla N, Armishaw C, Di Girolamo N, Wang HW, Hampartzoumian T, Simpson JL, Gibson PG, Hunt J, Hart P, Hughes JM, Perry MA, Alewood PF, Geczy CL: S100A12 provokes mast cell activation: a potential amplification pathway in asthma and innate immunity. J Allergy Clin Immunol. 2007, 119: 106-114. 10.1016/j.jaci.2006.08.021.
Mori Y, Kosaki A, Kishimoto N, Kimura T, Iida K, Fukui M, Nakajima F, Nagahara M, Urakami M, Iwasaka T, Matsubara H: Increased plasma S100A12 (EN-RAGE) levels in hemodialysis patients with atherosclerosis. Am J Nephrol. 2008, 29: 18-24. 10.1159/000148646.
Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM: RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999, 97: 889-901. 10.1016/S0092-8674(00)80801-6.
Valencia JV, Mone M, Koehne C, Rediske J, Hughes TE: Binding of receptor for advanced glycation end products (RAGE) ligands is not sufficient to induce inflammatory signals: lack of activity of endotoxin-free albumin-derived advanced glycation end products. Diabetologia. 2004, 47: 844-852. 10.1007/s00125-004-1392-9.
Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA: Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol. 2003, 170: 3233-3242.
Okutomi T, Tanaka T, Yui S, Mikami M, Yamazaki M, Abe S, Yamaguchi H: Anti-Candida activity of calprotectin in combination with neutrophils or lactoferrin. Microbiol Immunol. 1998, 42: 789-793.
Kerkhoff C, Eue I, Sorg C: The regulatory role of MRP8 (S100A8) and MRP14 (S100A9) in the transendothelial migration of human leukocytes. Pathobiology. 1999, 67: 230-232. 10.1159/000028098.
Ehlermann P, Eggers K, Bierhaus A, Most P, Weichenhan D, Greten J, Nawroth PP, Katus HA, Remppis A: Increased proinflammatory endothelial response to S100A8/A9 after preactivation through advanced glycation end products. Cardiovasc Diabetol. 2006, 5: 6-10.1186/1475-2840-5-6.
Harrison CA, Raftery MJ, Walsh J, Alewood P, Iismaa SE, Thliveris S, Geczy CL: Oxidation regulates the inflammatory properties of the murine S100 protein S100A8. J Biol Chem. 1999, 274: 8561-8569. 10.1074/jbc.274.13.8561.
Lim SY, Raftery M, Cai H, Hsu K, Yan WX, Hseih HL, Watts RN, Richardson D, Thomas S, Perry M, Geczy CL: S-nitrosylated S100A8: novel anti-inflammatory properties. J Immunol. 2008, 181: 5627-5636.
McCormick MM, Rahimi F, Bobryshev YV, Gaus K, Zreiqat H, Cai H, Lord RS, Geczy CL: S100A8 and S100A9 in human arterial wall. Implications for atherogenesis. J Biol Chem. 2005, 280: 41521-41529. 10.1074/jbc.M509442200.
Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, Kalea AZ, Lu Y, Rosario RH, Oruganti S, Nikolla Z, Belov D, Lalla E, Ramasamy R, Yan SF, Schmidt AM: Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE-/- mice. J Clin Invest. 2008, 118: 183-194. 10.1172/JCI32703.
Falcone C, Emanuele E, D'Angelo A, Buzzi MP, Belvito C, Cuccia M, Geroldi D: Plasma levels of soluble receptor for advanced glycation end products and coronary artery disease in nondiabetic men. Arterioscler Thromb Vasc Biol. 2005, 25: 1032-1037. 10.1161/01.ATV.0000160342.20342.00.
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, Medsger TA, Mitchell DM, Neustadt DH, Pinals RS, Schaller JG, Sharp JT, Wilder RL, Hunder GG: The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31: 315-324. 10.1002/art.1780310302.
Alpert JS, Thygesen K, Antman E, Bassand JP: Myocardial infarction redefined – a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000, 36: 959-969. 10.1016/S0735-1097(00)00804-4.
Wilson PW, D'Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB: Prediction of coronary heart disease using risk factor categories. Circulation. 1998, 97: 1837-1847.
Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC, Spertus JA, Costa F: Diagnosis and management of the metabolic syndrome. An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Executive summary. Cardiol Rev. 2005, 13: 322-327.
Cockcroft DW, Gault MH: Prediction of creatinine clearance from serum creatinine. Nephron. 1976, 16: 31-41. 10.1159/000180580.
Heijde van der D: How to read radiographs according to the Sharp/van der Heijde method. J Rheumatol. 2000, 27: 261-263.
Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M, Eisenbarth GS, Comings D, Mustelin T: A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. 2004, 36: 337-338. 10.1038/ng1323.
Gregersen PK, Silver J, Winchester RJ: The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30: 1205-1213. 10.1002/art.1780301102.
Gregersen PK, Silver J, Winchester RJ: The shared epitope hypothesis: an approach to understanding the molecular genetics of suseptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30: 1205-1213. 10.1002/art.1780301102.
Nakamura K, Yamagishi SI, Adachi H, Kurita-Nakamura Y, Matsui T, Yoshida T, Sato A, Imaizumi T: Elevation of soluble form of receptor for advanced glycation end products (sRAGE) in diabetic subjects with coronary artery disease. Diabetes Metab Res Rev. 2007, 23: 368-371. 10.1002/dmrr.690.
Ansell BJ, Navab M, Hama S, Kamranpour N, Fonarow G, Hough G, Rahmani S, Mottahedeh R, Dave R, Reddy ST, Fogelman AM: Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation. 2003, 108: 2751-2756. 10.1161/01.CIR.0000103624.14436.4B.
Van Lenten BJ, Hama SY, de Beer FC, Stafforini DM, McIntyre TM, Prescott SM, La Du BN, Fogelman AM, Navab M: Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J Clin Invest. 1995, 96: 2758-2767. 10.1172/JCI118345.
Navab M, Berliner JA, Subbanagounder G, Hama S, Lusis AJ, Castellani LW, Reddy S, Shih D, Shi W, Watson AD, Van Lenten BJ, Vora D, Fogelman AM: HDL and the inflammatory response induced by LDL-derived oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2001, 21: 481-488.
McMahon M, Grossman J, FitzGerald J, Dahlin-Lee E, Wallace DJ, Thong BY, Badsha H, Kalunian K, Charles C, Navab M, Fogelman AM, Hahn BH: Proinflammatory high-density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 2006, 54: 2541-2549. 10.1002/art.21976.
Eue I, Pietz B, Storck J, Klempt M, Sorg C: Transendothelial migration of 27E10+ human monocytes. Int Immunol. 2000, 12: 1593-1604. 10.1093/intimm/12.11.1593.
Sviridov D, Mukhamedova N, Remaly AT, Chin-Dusting J, Nestel P: Antiatherogenic functionality of high density lipoprotein: how much versus how good. J Atheroscler Thromb. 2008, 15: 52-62.
de Seny D, Fillet M, Ribbens C, Maree R, Meuwis MA, Lutteri L, Chapelle JP, Wehenkel L, Louis E, Merville MP, Malaise M: Monomeric calgranulins measured by SELDI-TOF mass spectrometry and calprotectin measured by ELISA as biomarkers in arthritis. Clin Chem. 2008, 54: 1066-1075. 10.1373/clinchem.2007.099549.
Bovin LF, Rieneck K, Workman C, Nielsen H, Sorensen SF, Skjodt H, Florescu A, Brunak S, Bendtzen K: Blood cell gene expression profiling in rheumatoid arthritis. Discriminative genes and effect of rheumatoid factor. Immunol Lett. 2004, 93: 217-226. 10.1016/j.imlet.2004.03.018.
Vencovsky J, Machacek S, Sedova L, Kafkova J, Gatterova J, Pesakova V, Ruzickova S: Autoantibodies can be prognostic markers of an erosive disease in early rheumatoid arthritis. Ann Rheum Dis. 2003, 62: 427-430. 10.1136/ard.62.5.427.
Youssef P, Roth J, Frosch M, Costello P, Fitzgerald O, Sorg C, Bresnihan B: Expression of myeloid related proteins (MRP) 8 and 14 and the MRP8/14 heterodimer in rheumatoid arthritis synovial membrane. J Rheumatol. 1999, 26: 2523-2528.
Hsu K, Passey RJ, Endoh Y, Rahimi F, Youssef P, Yen T, Geczy CL: Regulation of S100A8 by glucocorticoids. J Immunol. 2005, 174: 2318-2326.
Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI: Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008, 205: 2235-2249. 10.1084/jem.20080132.
Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, Poll van der T, Sorg C, Roth J: Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007, 13: 1042-1049. 10.1038/nm1638.
Wittkowski H, Hirono K, Ichida F, Vogl T, Ye F, Yanlin X, Saito K, Uese K, Miyawaki T, Viemann D, Roth J, Foell D: Acute Kawasaki disease is associated with reverse regulation of soluble receptor for advance glycation end products and its proinflammatory ligand S100A12. Arthritis Rheum. 2007, 56: 4174-4181. 10.1002/art.23042.
Basta G, Sironi AM, Lazzerini G, Del Turco S, Buzzigoli E, Casolaro A, Natali A, Ferrannini E, Gastaldelli A: Circulating soluble receptor for advanced glycation end products is inversely associated with glycemic control and S100A12 protein. J Clin Endocrinol Metab. 2006, 91: 4628-4634. 10.1210/jc.2005-2559.
Kosaki A, Hasegawa T, Kimura T, Iida K, Hitomi J, Matsubara H, Mori Y, Okigaki M, Toyoda N, Masaki H, Inoue-Shibata M, Nishikawa M, Iwasaka T: Increased plasma S100A12 (EN-RAGE) levels in patients with type 2 diabetes. J Clin Endocrinol Metab. 2004, 89: 5423-5428. 10.1210/jc.2003-032223.
Bouma G, Lam-Tse WK, Wierenga-Wolf AF, Drexhage HA, Versnel MA: Increased serum levels of MRP-8/14 in type 1 diabetes induce an increased expression of CD11b and an enhanced adhesion of circulating monocytes to fibronectin. Diabetes. 2004, 53: 1979-1986. 10.2337/diabetes.53.8.1979.
Gerli R, Goodson NJ: Cardiovascular involvement in rheumatoid arthritis. Lupus. 2005, 14: 679-682. 10.1191/0961203305lu2199oa.
Solomon DH, Curhan GC, Rimm EB, Cannuscio CC, Karlson EW: Cardiovascular risk factors in women with and without rheumatoid arthritis. Arthritis Rheum. 2004, 50: 3444-3449. 10.1002/art.20636.
Ridker PM: High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation. 2001, 103: 1813-1818.
Koyama H, Yamamoto H, Nishizawa Y: RAGE and soluble RAGE: potential therapeutic targets for cardiovascular diseases. Mol Med. 2007, 13: 625-635. 10.2119/2007-00087.Koyama.
Altwegg LA, Neidhart M, Hersberger M, Muller S, Eberli FR, Corti R, Roffi M, Sutsch G, Gay S, von Eckardstein A, Wischnewsky MB, Luscher TF, Maier W: Myeloid-related protein 8/14 complex is released by monocytes and granulocytes at the site of coronary occlusion: a novel, early, and sensitive marker of acute coronary syndromes. Eur Heart J. 2007, 28: 941-948. 10.1093/eurheartj/ehm078.
Mori Y, Kosaki A, Kishimoto N, Kimura T, Iida K, Fukui M, Nakajima F, Nagahara M, Urakami M, Iwasaka T, Matsubara H: Increased plasma S100A12 (EN-RAGE) levels in hemodialysis patients with atherosclerosis. Am J Nephrol. 2009, 29: 18-24. 10.1159/000148646.
Acknowledgements
Supported by grants from the PA Hospital Foundation, Australian Rotary Health Research Fund, the National Health and Medical Research Council of Australia, and an Australian Postgraduate Scholarship. Ranjeny Thomas is supported by Arthritis Queensland. We thank Joyce Cotterill for clinical support and Dr. Mark Jones for advice on statistical modeling.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YC, CG, MB and RT were involved in conception, design, acquisition, analysis and interpretation of data. WY and CG carried out S100A8, S100A9 and S00A12 assays. YC, CG, MB and RT wrote the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Chen, YS., Yan, W., Geczy, C.L. et al. Serum levels of soluble receptor for advanced glycation end products and of S100 proteins are associated with inflammatory, autoantibody, and classical risk markers of joint and vascular damage in rheumatoid arthritis. Arthritis Res Ther 11, R39 (2009). https://doi.org/10.1186/ar2645
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar2645