
Abstract
Background
Metastasis of breast cancer to distal organs is fatal. However, few studies have identified biomarkers that are associated with distant metastatic breast cancer. Furthermore, the inability of current biomarkers, such as HER2, ER, and PR, to differentiate between distant and nondistant metastatic breast cancers accurately has necessitated the development of novel biomarker candidates.
Methods
An integrated proteomics approach that combined filter-aided sample preparation, tandem mass tag labeling (TMT), high pH fractionation, and high-resolution MS was applied to acquire in-depth proteomic data from FFPE distant metastatic breast cancer tissues. A bioinformatics analysis was performed with regard to gene ontology and signaling pathways using differentially expressed proteins (DEPs) to examine the molecular characteristics of distant metastatic breast cancer. In addition, real-time polymerase chain reaction (RT-PCR) and invasion/migration assays were performed to validate the differential regulation and function of our protein targets.
Results
A total of 9441 and 8746 proteins were identified from the pooled and individual sample sets, respectively. Based on our criteria, TUBB2A was selected as a novel biomarker candidate. The metastatic activities of TUBB2A were subsequently validated. In our bioinformatics analysis using DEPs, we characterized the overall molecular features of distant metastasis and measured differences in the molecular functions of distant metastatic breast cancer between breast cancer subtypes.
Conclusions
Our report is the first study to examine the distant metastatic breast cancer proteome using FFPE tissues. The depth of our dataset allowed us to discover a novel biomarker candidate and a proteomic characteristics of distant metastatic breast cancer. Distinct molecular features of various breast cancer subtypes were also established. Our proteomic data constitute a valuable resource for research on distant metastatic breast cancer.
Similar content being viewed by others
Background
Breast cancer is one of the most prevalent and lethal cancers in women worldwide [1]. In particular, its annual incidence—currently 17 million cases—is increasing at an alarming rate [2, 3]. There are approximately 232,000 new cases of invasive breast cancer each year in the US, and approximately 40,000 women die each year from the disease; furthermore, roughly 90% of these deaths are caused by the most malignant form of breast cancer: distant metastatic breast cancer [2, 4]. Distant metastatic breast cancer, which preferentially metastasizes to distal organs, such as the bone, liver, lung, and brain, has a poor prognosis [5, 6]. In addition, this type of breast cancer causes various complications at the affected sites, such as pericardial effusion, pleural effusion, bone fracture, hypercalcemia, and red blood cell anemia, which worsens survival outcomes [7,8,9].
Distant metastatic breast cancer is assessed, based on various factors, such as tumor size, lymphovascular invasion, histological grade, nodal involvement, and hormone receptor status—all of which are independent risk factors for distant metastatic breast cancer [10,11,12,13]. Among these factors, breast cancer molecular subtypes are associated with various patterns of distant metastatic spread and related to differences in survival outcomes [10, 14]. For instance, the most widely known molecular subtypes, such as the luminal A, luminal B, HER2, and basal-like (triple-negative) groups, have site-specific, cumulative metastatic incidence rates, demonstrating substantial differences in the distant metastatic behavior of and overall survival between breast cancer subtypes [10].
Although various risks and molecular characteristics of distant metastatic breast cancer have been established, the prediction and diagnosis of distant metastasis in breast cancer with molecular biomarkers remain largely unexamined [4,5,6, 10,11,12,13]. Thus, characterizing the molecular signatures that are associated with distant metastasis using omics-based approaches, such as genomics, transcriptomics, and proteomics, might identify previously overlooked biomarker candidates.
Many genomic or transcriptomic studies have examined the molecular characteristics of distant metastatic breast cancer—for instance, genes that are associated with lung, brain, and bone metastasis from breast tumor [15,16,17,18, 20, 21]. In addition, genetic signatures that predict distant metastasis in breast cancer have been established through genomic profiling [19]. However, given the relatively low correlation between gene expression and protein expression, it is difficult to assume that the tendencies in genomic data will translate fully to proteomic data without verification [22, 23]. Similarly, considering that transcriptomic and proteomic data have a moderate correlation, the molecular characteristics of the transcriptome could not perfectly represent those of the proteome [24,25,26]. In the case of breast cancer, recent large dataset-based proteomic approaches have reported an intermediate correlation between the breast tumor proteome and the corresponding transcript levels [27, 28]. Furthermore, a recent report has described a low correlation between proteomes and transcriptomes in human breast cancer tissues, suggesting that a proteomic approach to human BC tissues could complement a transcriptomic method [29].
Although proteomic studies have been performed for various diseases, including breast cancer, none has investigated the overall characteristics of distant metastatic breast cancer [29,30,31,32,33,34,35,36,37, 44]. Proteomic research is expected to provide greater insight into the pathogenesis of distant metastatic breast cancer, generating novel information about the molecular features of distant metastasis—for example, by discovering novel protein biomarkers for the prediction or diagnosis of distant metastatic breast cancer. Thus, an in-depth proteomic analysis is important for yielding valuable resources in distant metastatic breast cancer—data that have not been found in genomic and transcriptomic analyses.
Recent advances in mass spectrometry (MS)-based proteomics have accelerated the development of high-throughput techniques for proteomic quantification [38, 39]. In addition, a tandem mass tag (TMT)-based strategy has facilitated relative protein quantification by comparing the reporter ion intensities that are obtained by MS/MS. Because this approach can quantify thousands of proteins precisely with high sensitivity, TMT-based techniques have been used widely to generate substantial datasets [40,41,42,43]. With a 6-plex TMT quantification technique, in combination with high-resolution MS, we constructed an in-depth proteomic map of distant metastatic breast cancer.
In this study, we hypothesized that in-depth proteomic data would supply important proteins to profile the molecular signatures of distant metastatic breast cancer. Using our proteomic techniques, we identified by far the largest number of proteins from FFPE distant and nondistant metastatic breast cancer tissues. Furthermore, we determined important protein targets to validate distant metastatic potential of breast cancer. The function of these targets was determined using several approaches, including RT-PCR and invasion/migration assays.
Through our criteria to narrow down the important proteins, we discovered a novel protein biomarker candidate differentially expressed in distant metastatic breast cancer. Furthermore, we examined the distinct biological functions of distant metastatic breast cancer between molecular subtypes. In summary, we have proposed the first protein biomarker candidate that potentially be able to distinguish distant metastasis, derived from primary breast tumors using FFPE tissue samples. We performed the initial examination of its molecular features at the protein level, providing insights into the pathogenesis of distant metastatic breast cancer.
Methods
Materials and reagents
Sodium dodecyl sulfate (SDS) and Trizma base were purchased from USB (Cleveland, OH), and sequencing-grade modified trypsin was purchased from Promega Corporation (Madison, WI). Dithiothreitol (DTT) and urea were obtained from AMRESCO (Solon, OH). POROS20 R2 beads were purchased from Applied Biosystems (Foster City, CA). High-purity (> 97%) mass spectrometry (MS)-grade ovalbumin was obtained from Protea (Morgantown, WV), and HLB OASIS columns were purchased from Waters (Milford, MA). Tandem mass tag (TMT) 6-plex isobaric reagents; a bicinchoninic acid (BCA) assay kit; LC/MS-grade solvents, such as acetone, acetonitrile (ACN), and water; and reducing agents, such as tris (2-carboxyethyl) phosphine (TCEP), were purchased from Thermo Fisher Scientific (Waltham, MA). All other reagents, if not noted otherwise, were obtained from Sigma-Aldrich (St. Louis, MO).
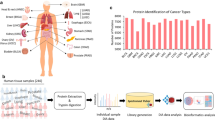
Sample selection
All clinical samples were collected from the Department of Pathology, Seoul National University Hospital (Seoul, South Korea). The distant metastasis group (dis-meta) was defined as patients who developed distant metastasis with or without lymph node metastasis. The nondistant metastasis group (nondis-meta) comprised patients who were not diagnosed as having distant metastasis with or without lymph node metastasis. All clinical specimens were collected from 18 patients with dis-meta and 18 patients with nondis-meta. The 18 patients in each group were divided into 3 breast cancer molecular subtypes (HER2, TNBC, and luminal). Tissue samples for distant and nondistant metastatic breast cancer were derived from the primary breast tumor. Clinical information on the patient samples is detailed in Additional file 1: Table S1. All patients consented to participate in the study per institutional review board guidelines (IRB No.1612-011-811).
Sample preparation of FFPE tissues for proteomic analysis
FFPE sections (10 μm) were incubated twice in xylene (Sigma-Aldrich, St. Louis, MO)—once each for 5 and 2 min—and then twice in 100% (v/v) ethanol for 90 s. The sections were then hydrated in 75% (v/v) ethanol for 90 s and distilled water for 90 s [33, 44]. Next, the tissues were scraped off the glass slides into microfuge tubes, after which protein extraction buffer (4% SDS; 0.3 M Tris, pH 8.5; 2 mM TCEP) was added. Following sonication, the samples were incubated at 100 °C for 2.5 h. Protein concentrations were measured using a bicinchoninic acid (BCA) reducing agent-compatible kit (Thermo Fisher Scientific, Waltham, MA).
Protein digestion was performed using a combination of acetone precipitation and filter-aided sample preparation (FASP) [45, 46]. Before the digestion step, 250 μg of extracted protein was precipitated with cold acetone at a buffer: acetone ratio of 1:5 and incubated at – 20 °C for 18 h. Next, the pellet was washed with 500 μl cold acetone, centrifuged at 15,000 rpm for 15 min, and air-dried for 1.5 h. The proteins that had precipitated were dissolved in 35 μl denaturation buffer (4% SDS and 100 mM DTT in 0.3 M TEAB pH 8.5).
After being heated at 100 °C for 35 min, the denatured proteins were loaded onto 30 kDa spin filters (Merck Millipore, Darmstadt, Germany). The buffer was exchanged 3 times with UREA solution (8 M UREA in 0.1 M TEAB, pH 8.5). After SDS was removed, cysteine residues were treated with alkylation buffer (50 mM IAA, 8 M UREA in 0.1 M TEAB, pH 8.5) for 1 h at room temperature in the dark. UREA buffer was exchanged with TEAB buffer (40 mM TEAB, pH 8.5). The proteins were digested with trypsin (enzyme-to-substrate ratio [w/w] of 1:50) and 4% ACN at 37 °C for 18 h. The digested peptides were eluted by centrifugation, and their concentrations were measured, based on the fluorescence emission of tryptophan at 350 nm, using an excitation wavelength of 295 nm [47]. The external standard sample, ovalbumin, was digested in the same manner.
6-Plex tandem mass tag (TMT) labeling
Because the number of samples exceeded that of the TMT channels, 2 independent TMT 6-plex labeling experiments—using a pooled sample set and individual sample set—were performed. Each TMT experiment consisted of 18 samples that were divided into 2 groups (dis-meta and non dis-meta). For the pooled sample set, equal amounts of 3 samples with identical molecular subtypes in each group were pooled, generating 6 pooled samples. Next, they were labeled with TMT 6-plex: 126-non dis-meta (HER2), 127-non dis-meta (TNBC), 128-non dis-meta (Luminal), 129-dis-meta (HER2), 130-dis-meta (TNBC), and 131-dis-meta (Luminal). At this step, several technical replicates of the sample sets were prepared. For the individual sample set, 18 individual patients were positioned in 3 TMT 6-plex sets: 126-non dis-meta (HER2), 127-non dis-meta (TNBC), 128-non dis-meta (Luminal), 129-dis-meta (HER2), 130-dis-meta (TNBC), and 131-dis-meta (Luminal). The detailed experimental workflow is described in Additional file 2: Fig. S1.
Prior to the TMT labeling step, 45 μg of each peptide sample was mixed with an equivalent volume of ovalbumin. Then, 40 mM TEAB buffer was added to each sample to equalize the volume. Next, TMT reagents were reconstituted in 110 μl anhydrous ACN. Each sample was labeled using 25 μl of the reconstituted TMT reagent. Then, 45 μl ACN was added in varying volumes to a final concentration of 30% and incubated at room temperature (25 °C) for 1.25 h. Hydroxylamine was added in various volumes to a concentration of 0.3% (v/v) to quench the reaction. TMT-labeled samples for each set were pooled at a ratio of 1:1. The pooled sample was lyophilized and desalted.
Desalting and high-pH reversed-phase (HPRP) peptide fractionation
The TMT-labeled samples were desalted on an HLB OASIS column per the manufacturer’s instructions. High-pH reversed-phase (HPRP) peptide fractionation was performed on an Agilent 1260 bioinert HPLC instrument (Agilent, Santa Clara, CA) with an Agilent 300 Extended-C18 column (4.6 mm I.D × 15 cm long, 5-μm C18 particle). TMT-labeled peptide samples were prefractionated at a flow rate of 1 mL/min for 60 min on a linear gradient, which ranged from 5% to 40% ACN with 15 mM ammonium hydroxide. The sample was separated into 96 fractions, which were then assembled into 12 fractions. The 12 fractions were lyophilized and stored at − 80 °C before MS analysis.
Sample preparation of breast cancer cells for proteomic analysis
MDA-MB-231 breast cancer cells were cultured in DMEM, and T47D cells were cultured in RPMI, containing 10% FBS and 1% penicillin and streptomycin. The cells were seeded in 75-cm2 culture plates. After a 24-h incubation at 37 °C with 5% CO2, the cells were scraped using a cell scraper and washed 3 times with 1 × PBS. The scraped cell pellets were centrifuged and washed again 3 times with 1 x PBS. The pellets were then transferred to microfuge tubes and mixed with protein extraction buffer (4% SDS; 0.3 M Tris, pH 7.5; 2 mM TCEP). Following sonication, the samples were incubated at 100 °C for 30 min. After protein extraction, the subsequent experimental procedures, such as protein digestion, TMT labeling, desalting, and peptide fractionation, were performed in the same manner as the FFPE tissues.
Reversed-phase (RP)-nano LC–ESI–MS/MS analysis
The prefractionated peptides were analyzed on an LC–MS system with an Easy-nLC 1000 (Thermo Fisher Scientific, Waltham, MA) that was equipped with a nanoelectrospray ion source (Thermo Fisher Scientific, Waltham, MA) and coupled to a Q-Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA), as described in our previous studies [45, 46]. The peptide samples were separated on a 2-column system, comprising a trap column (Thermo Fisher Scientific, 75 μm I.D. x 2 cm long, 3-μm Acclaim PepMap100 C18 beads) and an analytical column (Thermo Fisher Scientific, 75 μm I.D. x 50 cm long, 3-μm ReproSil-Pur C18-AQ beads). Lyophilized peptide samples were dissolved in Solvent A (0.1% formic acid water and 2% ACN) prior to injection.
The peptides were separated on a 180-min linear gradient, ranging from 6 to 26% Solvent B (100% ACN and 0.1% formic acid) for all peptide samples. The spray voltage was set to 2.2 kV in positive ion mode, and the heated capillary temperature was set to 320 °C. Mass spectra were collected in data-dependent acquisition (DDA) mode by top 20 method. Xcaliber (version 2.5) was used to set the mass spectrometer parameters as follows: mass range to 350–1650 m/z, resolution of 70,000 at 200 m/z for detected precursor ions, automatic gain control (AGC) at 3 x 106, isolation window for MS2 at 1.2 m/z, automatic gain control (AGC) for MS2 at 2 x 105, higher-energy collisional dissociation (HCD) scans at a resolution of 35,000, and normalized collision energy (NCE) of 32. The maximum ion injection time (maximum IT) for the full-MS and MS2 scans was 30 ms and 120 ms, respectively. Dynamic exclusion with an exclusion time of 40 s was used.
MS data search
Proteome Discoverer, version 2.2 (Thermo Fisher Scientific, Waltham, MA) was used to search the resulting RAW files. The full-MS and MS/MS spectra search was conducted using the SEQUEST HT algorithm against a modified version of the Uniprot human database (December 2014, 88,717 protein entries; http://www.uniprot.org), which included chicken ovalbumin. The database search was performed using the target-decoy strategy. The search parameters were as follows: a precursor ion mass tolerance value of 20 ppm (monoisotopic mass); a fragment ion mass tolerance value of 0.02 Da (monoisotopic mass); full enzyme digest with trypsin (after KR/−) and up to 2 missed cleavages; static modification values of 229.163 Da for lysine residues and peptide N-termini for TMT labeling and 57.02 Da for cysteine residues with carbamidomethylation; and dynamic modification values of 42.01 Da for protein N-terminal acetylation, 0.984 Da for asparagine deamidation, and 15.99 Da for methionine oxidation.
A false discovery rate (FDR) of less than 1% at the peptide and protein levels was used as the confidence criteria. Proteins were quantified by computing reporter ion relative intensities with the “Reporter Ions Quantifier” node in Proteome Discoverer. The co-isolation threshold value was 70%. The mass spectrometry-based proteome data lists of all identified proteins and peptides have been deposited into ProteomeXchange (http://proteomecentral.proteomexchange.org) through the PRIDE partner repository: dataset identifier PXD016061 [48, 69,70,71].
Quantification of protein abundance and statistical analysis
Protein levels were normalized, based on the ovalbumin content in each TMT channel. Fold-change values were calculated by dividing the average value of the normalized protein abundance in the dis-meta group by that of the non dis-meta group. Statistical analysis for the proteomic data was performed for the normalized protein levels using Perseus (version 1.5.8.5). Student’s t-test was used to identify differentially expressed proteins (DEPs) for selecting biomarker candidates that differentiate distant metastasis from nondistant metastasis of breast cancer. The statistical cutoff for the student’s t-test was a p-value < 0.05. In addition, ANOVA was used to determine DEPs for analyzing the molecular characteristics of distant metastatic breast cancer between molecular subtypes using bioinformatic tools. Specifically, 9 samples in each group were classified as HER2, TNBC, and luminal, resulting in 6 subtype groups (HER2 nondis-meta, TNBC nondis-meta, luminal nondis-meta, HER2 dis-meta, TNBC dis-meta, and luminal dis-meta). Next, the quantified proteins in these groups were analyzed to detect statistically significant proteins. The statistical cutoff for the ANOVA was p-value < 0.05. Receiver operating characteristic (ROC) analyses of biomarker performance were performed using MedCalc (version 12.5.0) and Prism (version 6.0).
Bioinformatics analysis
The Gene Ontology (GO) of the proteins was classified using the DAVID bioinformatics tool (version 6.8). GO classification was assessed by Fisher’s exact test to obtain a series of p-values that were filtered, based on a statistical significance of 0.05. Canonical pathways and downstream biological functions were enriched by Ingenuity Pathway Analysis (IPA, QIAGEN, Redwood City, CA). The analytical algorithms in IPA were used to predict the downstream effects on known biological pathways and functions, based on the inputted list of DEPs. IPA allocates activation scores on activated or inhibited status to biological functions and pathways that underlie the quantitative values of proteins. Fisher’s exact test was used to acquire p-values, whereas the degree of activation was measured using Z-scores. The p-value cutoff was set to 0.05, and the predictive activation Z-score cutoff was set to a magnitude of 1.
RNA extraction and real-time polymerase chain reaction (RT-PCR)
Total RNA was isolated from the following breast cancer cell lines using TRIzol (Invitrogen, Carlsbad, CA, USA) per the manufacturer’s instructions: MCF10A, MCF7, T47D, BT474, skBR3, MDA-MB-453, BT-20, MDA-MB-468, HCC70, HCC38, MDA-MB-157, MDA-MB-436, MDA-MB-231, and HS578T. Two micrograms of total RNA from each cell line was used for the reverse-transcription reaction. First-strand cDNA was synthesized by standard random priming with RNA inhibitor (Promega, Madison, WI) and Moleney murine leukemia virus reverse transcripts (Promega, Madison, WI). Following cDNA synthesis, target genes were amplified using specific primers and HIPI plus Master mix (ElpisBio, Daejeon, Korea).
Cell lines and culture conditions for invasion and migration assays
The MDA-MB-231 and Hs578T cell lines were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA) and the Korean Cell Line Bank (KCLB, Seoul, Korea), respectively. The cells were cultured in DMEM (Gibco, CA, USA), containing 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) and 1% penicillin/streptomycin (Gibco, CA, USA). The cells were maintained at 37 °C in a humidified atmosphere of 95% air and 5% CO2 and screened periodically for mycoplasma contamination. Both cell lines were confirmed by DNA profiling of short tandem repeats (STRs) by the KCLB (Seoul, Korea).
Small interfering RNA (siRNA) transfection
siRNAs that targeted LTF and TUBB2A and AccuTarget Negative Control siRNA were purchased from Bioneer (Daejeon, Korea). The siRNA sequences for LTF and TUBB2A were as follows: siLTF-1, 5′-GAGAUCAGACACUACCUU-3′; siLTF-2, 5′-CACACUGUUGAUGUAAUGA-3′; siTUBB2A-1,′-CUCAAGCAUGGUCUUUCA-3′; siTUBB2A-2, 5′-CACACUGUUGAUGUAAUGA-3′. Cells were transfected using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) per the manufacturer’s instructions. After a 48-h incubation, silencing of LTF and TUBB2A was confirmed by measuring their respective mRNA levels.
Cell migration and invasion assays
Quantitative cell migration and invasion were assessed using 24-well inserts (Corning Incorporated, NY, USA) with 8-μm pores according to the manufacturer’s instructions. In brief, for the transwell migration assay, transfected cells (5 × 104 cells) were seeded into the upper chamber, and medium that contained 10% FBS was added to the lower chamber. After a 24-h incubation, the cells on the top of the membrane were removed using a cotton swab. The remaining migrant cells were washed with PBS, fixed in 4% paraformaldehyde, stained with 1% crystal violet for 10 min, and imaged and counted in 3 randomly selected fields under a microscope (Nikon, Tokyo, Japan). These experiments were performed in triplicate.
For the in vitro invasion assay, the upper wells of Boyden chambers were coated with 2 mg/ml of Matrigel (Corning Incorporated, NY, USA) at 37 °C in a 5% CO2 incubator for 2 h. The cells (1 × 105 cells) were seeded into the upper chamber, and medium that contained 10% FBS was added to the lower chamber. The rest of the assay was performed as described above.
Results
Construction of distant metastatic breast cancer proteomic datasets
In the pooled sample set, 9441 proteins were identified, and 7179 proteins were quantified across all samples. In the individual sample set, 8746 proteins were identified, and 6642 proteins were quantified in all samples (Fig. 1, Additional file 2: Fig. S2a). In addition, the number of identifications in each sample was calculated, resulting in a range from 7515 to 7798 identified proteins in the individual sample set and 8287 to 8309 proteins in the pooled sample set. Overall, the numbers of proteins in the samples of each sample set were similar (Additional file 2: Fig. S2b–c).

Schematic of overall proteomic results of the TMT-based proteomic analysis. Number of identified proteins; pooled sample set: 9441, individual sample set: 8746, and cell line set: 7823. Number of DEPs by statistical analysis and the steps for selection of protein targets. Validation phase of protein targets; real-time polymerase chain reaction (RT-PCR) and migration/invasion assay
Our proteomic platform enabled us to perform an in-depth analysis of the distant metastatic breast cancer proteome, as evidenced by a dynamic range that spanned over 6 orders of magnitude (Additional file 2: Fig. S3). This comprehensive dataset included many established biomarkers for breast cancer, including the receptor tyrosine kinase erbB-2 (HER2), estrogen receptor (ESR1), progesterone receptor (PGR), and androgen receptor (AR). Notably, established protein biomarkers for metastatic breast cancer, such as EGFR, HSPD1, PRDX6, and TPM4, which are related to lymph node and regional metastasis, were also detected [50]. Moreover, this proteome encompassed most of the identified proteins in our previous study and included an additional 3757 and 3126 newly identified proteins in the pooled and individual sample sets, respectively (Additional file 2: Fig. S4) [44]. Consequently, our in-depth proteomic profiling generated a comprehensive dataset that is suitable for biomarker discovery and analysis with regard to determining the underlying mechanisms of distant metastasis in breast cancer. All identified proteins of each sample set are listed in Additional file 3: Table S2.
Quality assessment of proteomic data
The multiplexing feature of the TMT-based strategy allowed us to examine the quantitative variation within and between our samples. Interbatch and intrabatch variation was assessed using an internal standard, ovalbumin. As a result, the interbatch and intrabatch normalization produced coefficients of variation of 4.17% and 6.7% in the pooled and individual sample sets, respectively (Additional file 2: Fig. S5a). Although the variation in non-normalized intensities reflected excellent reproducibility, a slight improvement in reproducibility was observed when the levels of proteins were normalized to ovalbumin (Additional file 2: Fig. S5b–c).
Next, correlation values were calculated to assess the variation between technical replicates in the pooled sample set. MS analysis of the pooled sample set showed excellent correlation, with Pearson’s correlation values ranging from 0.993 to 0.994 and averaging 0.993 (Additional file 2: Fig. S5d). In addition, the correlation between the quantitative levels of all samples was calculated to assess the variation across individual samples. MS analysis of the individual sample set revealed a wider range of correlation values than that of the pooled sample set, with Pearson’s correlation values ranging from 0.647 to 0.988 and averaging 0.927 (Additional file 2: Fig. S5e). One sample, a HER2 type in the non dis-meta group, had low correlation values when paired with other individual samples, resulting in a range of 0.647 to 0.778. Slight differences in protein abundance between individual samples were observed.
Determination of protein targets to validate distant metastatic potential
To select important protein targets to verify distant metastatic potential of breast cancer, the quantified proteins in the BC FFPE tissues datasets (i.e., the pooled and individual sample sets) were examined separately by statistical analysis. For the proteomic datasets of BC FFPE tissues, student’s t-test was performed to determine differentially expressed proteins (DEPs) between the nondistant metastasis and distant metastasis groups. When a Benjamini–Hochberg false discovery rate (BH-FDR) cutoff of 0.05 was applied to the proteins in the pooled and individual sample sets respectively, however, none of the proteins in nondis-meta and dis-meta was significantly differentially expressed. Nonetheless, to determine protein targets for validation of distant metastatic breast cancer, alternative criteria were applied to the datasets.
The criteria were as follows: 1. The quantified proteins in our BC FFPE tissue proteomic datasets must pass a p-value (unadjusted for multiple comparison) cutoff of 0.05 by student’s t-test for determining DEPs in nondis-meta versus dis-meta. 2. Overlapping DEPs in both BC FFPE tissue datasets were selected. 3. Overlapping DEPs that were also identified in the BC cell line proteomic dataset and demonstrated a consistent expression pattern in all 3 datasets were selected. 4. Overlapping DEPs that passed a fold-change cutoff of 1.2 were selected. 5. The most highly up-regulated and down-regulated DEPs were selected. Therefore, DEPs that satisfied all of the requirements were selected as protein targets for validation of distant metastatic potential (Fig. 1).
Specifically, a total of 180 and 96 proteins were initially selected as DEPs by student’s t-test (p-value < 0.05) in the pooled and individual sample sets, respectively (Fig. 1, Additional file 4: Table S3). Next, overlapping proteins in DEPs of each sample set were selected.
As a result, 17 overlapping DEPs in both sets were selected. The results of the statistical analysis for these proteins are listed in Table 1. Of the 17 proteins, 5 (HSPA9, PSMB4, CTNNA1, XPO5, and PAFAH1B3) functioned in the growth, proliferation, metastasis, and recurrence of cancer [51,52,53,54,55,56]. Specifically, HSPA9 was associated with metastasis of hepatocellular carcinoma (HCC), and overexpression of HSPA9 increased the malignancy and aggressive behavior of HCC [51, 52]. Overexpression of PSMB4 increases cellular growth and the viability of breast cancer and ovarian cancer, leading to a poor prognosis [53, 54]. The deletion of CTNNA1 effects the loss of cell-to-cell adhesion, enhancing the growth and mobility of breast cancer cells [55]. XPO5 exports pre-miRNAs through the nuclear membrane to the cytoplasm and is thus important in breast cancer tumorigenesis [56]. PAFAH1B3 is a critical driver of the pathogenicity of breast cancer by inhibiting tumor-suppressing signaling lipids [72]. These 5 proteins were upregulated in our distant metastasis group, which we propose stimulate the distant metastatic potential of breast cancer.
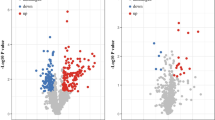
Subsequently, we examined whether the overlapping 17 proteins were also differentially expressed in the proteomic dataset of BC cell lines, comparing less-invasive T47D and highly invasive MDA-MB-231 cells. This examination was performed to identify proteins that might have molecular features that are related to the distant metastasis of breast cancer by comparing the BC FFPE and BC cell line proteomes. Five proteins had consistent expression patterns between all proteomic datasets: tubulin beta-2A chain (TUBB2A); lactotransferrin (LTF); acyl-coenzyme a dehydrogenase, C-4 to C-12 straight chain, isoform CRA_a (ACADM); proteasome subunit beta type-4 (PSMB4); and mitotic checkpoint protein BUB3 (BUB3) (Table 1). Next, with regard to the five proteins, the fold-change in expression between nondistant metastatic and distant metastatic groups was calculated. When the fold-change cutoff was set to 1.2, two proteins were selected: LTF was the most extensively downregulated protein, whereas TUBB2A was the most highly upregulated (Fig. 1, Table 1). The normalized abundance of LTF and TUBB2A distinguished the 2 sample groups significantly (Fig. 2a). Based on the criteria, LTF and TUBB2A were selected as important protein targets for validation of their function in relation to distant metastasis of breast cancer.

Validation of TUBB2A and LTF as protein targets. a Protein expression patterns of TUBB2A and LTF by mass spectrometry; expression pattern of reporter ion intensity of TUBB2A (upper panel) and LTF (lower panel) in pooled sample set (left panel) and individual sample set (right panel), respectively. The data in the interquartile range are displayed as black dots (* < p-value 0.05; **** < p-value 0.0001).b Expression patterns of TUBB2A and LTF in various breast cancer cell lines by RT PCR. Higher expression levels are lighter than lower levels (red line; higher invasive BC cell lines, blue line; lower invasive BC cell lines). c Results of invasion and migration assays for TUBB2A using Hs578T and MDA-MB-231 BC cell lines. RT-PCR of TUBB2A, downregulated by siRNA transfection in both cell lines (upper panel). Images of invading and migrating cells (lower left panel) and percentage (%) of invading and migrating cells (lower right panel) (*** < p-value 0.001)
Expression levels of TUBB2A and LTF verified by RT-PCR
The difference in the expression of TUBB2A and LTF was validated by RT-PCR in 1 normal breast cell line and 13 breast cancer cell lines, the relative invasiveness of which was determined per other studies [74,75,76,77,78,79,80,81]. The expression of LTF was lower in the higher invasive group than in the lower invasive group, except in 3 cell lines (BT20, MDA-MB-368, and HCC70). In particular, HCC70 expressed the most LTF (Fig. 2b). The level of TUBB2A was generally higher in the higher invasive group compared with the lower invasive group. Specifically, MDA-MB-231 had the highest expression of TUBB2A (Fig. 2b). The expression level of TUBB2A by MS was consistent with that by RT-PCR. The patterns of LTF by MS were not consistent with the RT-PCR results.
Distant metastatic potential of TUBB2A
The correlation between TUBB2A and metastatic characteristics was validated by invasion and migration assay. Two highly invasive BC cell lines (Hs578T and MDA-MB-231) were used to examine invasion and migration, based on the levels of TUBB2A. As a result, by siRNA transfection, TUBB2A was downregulated in both cell lines by RT-PCR. The number of invading cells fell significantly by over 50% when TUBB2A was knocked down compared with the control group (siControl), as did the number of migrating cells (Fig. 2c). Conversely, because the relative cell proliferation did not differ significantly on the day when the invasion and migration assays were conducted (Additional file 2: Fig. S6), the decreased invasiveness of the cells did not result from the altered cell proliferation. Thus, the distant metastatic potential of TUBB2A was verified, independent of the influence of cell proliferation.
To determine the ability of TUBB2A as a novel protein biomarker candidate of distant metastatic breast cancer, its performance was evaluated in the individual sample set. The sensitivity, specificity, and positive predictive value (PPV) by receiver operating characteristic (ROC) analysis were 78%, 100%, and 88%, respectively. Furthermore, the area under curve (AUC) value was 0.852, based on the ROC curve, and the threshold value, expressed as reporter ion intensity, that corresponded to the highest Youden’s index was 13,178 (Additional file 2: Fig. S7). Based on these results, we expected TUBB2A to perform well in the diagnosis and prediction of distant metastatic breast cancer.
Biological functions of distant metastatic breast cancer
To examine the functional signatures of distant metastatic breast cancer, we performed a bioinformatics analysis using 259 DEPs from the 2 sample sets. By gene ontology (GO) enrichment analysis, the 177 upregulated proteins in the distant metastasis group were assigned to various biological processes, such as cell–cell adhesion, proteolysis during cellular protein catabolism, NIK/NK-kappa B signaling, microtubule-based processes, and retrograde vesicle-mediated transport,- Golgi-to-ER (Fisher’s exact test p-value < 0.05) (Additional file 2: Fig. S8a, Additional file 5: Table S4). The most significant biological process in upregulated proteins was the regulation of mRNA stability (p-value = 7.82E−07). Conversely, the 82 downregulated proteins were allocated to various biological processes, including oxidation–reduction, organization of actin cytoskeleton, response to hydrogen peroxide, thrombin receptor signaling, sequestering of actin monomers, and positive regulation of toll-like receptor 4 signaling (Fisher’s exact test p-value < 0.05) (Additional file 2: Fig. S8b, Additional file 5: Table S4). The most significant biological process in downregulated proteins was oxidation–reduction (p-value = 2.89E–04).
In the enrichment of biological functions and pathways, the 259 DEPs were assigned to 6 canonical pathways and 11 downstream biological functions (Fisher’s exact test p-value < 0.05, and Z-score > 1). Canonical pathways included acute phase response signaling, ILK signaling, actin cytoskeletal signaling, leukocyte extravasation signaling, and tRNA charging (Additional file 2: Fig. S9a, Additional file 6: Table S5). The most significant and activated canonical pathway was glycolysis I (p-value = 1.74E−06, and activation Z-score = 2.236). Biological functions included polarization of tumor cell lines, orientation of cells, adhesion of BC cell lines, binding of NFkB sites, glycolysis in tumor cell lines, and proliferation of tumor/carcinoma cell lines (Additional file 2: Fig. S9b, Additional file 6: Table S5). The most significant and activated biological function was cell proliferation of tumor cell lines (p-value = 1.69E−08, and activation Z-score = 2.451). Based on our results, we propose that the interaction of various biological functions induces distant metastatic breast cancer.
Of the 2 protein targets, the result showed that the TUBB2A has association with the proliferation of tumor/carcinoma cell lines, microtubule-based processes, epithelial adherens junction signaling, 14-3-3-mediated signaling, and phagosome maturation. The most significant function of TUBB2A was cell proliferation of tumor cell lines (p-value = 1.69E−08). LTF was involved in the binding of NFkB sites, negative regulation of apoptotic process, positive regulation of I-KappaB kinase/NF-kappaB signaling, negative regulation of ATPase activity, and positive regulation of toll-like receptor 4 signaling pathway. Binding of NFkB sites was the most significant function (p-value = 2.17E−04) (Additional file 2: Fig. S10, Additional file 7: Table S6). Thus, these candidates had distinct and independent biological characteristics.
Proteomic alterations in distant metastatic breast cancer between molecular subtypes
According to the results of a previous study, pooling biological groups can reduce the variation that originates from the sample while retaining the defining features of the group itself [57]. We expected our pooled samples for each molecular subtype to reveal distinct information on the molecular characteristics between the HER2, TNBC, and luminal groups. For these reasons, a pooled sample set was used to identify the changes in proteins between distinct breast cancer molecular subtypes in the distant metastasis and nondistant metastasis groups.
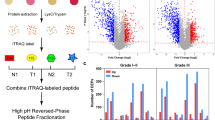
By ANOVA, 1086 proteins were differentially expressed between breast cancer molecular subtypes (p-value < 0.05) (Fig. 3a, Additional file 8: Table S7). These DEPs were then analyzed by hierarchical clustering to determine their expression patterns between breast cancer molecular subtypes, resulting in 6 groups: upregulated proteins in HER2-non-distant metastasis (cluster 1; 176 DEPs), upregulated proteins in HER2-distant metastasis (cluster 2; 124 DEPs), upregulated proteins in TNBC-non-distant metastasis (cluster 3; 193 DEPs), upregulated proteins in TNBC-distant metastasis (cluster 4; 342 DEPs), upregulated proteins in luminal-non-distant metastasis (cluster 5; 29 DEPs), and upregulated proteins in luminal-distant metastasis (cluster 6; 184 DEPs).

Proteomic alteration in distant metastatic breast cancer between molecular subtypes. a Hierarchical clustering of differentially expressed proteins (DEPs) between distant metastatic breast cancer molecular subtypes (ANOVA, p-value < 0.05). The DEPs (1086) from the pooled sample set were divided into 6 groups. Clusters of upregulated proteins are marked in red. b–d Canonical pathway enrichment of clusters 2, 4, and 6. The significant pathways (Fisher’s exact test p-value < 0.05) were deduced using Ingenuity Pathway Analysis (IPA), and their activation and inhibition states are expressed as Z-scores
Biological functions of distant metastatic breast cancer between molecular subtypes
To gain greater insight into the molecular features of distant metastatic breast cancer between molecular subtypes, pathway enrichment analysis was conducted for clusters 2, 4, and 6, which comprised proteins that were upregulated in the distant metastasis group of each molecular subtype. By Ingenuity Pathway Analysis (IPA), 2 canonical pathways were derived for cluster 2, versus 14 for cluster 4 and 11 for cluster 6 (p-value < 0.05, Z-score > 1) (Fig. 3b–d, Additional file 9: Table S8). Specifically, in cluster 2, only PI3K/AKT signaling and BAG signaling were deduced and activated between three subtypes. PI3K/AKT signaling was the most highly activated pathway (Z-score = 2) in the HER2 type (Fig. 3b, Additional file 9: Table S8). In cluster 4, all 14 pathways were activated—glycolysis 1, gluconeogenesis 1, and tRNA charging were extensively activated in the TNBC types (Fig. 3c, Additional file 9: Table S8). tRNA charging was the most highly activated pathway (Z-score = 2.828), whereas EIF2 signaling was the least activated (Z-score = 0.333) in TNBC types (Fig. 3c, Additional file 9: Table S8). In cluster 6, most pathways were activated, such as actin cytoskeleton signaling, acute phase response signaling, intrinsic prothrombin activation, and GP6 signaling, in the luminal type. Among them, GP6 signaling was the most highly activated (Z-score = 3.464). However, LXR/RXR signaling was inhibited in the luminal type (Z-score = − 0.707) (Fig. 3d, Additional file 9: Table S8). Based on our results, distinct activation states exist between the HER2, TNBC, and luminal types.
Discussion
One of the goals of our study was to discover novel protein biomarker candidates of distant metastatic breast cancer. Initially, we considered the potential problem with multiple comparisons, which can generate false positives if unaddressed, in selecting the protein targets. Therefore, we applied a multiple testing correction to our datasets. However, none of proteins was able to pass the BH FDR cutoff. Thus, we proposed alternative criteria to compensate for the statistically insufficient significance of proteins in determining the protein targets.
When the criteria were applied to our in-depth proteome data, LTF (p-value < 0.001) and TUBB2A (p-value < 0.05) appeared as important protein targets for validation of distant metastatic potential. TUBB2A was upregulated and LTF was downregulated in the distant metastasis group. TUBB2A was upregulated in more invasive breast cancer cell lines (i.e., BC cell lines in the higher invasive group), whereas the expression patterns of LTF were perturbed across breast cancer cell lines by RT-PCR. Considering the expression level of TUBB2A in the higher-invasiveness group and the high malignancy of distant metastatic breast cancer [4, 58, 59], the upregulation of TUBB2A might promote the invasion of breast cancer cells, inducing the potential of distant metastatic breast cancer. In addition, based on the results of the invasion and migration assay, we verified that the high expression of TUBB2A increases the mobility of breast cancer cells, providing further support for TUBB2A as a novel biomarker candidate of distant metastatic breast cancer.
Regarding performance of TUBB2A, TUBB2A could distinguish between distant metastasis and nondistant metastasis (i.e., 78% sensitivity, 100% specificity, and an AUC value of 0.852) and might predict distant metastasis (i.e., 88% PPV) in the individual sample set. However, because our TMT-based data were obtained from a small cohort (n = 36), future studies should evaluate the performance of TUBB2A by absolute quantitation in a large cohort to assess its clinical applicability, which lies beyond the scope of our current study. One possible design would be to quantify TUBB2A using targeted proteomic techniques, such as multiple reaction monitoring (MRM) and parallel reaction monitoring (PRM).
Another goal was to determine the overall biological functions that exist in distant metastatic breast cancer. Biological functions that are related to proliferation and movement of cancer cells were activated. Specifically, cell polarization/orientation was related to cell adhesion, and actin-based signaling was associated with migration [60,61,62]. NF-kappa B modulates the immune response, but its inhibition and dysregulation are linked to improper immune development [63, 64]. Thus, the inhibition of polarization of tumor cell lines and adhesion of BC cell lines might weaken the adhesion between cells in primary breast tumors, and the activation of actin cytoskeletal signaling and proliferation of tumor cell lines might enhance the movement of breast cancer cells. In addition, blocking NF kappa B binding sites might allow breast cancer cells to migrate to other distal sites without activating the immune system.
We noted proteins that were associated with distant metastatic breast cancer, based on our bioinformatics analysis. By GO analysis, ‘cell–cell adhesion’ terms were observed in upregulated and downregulated DEPs. However, each term consisted of different proteins. Furthermore, proteins in ‘adhesion of BC cell lines’ term did not overlap with those in the ‘cell–cell adhesion’ term. Thus, adhesion between breast cancer cells in primary tumors might be weakened, but that between breast cancer cells and cells in other organs could be strengthened, due to various proteins with potentially distinct functions in cell adhesion. In our pathway enrichment analysis, FN1 overlapped between activated leukocyte extravasation signaling and inhibited acute phase response signaling. Considering the opposing states of these pathways, the former might enhance the mobility of breast cancer cells to other organs, shuttling leukocytes out of the circulatory system. In parallel, inhibition of acute phase response signaling might suppress the immune response. Thus, FN1 might create a suitable microenvironment that is conducive to distant metastasis of breast cancer.
With regard to our protein targets, TUBB2A was associated with cellular proliferation, movement, and adhesion, and LTF was involved in cell death, the immune response, and metabolism. Based on these functions, TUBB2A might control the mobility of distant metastatic breast cancer by regulating the adhesion and proliferation of breast cancer cells, and LTF might govern the death of breast cancer cells and the immune system during distant metastasis. Thus, TUBB2A might be a key protein that controls the migration of breast cancer cells from a primary tumor. LTF might be an auxiliary protein that helps breast cancer cells survive during movement toward distal sites by disrupting the immune system.
Another goal was to determine the characteristics of distant metastatic breast cancer between molecular subtypes. In cluster 2, the most highly activated pathway was PI3K/AKT signaling in the HER2 type. A previous study that used transcriptome data revealed that PI3K/AKT kinases are expressed in circulating breast tumor cells and that the activation of this signal regulates their metastatic and malignant state [68]. Compared with our proteomic results, the activation states of PI3K/AKT signaling were consistent. Thus, our PI3K/AKT signaling proteins might be associated with the regulation of distant metastatic potential and function as targets for the eradication of HER2-type distant metastatic breast cancer.
In cluster 4, the most highly activated pathway was tRNA charging signaling in the TNBC type. The exact functions of this pathway in distant metastatic breast cancer have not been determined. However, based on a previous study, tRNA overexpression in breast tumor cells might increase the translational efficiency of genes that are related to the progression and development of breast cancer [67]. The tRNA charging-related proteins that we recorded might be upregulated and translationally modified products of such genes, influencing the distant metastatic potential and progression of breast cancer. Thus, these proteins might be targets for removal or suppression in slowing the malignancy of TNBC-type distant metastatic breast cancer.
In cluster 6, the most highly activated pathway was glycoprotein 6 (GP6) signaling in the luminal type. GP6 is a platelet membrane glycoprotein that functions as a receptor for collagen and regulates the collagen-induced activation and aggregation of platelets [65, 66]. The detailed functions of this pathway in distant metastatic breast cancer have not been described. However, based on its functions, breast cancer cells could migrate easily to distal sites, masking their aggregate forms with platelet-combined forms. Furthermore, breast cancer cell complexes might adhere to collagen and subsequently to platelets, leading to additional platelet aggregation. Thus, GP6 signaling and its factors might facilitate the circulation of breast cancer cells with little activation of the immune systems due to their disguised forms, allowing them to settle at distal sites. Furthermore, the expression level of these proteins could be used to monitor the progression of luminal-type distant metastatic breast cancer.
Although we performed pathway enrichment analysis using the upregulated DEPs in the 3 clusters, one of the benefits of our study was that it could have considered the downregulated DEPs in the remaining 3 clusters (clusters 1, 3, and 5) in the analysis. These proteins might be related to distinct biological activities that suppress the activation of distant metastatic breast cancer between subtypes. Consequently, our proteomic clusters might expand our understanding of the effects of molecular subtype on distant metastatic breast cancer.
Without our in-depth proteomic data, most of our DEPs might be unable to be identified or detected in other studies, because we are the first to collect proteomic data in distant metastatic breast cancer, analyzing clinical FFPE tissues from primary breast tumors. Our results indicate that the pathological relevance of our FFPE tissues in BC research is valid at the proteomic level and in severe breast cancer pathologies. Through our latent data, we discovered a novel protein biomarker candidate that has the potential to distinguish distant metastatic breast cancer and demonstrated distinct molecular features between BC subtypes. We expect that our biomarker candidate can be used to diagnose and predict distant metastatic breast cancer. Furthermore, our molecular pathways should provide insights into the relationship between molecular subtypes and distant metastatic breast cancer.
Conclusions
We have constructed a comprehensive proteome of distant metastatic breast cancer by analyzing FFPE tissue slides using TMT-based mass spectrometric techniques. Our study demonstrates that the TMT-based approach is beneficial, because its greater quantitative ability generates a larger selection of proteins from which to choose novel biomarker candidates. This finding was verified by our proteomic dataset, which comprised the largest number of proteins in distant metastatic breast cancer. Through our criteria, we selected 2 important protein targets for distant metastatic breast cancer and performed functional studies to validate them. Finally, we were able to propose a novel protein biomarker candidate. Furthermore, our bioinformatics analysis revealed specific molecular characteristics between molecular subtypes. Thus, our in-depth proteomic data and analyses can be an important resource for distant metastatic breast cancer research. In future studies, we hope to assemble a larger cohort of breast cancer FFPE samples to test the performance of our novel biomarker candidate using targeted proteomics techniques, such as parallel reaction monitoring (PRM) and multiple reaction monitoring (MRM).
Availability of data and materials
All datasets that were generated and analyzed during this study are included in this published article and its additional information files. The MS proteomic data in this study have been deposited into ProteomeXchange (http://proteomecentral.proteomexchange.org) through the PRIDE partner repository [43]: dataset identifier PXD016061. The datasets are available from the corresponding author on reasonable request. Username: reviewer41229@ebi.ac.uk, Password: 4ybAzlVM.
Abbreviations
- ACN:
-
Acetonitrile
- TUBB2A:
-
Tubulin beta 2A chain
- LTF:
-
Lactotransferrin
- HSPA9:
-
Stress-70 protein, mitochondrial
- PSMB4:
-
Proteasome subunit beta type-4
- CTNNA1:
-
Catenin alpha-1
- XPO5:
-
Exportin-5
- PAFAH1B3:
-
Platelet-activating factor acetylhydrolase IB subunit gamma
- TMSB10:
-
Thymosin beta-10
- TMSB4X:
-
Thymosin beta-4
- RAC1:
-
Ras-related protein-Rac1
- PFKM:
-
ATP-dependent 6-phosphofructokinase
- PKM:
-
Pyruvate kinase PKM
- FN1:
-
Fibronectin
- GP6:
-
Glycoprotein 6
- CV:
-
Coefficient of variation
- FDR:
-
False discovery rate
- DEP:
-
Differentially expressed protein
- FFPE:
-
Formalin-fixed paraffin-embedded
- MS:
-
Mass spectrometry
- TMT:
-
Tandem mass tag
- RT-PCR:
-
Real-time polymerase chain reaction
- Dis-meta:
-
Distant metastasis
- Non dis-meta:
-
Non-distant metastasis
- GO:
-
Gene ontology
- HCD:
-
Higher-energy collisional dissociation
- AGC:
-
Automatic gain control
- NCE:
-
Normalized collision energy
- DDA:
-
Data-dependent acquisition
- Maximum IT:
-
Maximum ion injection time
- HPRP:
-
High-pH reversed-phase
- RP:
-
Reverse-phase
- FFPE:
-
Formalin-fixed paraffin-embedded
- TMT:
-
Tandem mass tag
- BC:
-
Breast cancer
- MRM:
-
Multiple reaction monitoring
- PRM:
-
Parallel reaction monitoring
- BH-FDR:
-
Benjamini–Hochberg false discovery rate
References
Yanovich G, Agmon H, Harel M, Sonnenblick A, Peretz T, Geiger T. Clinical proteomics of breast cancer reveals a novel layer of breast cancer classification. Cancer Res. 2018;78:6001–10.
DeSantis CE, Ma J, Goding Sauer A, Newman LA, Jemal A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J Clin. 2017;67:439–48.
Anastasiadi Z, Lianos GD, Ignatiadou E, Harissis HV, Mitsis M. Breast cancer in young women: an overview. Updates Surg. 2017;69:313–7.
Fredholm H, Eaker S, Frisell J, Holmberg L, Fredriksson I, Lindman H. Breast cancer in young women: poor survival despite intensive treatment. PLoS ONE. 2009;4:e7695.
Hess KR, Varadhachary GR, Taylor SH, Wei W, Raber MN, Lenzi R, Abbruzzese JL. Metastatic patterns in adenocarcinoma. Cancer. 2006;106:1624–33.
Chang J, Clark GM, Allred DC, Mohsin S, Chamness G, Elledge RM. Survival of patients with metastatic breast carcinoma: importance of prognostic markers of the primary tumor. Cancer. 2003;97:545–53.
Coleman RE, Rubens RD. The clinical course of bone metastases from breast cancer. Br J Cancer. 1987;55:61–6.
Lobbezoo DJ, van Kampen RJ, Voogd AC, Dercksen MW, van den Berkmortel F, Smilde TJ, van de Wouw AJ, Peters FP, van Riel JM, Peters NA, et al. Prognosis of metastatic breast cancer: are there differences between patients with de novo and recurrent metastatic breast cancer? Br J Cancer. 2015;112:1445–51.
Horton J. Follow-up of breast cancer patients. Cancer. 1984;53:790–7.
Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, Nielsen TO, Gelmon K. Metastatic behavior of breast cancer subtypes. J Clin Oncol. 2010;28:3271–7.
Chia S, Norris B, Speers C, Cheang M, Gilks B, Gown AM, Huntsman D, Olivotto IA, Nielsen TO, Gelmon K. Human epidermal growth factor receptor 2 overexpression as a prognostic factor in a large tissue microarray series of node-negative breast cancers. J Clin Oncol. 2008;26:5697–704.
Alanko A, Heinonen E, Scheinin T, Tolppanen EM, Vihko R. Significance of estrogen and progesterone receptors, disease-free interval, and site of first metastasis on survival of breast cancer patients. Cancer. 1985;56:1696–700.
Kate RJ, Nadig R. Stage-specific predictive models for breast cancer survivability. Int J Med Inform. 2017;97:304–11.
Schnitt SJ. Classification and prognosis of invasive breast cancer: from morphology to molecular taxonomy. Mod Pathol. 2010;23(Suppl 2):S60–4.
Minn AJ, Gupta GP, Padua D, Bos P, Nguyen DX, Nuyten D, Kreike B, Zhang Y, Wang Y, Ishwaran H, et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc Natl Acad Sci USA. 2007;104:6740–5.
Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, Massague J. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–9.
Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–24.
Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–49.
Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–9.
Bellahcene A, Bachelier R, Detry C, Lidereau R, Clezardin P, Castronovo V. Transcriptome analysis reveals an osteoblast-like phenotype for human osteotropic breast cancer cells. Breast Cancer Res Treat. 2007;101:135–48.
Garcia M, Millat-Carus R, Bertucci F, Finetti P, Birnbaum D, Bidaut G. Interactome-transcriptome integration for predicting distant metastasis in breast cancer. Bioinformatics. 2012;28:672–8.
Geiger T, Cox J, Mann M. Proteomic changes resulting from gene copy number variations in cancer cells. PLoS Genet. 2010;6:e1001090.
Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–7.
Lundberg E, Fagerberg L, Klevebring D, Matic I, Geiger T, Cox J, Algenas C, Lundeberg J, Mann M, Uhlen M. Defining the transcriptome and proteome in three functionally different human cell lines. Mol Syst Biol. 2010;6:450.
Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–42.
Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol. 2011;7:548.
Jezequel P, Guette C, Lasla H, Gouraud W, Boissard A, Guerin-Charbonnel C, Campone M. iTRAQ-Based quantitative proteomic analysis strengthens transcriptomic subtyping of triple-negative breast cancer tumors. Proteomics. 2019;19:e1800484.
Johansson HJ, Socciarelli F, Vacanti NM, Haugen MH, Zhu Y, Siavelis I, Fernandez-Woodbridge A, Aure MR, Sennblad B, Vesterlund M, et al. Breast cancer quantitative proteome and proteogenomic landscape. Nat Commun. 2019;10:1600.
Bouchal P, Schubert OT, Faktor J, Capkova L, Imrichova H, Zoufalova K, Paralova V, Hrstka R, Liu Y, Ebhardt HA, et al. Breast cancer classification based on proteotypes obtained by SWATH mass spectrometry. Cell Rep. 2019;28(832–843):e837.
Shin J, Dan K, Han D, Kim JW, Kim KK, Koh Y, Shin DY, Hong J, Yoon SS, Park S, et al. Plasma-based protein biomarkers can predict the risk of acute graft-versus-host disease and non-relapse mortality in patients undergoing allogeneic hematopoietic stem cell transplantation. Blood Cells Mol Dis. 2019;74:5–12.
Kim YS, Han D, Kim J, Kim DW, Kim YM, Mo JH, Choi HG, Park JW, Shin HW. In-depth, proteomic analysis of nasal secretions from patients with chronic rhinosinusitis and nasal polyps. Allergy Asthma Immunol Res. 2019;11:691–708.
Duangkumpha K, Stoll T, Phetcharaburanin J, Yongvanit P, Thanan R, Techasen A, Namwat N, Khuntikeo N, Chamadol N, Roytrakul S, et al. Discovery and qualification of serum protein biomarker candidates for cholangiocarcinoma diagnosis. J Proteome Res. 2019;18:3305–16.
Lee H, Kim K, Woo J, Park J, Kim H, Lee KE, Kim H, Kim Y, Moon KC, Kim JY, et al. Quantitative proteomic analysis identifies AHNAK (neuroblast differentiation-associated protein AHNAK) as a novel candidate biomarker for bladder urothelial carcinoma diagnosis by liquid-based cytology. Mol Cell Proteomics. 2018;17:1788–802.
Jin J, Son M, Kim H, Kim H, Kong SH, Kim HK, Kim Y, Han D. Comparative proteomic analysis of human malignant ascitic fluids for the development of gastric cancer biomarkers. Clin Biochem. 2018;56:55–61.
Do M, Han D, Wang JI, Kim H, Kwon W, Han Y, Jang JY, Kim Y. Quantitative proteomic analysis of pancreatic cyst fluid proteins associated with malignancy in intraductal papillary mucinous neoplasms. Clin Proteomics. 2018;15:17.
Park J, Han D, Do M, Woo J, Wang JI, Han Y, Kwon W, Kim SW, Jang JY, Kim Y. Proteome characterization of human pancreatic cyst fluid from intraductal papillary mucinous neoplasm by liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2017;31:1761–72.
Geyer PE, Kulak NA, Pichler G, Holdt LM, Teupser D, Mann M. Plasma proteome profiling to assess human health and disease. Cell Syst. 2016;2:185–95.
Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–55.
Choudhary C, Mann M. Decoding signalling networks by mass spectrometry-based proteomics. Nat Rev Mol Cell Biol. 2010;11:427–39.
Murphy JP, Stepanova E, Everley RA, Paulo JA, Gygi SP. Comprehensive temporal protein dynamics during the diauxic shift in Saccharomyces cerevisiae. Mol Cell Proteomics. 2015;14:2454–65.
Kim DK, Park J, Han D, Yang J, Kim A, Woo J, Kim Y, Mook-Jung I. Molecular and functional signatures in a novel Alzheimer’s disease mouse model assessed by quantitative proteomics. Mol Neurodegener. 2018;13:2.
Christoforou A, Mulvey CM, Breckels LM, Geladaki A, Hurrell T, Hayward PC, Naake T, Gatto L, Viner R, Martinez Arias A, Lilley KS. A draft map of the mouse pluripotent stem cell spatial proteome. Nat Commun. 2016;7:8992.
Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang EC, Aicheler R, Murrell I, Wilkinson GW, et al. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell. 2014;157:1460–72.
Jin MS, Lee H, Woo J, Choi S, Do MS, Kim K, Song MJ, Kim Y, Park IA, Han D, Ryu HS. Integrated multi-omic analyses support distinguishing secretory carcinoma of the breast from basal-like triple-negative breast cancer. Proteomics Clin Appl. 2018;12:e1700125.
Han D, Moon S, Kim Y, Kim J, Jin J, Kim Y. In-depth proteomic analysis of mouse microglia using a combination of FASP and StageTip-based, high pH, reversed-phase fractionation. Proteomics. 2013;13:2984–8.
Han D, Jin J, Woo J, Min H, Kim Y. Proteomic analysis of mouse astrocytes and their secretome by a combination of FASP and StageTip-based, high pH, reversed-phase fractionation. Proteomics. 2014;14:1604–9.
Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11:319–24.
Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32:223–6.
Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–62.
Brinton LT, Brentnall TA, Smith JA, Kelly KA. Metastatic biomarker discovery through proteomics. Cancer Genomics Proteomics. 2012;9:345–55.
Yi X, Luk JM, Lee NP, Peng J, Leng X, Guan XY, Lau GK, Beretta L, Fan ST. Association of mortalin (HSPA9) with liver cancer metastasis and prediction for early tumor recurrence. Mol Cell Proteomics. 2008;7:315–25.
Piao HL, Yuan Y, Wang M, Sun Y, Liang H, Ma L. Alpha-catenin acts as a tumour suppressor in E-cadherin-negative basal-like breast cancer by inhibiting NF-kappaB signalling. Nat Cell Biol. 2014;16:245–54.
Liu R, Lu S, Deng Y, Yang S, He S, Cai J, Qiang F, Chen C, Zhang W, Zhao S, et al. PSMB4 expression associates with epithelial ovarian cancer growth and poor prognosis. Arch Gynecol Obstet. 2016;293:1297–307.
Wang H, He Z, Xia L, Zhang W, Xu L, Yue X, Ru X, Xu Y. PSMB4 overexpression enhances the cell growth and viability of breast cancer cells leading to a poor prognosis. Oncol Rep. 2018;40:2343–52.
Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest. 2002;109:987–91.
Leaderer D, Hoffman AE, Zheng T, Fu A, Weidhaas J, Paranjape T, Zhu Y. Genetic and epigenetic association studies suggest a role of microRNA biogenesis gene exportin-5 (XPO5) in breast tumorigenesis. Int J Mol Epidemiol Genet. 2011;2:9–18.
Diz AP, Truebano M, Skibinski DO. The consequences of sample pooling in proteomics: an empirical study. Electrophoresis. 2009;30:2967–75.
Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer. 2007;109:1721–8.
Weigelt B, Peterse JL, van ‘t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5:591–602.
Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9.
Altschuler SJ, Angenent SB, Wang Y, Wu LF. On the spontaneous emergence of cell polarity. Nature. 2008;454:886–9.
Wu J, Mlodzik M. A quest for the mechanism regulating global planar cell polarity of tissues. Trends Cell Biol. 2009;19:295–305.
Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–4.
Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62.
Ezumi Y, Uchiyama T, Takayama H. Molecular cloning, genomic structure, chromosomal localization, and alternative splice forms of the platelet collagen receptor glycoprotein VI. Biochem Biophys Res Commun. 2000;277:27–36.
Jandrot-Perrus M, Busfield S, Lagrue AH, Xiong X, Debili N, Chickering T, Le Couedic JP, Goodearl A, Dussault B, Fraser C, et al. Cloning, characterization, and functional studies of human and mouse glycoprotein VI: a platelet-specific collagen receptor from the immunoglobulin superfamily. Blood. 2000;96:1798–807.
Pavon-Eternod M, Gomes S, Geslain R, Dai Q, Rosner MR, Pan T. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 2009;37:7268–80.
Kallergi G, Agelaki S, Kalykaki A, Stournaras C, Mavroudis D, Georgoulias V. Phosphorylated EGFR and PI3K/Akt signaling kinases are expressed in circulating tumor cells of breast cancer patients. Breast Cancer Res. 2008;10:R80.
Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–50.
Perez-Riverol Y, Xu QW, Wang R, Uszkoreit J, Griss J, Sanchez A, Reisinger F, Csordas A, Ternent T, Del-Toro N, et al. PRIDE inspector toolsuite: moving toward a universal visualization tool for proteomics data standard formats and quality assessment of ProteomeXchange datasets. Mol Cell Proteomics. 2016;15:305–17.
Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, Campbell DS, Bernal-Llinares M, Okuda S, Kawano S, et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017;45:D1100–6.
Mulvihill MM, Benjamin DI, Ji X, Le Scolan E, Louie SM, Shieh A, Green M, Narasimhalu T, Morris PJ, Luo K, Nomura DK. Metabolic profiling reveals PAFAH1B3 as a critical driver of breast cancer pathogenicity. Chem Biol. 2014;21:831–40.
Dai X, Cheng H, Bai Z, Li J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J Cancer. 2017;8:3131–41.
Sommers CL, Byers SW, Thompson EW, Torri JA, Gelmann EP. Differentiation state and invasiveness of human breast cancer cell lines. Breast Cancer Res Treat. 1994;31:325–35.
Bae SN, Arand G, Azzam H, Pavasant P, Torri J, Frandsen TL, Thompson EW. Molecular and cellular analysis of basement membrane invasion by human breast cancer cells in Matrigel-based in vitro assays. Breast Cancer Res Treat. 1993;24:241–55.
Hughes L, Malone C, Chumsri S, Burger AM, McDonnell S. Characterisation of breast cancer cell lines and establishment of a novel isogenic subclone to study migration, invasion and tumourigenicity. Clin Exp Metastasis. 2008;25:549–57.
Quail DF, Maciel TJ, Rogers K, Postovit LM. A unique 3D in vitro cellular invasion assay. J Biomol Screen. 2012;17:1088–95.
Ziperstein MJ, Guzman A, Kaufman LJ. Breast cancer cell line aggregate morphology does not predict invasive capacity. PLoS ONE. 2015;10:e0139523.
Ribeiro AS, Albergaria A, Sousa B, Correia AL, Bracke M, Seruca R, Schmitt FC, Paredes J. Extracellular cleavage and shedding of P-cadherin: a mechanism underlying the invasive behaviour of breast cancer cells. Oncogene. 2010;29:392–402.
Li J, Wei J, Mei Z, Yin Y, Li Y, Lu M, Jin S. Suppressing role of miR-520a-3p in breast cancer through CCND1 and CD44. Am J Transl Res. 2017;9:146–54.
Chiu HW, Lin HY, Tseng IJ, Lin YF. OTUD7B upregulation predicts a poor response to paclitaxel in patients with triple-negative breast cancer. Oncotarget. 2018;9:553–65.
Acknowledgements
This work was supported by the Industrial Strategic Technology Development Program (#10079271 and #20000134), funded by the Ministry of Trade, Industry, and Energy (MOTIE, Korea); the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant Number: HI17C0048); the Basic Science Research Program through the Seoul National University Hospital Research Fund (26-2016-0020); and the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT & Future Planning (Grant Number: 2018R1A1A1A05077484).
Author information
Authors and Affiliations
Contributions
DS, JP, HR, and YK contributed to the study concept and design; DS and JP acquired the data; DS performed the statistical analysis; DH, JM, and HR provided administrative, technical, and material support; and DS, JP, and YK drafted this manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
All patients or legal guardians consented to participation in the study in accordance with Institutional Review Board guidelines (IRB No.1612-011-811) of Seoul National University Hospital, South Korea.
Consent for publication
All authors consent to the publication of this manuscript.
Competing interests
The authors declare no competing financial interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Clinical information on patients. Clinical information on all 36 patients is listed.
Additional file 2: Figure S1.
Detailed experimental workflow of TMT-based proteomic study. Graphical representation of the workflow for our TMT experiments. Three sample sets were analyzed using our TMT-based proteomic techniques. Figure S2. Identified and quantified proteins in TMT experiments. (a) The number of identified and quantified proteins in the pooled sample set, individual sample set, and cell line set. (B) The number of identified proteins in each sample of the individual sample set. (C) The number of identified proteins in each sample of the pooled sample set. Figure S3. Dynamic ranges of protein abundance in pooled sample set and individual sample set. The dynamic range of the pooled sample set is marked in yellow, and that of the individual sample set is marked in blue. Known metastatic biomarkers are indicated in red, and breast cancer markers are marked in black. Figure S4. Comparative analysis between our FFPE tissue proteome and those of our previous studies. (a) Comparison of identified proteins between our pooled sample proteome data and those of MS Jin et al. (b) Comparison of identified proteins between our individual sample proteome data and those of MS Jin et al. Figure S5. Quality assessment of MS analysis. (a) Abundance and technical variation of the external standard, ovalbumin. Ovalbumin was quantified in the middle-high abundance interval and had a CV of 4.2% and 6.7% in the pooled and individual sample sets in 18 TMT channels, respectively. (b), (c) The quantitative reproducibility of all proteins was improved slightly on normalization with the external standard, ovalbumin; the median CV value of the biological replicates of the pooled and individual sample sets decreased by 0.36% and 1.54%, respectively. (d) Cross-correlation analysis using the protein levels to confirm the repeatability of the MS analyses between experimental sets of the pooled sample set. (e) Variabilities in individual samples in our MS analysis are depicted in a multiscatter plot. Reproducibility between individual samples is represented by Pearson’s correlation value. Values of correlation with HER2 ND-2 are marked in red. (ND; non dis-meta, D; dis-meta, LU; luminal, - #; number of TMT set). Figure S6. Cell proliferation of MDA-MB 231 and Hs578T cell lines. Relative cell proliferation was observed for 3 days, when TUBB2A was knocked down, compared with the control group (siControl) (* < p-value 0.05; ** < p-value 0.01). The time point at which the migration and invasion assays were performed is indicated in the blue circle. Figure S7. Performance of the novel biomarker TUBB2A in the individual sample set. Table of summary statistics in ROC analysis, ROC curve with AUC = 0.852, and interactive dot diagram with sensitivity = 78%, specificity = 100%, and reporter ion intensity threshold = 13,178. Figure S8. Gene ontology analysis of all 259 DEPs in the two sample sets using The Database for Annotation, Visualization and Integrated Discovery (DAVID). (a) Biological process terms of 177 upregulated DEPs. (b) Biological process terms of 82 downregulated DEPs (Fisher’s exact test p-value < 0.05). Figure S9. IPA analysis of all 259 DEPs in the two sample sets regarding canonical pathway, and downstream biological functions. (a) Canonical pathway enrichment of all 259 DEPs in the two sample sets. (b) Hierarchical clustering of downstream biological functions assessed by IPA using the 259 DEPs. The significant pathways, and downstream biological functions (Fisher’s exact test p-value < 0.05) were deduced using Ingenuity Pathway Analysis (IPA), and their activation and inhibition states are expressed as Z-scores. Figure S10. Biological functions and canonical pathways related to the two protein targets by IPA and DAVID analysis. Biological functions and pathways of TUBB2A (upper panel) and LTF (lower panel) (Fisher’s exact test p-value < 0.05 for DAVID and IPA analysis).
Additional file 3: Table S2.
List of all identified proteins in this study. MS information on identified proteins is listed in the pooled and individual sample sets. Normalized protein levels of each sample were used for further statistical analysis.
Additional file 4: Table S3.
List of significantly differentially expressed proteins (DEPs) by student t-test. Statistically significant DEPs by student’s t-test (p-value < 0.05), fold-changes, p-values, and adjusted p-values (Benjamini–Hochberg FDR cutoff of 0.05) in the pooled sample set, individual sample set, and cell line set. These proteins were used to select protein targets for validation of distant metastatic potential and perform the bioinformatics analysis.
Additional file 5: Table S4.
GO analysis using the DAVID bioinformatics tool. Biological processes of upregulated and downregulated DEPs by student’s t-test are listed. The p-value (modified Fisher exact p-value) cutoff for the GO annotation was set to < 0.05. Genes that were associated with each GO term are represented as official gene symbols. ‘GO direct’ filters extensive GO terms, based on the measured specificity of each term.
Additional file 6: Table S5.
Downstream biological functions and canonical pathways of DEPs by student t-test by IPA analysis. Downstream biological functions and canonical pathways were examined using the IPA informatics tool. The p-value cutoff was set to < 0.05, and the activation Z-score was set to > 1. Proteins in each biological function and pathway are listed. P-values and Z-scores of biological functions and pathways are shown.
Additional file 7: Table S6.
Biological functions of TUBB2A and LTF. Biological functions of TUBB2A and LTF were examined using the IPA and DAVID bioinformatics tools. Biological processes and canonical pathways of TUBB2A and LTF are listed. The p-value cutoff was set to < 0.05 for the IPA analysis. The p-value (modified Fisher exact p-value) cutoff for the GO annotation was set to < 0.05. Proteins in each biological function are listed.
Additional file 8: Table S7.
List of significantly differentially expressed proteins (DEPs) by ANOVA. Statistical significance and p-values for DEPs by ANOVA (p-value < 0.05) in the pooled and individual sample sets. Significantly differentially expressed proteins in six clusters in each sample set are listed. The proteins in clusters 2, 4, and 6 of the pooled sample set were used to perform a bioinformatics analysis of the molecular characteristics of distant metastatic breast cancer between molecular subtypes. Adjusted p-values for DEPs in the pooled sample set are listed (Benjamini-Hochberg FDR cutoff of 0.05).
Additional file 9: Table S8.
Canonical pathways of clusters enriched by IPA analysis. Canonical pathways in clusters 2, 4, and 6 of the pooled sample set were investigated using the IPA informatics tool. Canonical pathways between molecular subtypes are listed. The p-value cutoff was set to < 0.05, and the activation Z-score was set to > 1. P-values and Z-scores of the canonical pathways are listed.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shin, D., Park, J., Han, D. et al. Identification of TUBB2A by quantitative proteomic analysis as a novel biomarker for the prediction of distant metastatic breast cancer. Clin Proteom 17, 16 (2020). https://doi.org/10.1186/s12014-020-09280-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12014-020-09280-z