
Abstract
Co3O4 nanoparticles were prepared using [Co(NH3)6](NO3)2 as a starting material via a solid-state thermal decomposition route at low temperature (150°C). The product was characterized by thermal analysis (thermogravimetric/derivative thermogravimetric/differential thermal analysis), X-ray diffraction, Fourier transform infrared spectroscopy, Raman spectroscopy, Brunauer-Emmett-Teller specific surface area measurement, UV-visible spectroscopy, transmission electron microscopy (TEM), energy-dispersive X-ray spectroscopy, and magnetic measurements. The results confirmed that the nanoparticles are highly pure Co3O4 with weak ferromagnetic properties. TEM images showed that the Co3O4 nanoparticles have an average diameter size of around 13 nm. The optical spectrum indicated two direct bandgaps at 2.3 and 3.2 eV which are blueshifted relative to reported values for the bulk sample. By this fast and simple method, Co3O4 nanoparticles can be produced without expensive and toxic solvents or complicated equipment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Transition metal oxide nanoparticles represent a broad class of materials that have been investigated extensively due to their interesting catalytic, electronic, and magnetic properties relative to those of the bulk counterparts, and the wide scope of their potential applications[1–4]. Among metal oxides, special attention has been focused on the synthesis and properties of spinel-type Co3O4 which is important as a heterogeneous catalyst, solid-state sensor, anode material in Li-ion rechargeable batteries, pigment, electrochromic sensor, and magnetic material and in solar energy storage[5–12]. For these diverse applications, it is of great importance to prepare Co3O4 with well-controlled dimensionality, sizes, and crystal structure. Up to now, several methods have been reported in order to synthesize Co3O4 nanoparticles, including sol–gel method[13], hydrothermal method[14, 15], combustion method[16], microemulsion method[17], chemical spray pyrolysis[18], chemical vapor deposition[19, 20], thermal decomposition of cobalt precursors[21–26], sonochemical route[27, 28], co-precipitation[29], microwave irradiation[30], and mechanochemical processing[31]. However, most of these methods have one or more drawbacks, such as prolonged reaction times, the use of toxic and expensive solvents/reagents, complicated synthetic steps, the use of expensive equipment, and high synthetic temperatures. Therefore, the development of simple, inexpensive, and nontoxic methods for the preparation Co3O4 nanoparticles at relatively low temperature is still demanded.
Among the numerous methods developed for preparing metal oxide nanomaterials, the molecular precursor route has been regarded as one of the most convenient and practical techniques because it not only enables to avoid special instruments, complicated processes, and severe preparation conditions but also provides good control over purity, homogeneity, composition, phase, and microstructure of the resultant products[32–39]. By choosing a proper molecular precursor, coupled with a rational calcining procedure or other decomposition processes[40–43], nanocrystalline products could be obtained usually under the conditions significantly milder than those employed in the conventional solid-state synthesis.
Herein, we report on the preparation of Co3O4 nanoparticles by direct solid-state thermal dissociation of the labile [Co(NH3)6](NO3)2 complex as a new precursor, as well as the characterization of the obtained product by X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FT-IR), Raman spectroscopy, UV-visible (UV–vis) spectroscopy, Brunauer-Emmett-Teller (BET) specific surface area measurement, energy-dispersive X-ray spectroscopy (EDX), transmission electron microscopy (TEM), thermal analysis (thermogravimetric(TG)/derivative thermogravimetric (DTG)/differential thermal analysis (DTA)), and magnetic measurements.
Results and discussion
First, the thermal behavior of the [Co(NH3)6](NO3)2 precursor was studied by thermal analysis. Figure1 shows TG, DTG, and DTA curves recorded for [Co(NH3)6](NO3)2 at a constant heating rate of 10°C min−1in the temperature range of 25°C to 600°C. The TG and DTG curves show that the decomposition of the complex proceeds in two main stages. The first stage that occurred at about 100°C shows 12.20% weight loss which is consistent with the theoretical value of 11.95% caused by the loss of two molecules of NH3 per one molecule of the complex. In the second stage, an extensive weight loss (60%) is observed at about 150°C, which related to the decomposition of the residue [Co(NH3)4](NO3)2 complex. Above 150°C, the weight remained constant, confirming the complete decomposition of the complex. The weight loss of all steps is about 72% which is consistent with the theoretical value (71.85%) calculated for the formation of Co3O4 from the complex. The DTA curve in the inset of Figure1 shows two characteristic peaks. The small endothermic peak at about 100°C can be explained by freeing two NH3 molecules in consistent with TG and DTG data. The residue gives a sharp exothermic peak at about 150°C. This exothermic peak related to the explosive decomposition of the complex via an intramolecular redox process occurring between the reductants (NH3 ligands) and the oxidants (NO3−). The explosive decomposition of the complex resulted in the solid Co3O4 and gaseous products, i.e., NH3, N2, NO, N2O, and H2O. Although not all of the reaction products were determined in the analyses, the decomposition of the complex and formation of the Co3O4 nanoparticles can be expressed as follows:

TG, DTG, and DTA curves of the [Co(NH 3 ) 6 ](NO 3 ) 2 complex.
9[Co(NH3)6](NO3)2(s) → 3Co3O4(s) + 30NH3(g) + 4NO(g) + 2N2O(g) + 17N2(g) + 36H2O(g)
Figure2 shows FT-IR spectra of the [Co(NH3)6](NO3)2 complex and its decomposition products at different temperatures. In Figure2(spectrum a), the characteristic stretching bands of NH3 and NO3 groups are observed at about 3,500 to 3,000, 1,600, 1,350, and 800 cm−1[44]. As shown in Figure2(spectrum b), all the bands associated with the complex clearly disappeared when the complex was decomposed at 150°C. At this temperature, only two characteristic strong bands of the spinel-type Co3O4 structure at about 663.47 and 570.89 cm−1 are observed[45], confirming that the complex was decomposed completely at 150°C to the Co3O4 phase as indicated by the TG/DTA results. In Figure2(spectrum c), the FT-IR spectrum of the sample decomposed at 175°C shows the strong bands related to Co3O4 without obvious change.

FT-IR spectra of (a) the [Co(NH 3 ) 6 ](NO 3 ) 2 complex and its decomposed products at (b) 150°C and (c) 175°C. The black-filled square indicates the Co3O4 phase.
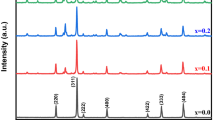
Figure3 presents the XRD patterns of the decomposition products of the [Co(NH3)6](NO3)2 complex at 150°C and 175°C. Well-defined diffraction peaks at about 19.52°, 31.50°, 37.05°, 38.77°, 44.96°, 55.80°, 59.53°, 65.30°, 74.55°, 77.50°, and 78.60° are observed, corresponding to the (111), (220), (311), (222), (400), (422), (511), (440), (620), (533), and (622) planes of Co3O4 crystals, respectively (JCPDS card no. 76–1802). This result confirms that the complex was decomposed completely into the Co3O4 phase at 150°C, in good agreement with the thermal analysis and FT-IR results. No impurity peaks were found in the XRD patterns, confirming that the product is well-crystallized Co3O4 with high purity. As can be seen, the diffraction peaks are broadened because of the small size effect of the nanoparticles. The average crystallite size (d) was calculated to be approximately 13 nm by the Scherrer relation[46]: d = 0.9λ/(B cosθ), where λ is the wavelength of Cu Kα radiation, B is the corrected full width at half maximum of the diffraction peak, and θ is the Bragg angle. As shown in Figure3(pattern b), no new phase was observed when the decomposition temperature increased to 175°C, but the width of the Co3O4 peaks decreased because of crystallite growth.

XRD patterns of the [Co(NH 3 ) 6 ](NO 3 ) 2 complex decomposed at (a) 150°C and (b) 175°C.
Figure4 displays the Raman spectrum of the Co3O4 nanoparticles. As shown in Figure4, the Raman spectrum of the Co3O4 nanoparticles in the range of 300 to 800 nm shows four obvious peaks located at around 468, 510, 605, and 670 cm−1, corresponding to the four Raman-active modes (A1g, Eg, and 2F2g) of Co3O4. The Raman shifts are consistent with those of pure crystalline Co3O4[47, 48], indicating that the Co3O4 nanoparticles have a similar crystal structure to bulk Co3O4. However, compared with those for bulk Co3O4, the peak positions of the four active modes shift to low wavenumbers by about 10 to 20 cm−1[49]. This phenomenon is attributed to the optical phonon confinement effect in nanostructures, which can cause uncertainty in the phonon wave vectors and thus a downshift in the Raman peaks[50]. This result further confirms the formation of the Co3O4 nanoparticles.

Raman spectrum of the Co 3 O 4 nanoparticles.
Figure5 shows TEM images of the Co3O4 powder prepared by thermal decomposition of the [Co(NH3)6](NO3)2 complex at 150°C. The TEM sample was prepared by dispersing the powder in ethanol by ultrasonic vibration. It can be seen that the product was formed from extremely fine spherical particles which were loosely aggregated. The uniform Co3O4 particles have sphere-like shapes with weak agglomeration. As can be seen in the inset of Figure5, the particle sizes possess a narrow distribution in a range of 6 to 20 nm, and the mean particle diameter is about 13 nm. In fact, the mean particle size determined by TEM is very close to the average particle size calculated by the Debye-Scherer formula from the XRD pattern.

TEM images of the Co 3 O 4 nanoparticles in two different magnifications. The inset shows the particle size distribution of Co3O4 nanoparticles.
The EDX spectrum of the product in Figure6 reveals the presence of only cobalt and oxygen peaks, with no other relevant elements present; the large Si peak can be attributed to the Si wafer upon which the nanoparticles were deposited prior to analysis. The atomic percentages of Co and O were found to be 43.15% and 56.95%, respectively. The atomic ratio of Co and O is 3:3.96, which approaches the theoretical value for Co3O4. This observation further confirms that the final product is only highly pure Co3O4 nanoparticles.

EDX analysis of the Co 3 O 4 nanoparticles.
The surface area of the Co3O4 nanoparticles obtained from the decomposition of the complex was measured by the BET method. The specific surface area of the sample was 77.50 m2/g. Assuming that the nanoparticles are almost spherical, as confirmed by TEM, the surface area can be used to estimate the particle size according to DBET = 6,000/(ρ × SBET), where DBET is the diameter of a spherical particle (nm), ρ is the theoretical density of Co3O4 (6.05 g/cm3), and SBET is the specific surface area of Co3O4 powder (m2/g). The particle size calculated from the surface area data is about 12.80 nm, which is in good agreement with the XRD and TEM results.
The optical absorption properties of the as-prepared Co3O4 nanoparticles were investigated at room temperature by UV–vis spectroscopy (Figure7). There are two obvious absorption peaks at 281 and 531 nm. Co3O4 is a p-type semiconductor, and its optical bandgap can be obtained from the following equation[16]: (Ahv)n = B(hv − Eg), where hv is the photon energy (eV), A is the absorption coefficient, B is a constant relative to the material, Eg is the bandgap, and n is either 1/2 for an indirect transition or 2 for a direct transition. The (Ahv)2 versus hv curve for the product is shown in the inset of Figure7. The value of hv extrapolated to (Ahv)2 = 0 gives the absorption bandgap energy. Two regions with a linear relationship are observed in the ranges of 3.8 to 6.1 and 1.9 to 2.5, respectively, giving two Eg values of 3.2 and 2.3 eV. The bandgap of 2.3 eV can be associated with the OII−→CoII charge transfer process (valence to conduction band excitation), while the 3.2 eV bandgap relates to the OII−→CoIII charge transfer (with the CoIII level located below the conduction band)[51]. As has been investigated in the literatures[27, 52], the Eg values of Co3O4 nanoparticles are greater than those of bulk Co3O4 (Eg=1.77 and 3.17 eV, respectively). The increase in the bandgap of the Co3O4 nanoparticles may ascribe to the quantum confinement effects of nanomaterials.

UV–vis spectrum and ( Ahv ) 2 - hv curve (inset) of the Co 3 O 4 nanoparticles prepared at 150°C.
Figure8 shows the magnetic properties of the Co3O4 nanoparticles. The fine shape of the hysteresis loops is a characteristic of a weak ferromagnetic behavior, although bulk Co3O4 is antiferromagnetic[53]. From the inset, the coercive field (Hc) and the remanent magnetization (Mr) are estimated to be 0.015 kOe and 0.002 emu/g, respectively. The low coercive fields and remanent magnetizations confirm that the Co3O4 nanoparticles have weak ferromagnetic properties. The maximum applied field, 8 kOe, does not saturate the magnetizations which should be attributed to weak ferromagnetic ordering of the spins in the nanoparticles. Co3O4 nanoparticles consist of small magnetic domains, each characterized by its own randomly oriented magnetic moment. The total magnetic moment of the nanoparticles is the sum of these magnetic domains coupled by dipolar interactions. The ferromagnetic behavior of the Co3O4 nanoparticles can be explained as follows: bulk Co3O4 has a normal spinel structure with antiferromagnetic exchange between ions occupying tetrahedral and octahedral sites[53]. It has zero net magnetization due to the complete compensation of sublattice magnetizations. Hence, the change from an antiferromagnetic state for bulk Co3O4 to a weakly ferromagnetic state for Co3O4 nanoparticles can be ascribed to uncompensated surface spins and/or finite size effects[54, 55].

Magnetization vs. field ( M - H ) of Co 3 O 4 nanoparticles prepared from [Co(NH 3 ) 6 ](NO 3 ) 2 precursor at 150°C. The inset shows its expansion of magnetization vs. field near the lower applied field.
Conclusions
In this work, we presented a simple and low-temperature method of synthesizing spinel-type Co3O4 nanoparticles with an average particle size of 13.5 nm through thermolysis of the [Co(NH3)6](NO3)2 at low temperature (150°C). Co3O4 nanoparticles are formed from this complex via an explosive redox reaction between NH3 ligands as the reducing agent and the NO3− ions acting as the oxidizing agent. This method yields sphere-like Co3O4 nanoparticles with weak agglomeration, a narrow size distribution, and weak ferromagnetic behavior. The estimated optical absorption bandgaps of the Co3O4 nanoparticles were about 2.3 and 3.2 eV, which are relatively blueshifted, compared with the values for the bulk sample. This approach provides a one-step, general, and inexpensive route for the preparation of Co3O4 nanoparticles with high purity for industrial and high-technology applications.
Methods
Preparation of Co3O4 nanoparticles
Initially, the [Co(NH3)6](NO3)2 complex was synthesized via a simple reaction of an aqueous solution of [Co(H2O)6](NO3)2 with concentrated ammonia solution according to the reported method[56]. By adding acetone to the reaction mixture, the complex was precipitated as an orange precipitate. The composition of the complex was confirmed by thermal analysis, FT-IR, and elemental analysis: anal. calc. for [Co(NH3)6](NO3)2: Co, 20.68; H, 6.32; N, 39.30; found: Co, 21.10; H, 6.25; N, 39.89. To prepare Co3O4 nanoparticles, the [Co(NH3)6](NO3)2 complex was decomposed at selected temperatures for 1 h in an electric furnace under ambient air. The temperatures were selected according to the thermoanalytical data. The decomposition product was collected for characterization.
Characterization techniques
The XRD patterns were recorded by a Rigaku D-max C III X-ray diffractometer (Rigaku Corporation, Shibuya-ku, Japan) using Ni-filtered Cu Kα radiation (λ = 1.5418 Å) in order to determine the phases present in the decomposed samples. Infrared spectra were recorded on a Schimadzu system 160 FT-IR spectrophotometer (Shimadzu Corporation, Kyoto, Japan) using KBr pellets. The Raman spectra were measured on a Spex 1403 Raman spectrometer. Thermal analysis was conducted with a Netzsch STA 409 PC/PG thermal analyzer at a heating rate of 5°C min−1 in air. The optical absorption spectrum was recorded on a Shimadzu 1650PC UV–vis spectrophotometer in a wavelength range of 200 to 700 nm at room temperature. The sample for UV–vis studies was well dispersed in distilled water to form a homogeneous suspension by sonication for 25 min. The particle size was determined using a transmission electron microscope (Philips CM10, Philips, Amsterdam, The Netherlands) equipped with an energy dispersive X-ray analyzer. For the TEM measurements, the powders were ultrasonicated in ethanol, and a drop of the suspension was dried on a carbon-coated microgrid. The specific surface area of the product was measured by the BET method using N2 adsorption-desorption isotherm carried out at −196°C on a surface area analyzer (Micromeritics ASAP 2010, Micromeritics, Norcross, GA, USA). Before each measurement, the sample was degassed at 150°C for 2 h. A vibrating sample magnetometer (Meghnatis Daghigh Kavir Co., Kashan, Iran) was used to measure the magnetic properties of Co3O4 nanoparticles.
Authors’ information
SF is a professor at the Department of Chemistry, Lorestan University, Iran. He received his BSc degree in Chemistry from Shahid Chamran University in 1991, his MSc degree in Inorganic Chemistry from Tehran University in 1994, and his Ph.D. in Inorganic Chemistry from Isfahan University, Iran, in 2000. His recent research has concentrated on synthesis and characterization of metal and metal oxides nanostructures, and their catalytic applications. KP obtained her BSc and MSc degrees in Chemistry from the Faculty of Science, Lorestan University, Iran, in 2008 and 2012, respectively. During her MSc course, she has been involved in synthesizing cobalt oxide nanostructures through thermal decomposition of some ammine complexes. Her research interest is in the area of nanomaterial preparation. SS obtained her BSc degree in Applied Chemistry in 2009 from the Faculty of Science, Arak University, Iran. She earned her MSc degree in Inorganic Chemistry in 2012 with the thesis entitled ‘Preparation and characterization of silver nanoparticles via chemical reduction with hydrogen peroxide (H2O2),’ from the Faculty of Science, Lorestan University, Iran. Her research area interests include metals and metal oxide nanomaterials.
References
He JH, Wu TH, Hsin CL, Li KM, Chen LJ, Chueh YL: Beak-like SnO 2 nanorods with strong photoluminescent and field-emission properties. Small 2006, 2: 116–120. 10.1002/smll.200500210
Xu LP, Sithambaram S, Zhang YS, Chen CH, Jin L, Joesten R, Suib SL: Novel urchin-like CuO synthesized by a facile reflux method with efficient olefin epoxidation catalytic performance. Chem. Mater. 2009, 21: 1253–1259. 10.1021/cm802915m
Hu CC, Wu YT, Chang KH: Low-temperature hydrothermal synthesis of Mn 3 O 4 and MnOOH single crystals: determinant influence of oxidants. Chem. Mater. 2008, 20: 2890–2894. 10.1021/cm703245k
Zhou J, Ding Y, Deng SZ, Gong L, Xu NS, Wang ZL: Three-dimensional tungsten oxide nanowire networks. Adv. Mater. 2005, 17: 2107–2110. 10.1002/adma.200500885
Lou XW, Deng D, Lee JY, Feng J, Archer LA: Self-supported formation of needlelike Co 3 O 4 nanotubes and their application as lithium-ion battery electrodes. Adv. Mater. 2008, 20: 258–262. 10.1002/adma.200702412
Casas-Cabanas M, Binotto G, Larcher D, Lecup A, Giordani V, Tarascon JM: Defect chemistry and catalytic activity of nanosized Co 3 O 4 . Chem. Mater. 2009, 21: 1939–1947. 10.1021/cm900328g
Li W-Y, Xu L-N, Chen J: Co 3 O 4 nanomaterials in lithium-ion batteries and gas sensors. Adv. Funct. Mater. 2005, 15: 851–857. 10.1002/adfm.200400429
Chou S-L, Wang J-Z, Liu H-K, Dou S-X: Electrochemical deposition of porous Co 3 O 4 nanostructured thin film for lithium-ion battery. J. Power. Sources 2008, 182: 359–364. 10.1016/j.jpowsour.2008.03.083
Askarinejad A, Bagherzadeh M, Morsali A: Catalytic performance of Mn 3 O 4 and Co 3 O 4 nanocrystals prepared by sonochemical method in epoxidation of styrene and cyclooctene. Appl. Surface Sci. 2010, 256: 6678–6682. 10.1016/j.apsusc.2010.04.069
Li YG, Tan B, Wu YY: Mesoporous Co 3 O 4 nanowire arrays for lithium ion batteries with high capacity and rate capacity. Nano Lett. 2008, 8: 265–270. 10.1021/nl0725906
Mate VR, Shirai M, Rode CV: Heterogeneous Co 3 O 4 catalyst for selective oxidation of aqueous veratryl alcohol using molecular oxygen. Catal. Commun. 2013, 33: 66–69.
Makhlouf SA: Magnetic properties of Co 3 O 4 nanoparticles. J. Magn. Magn. Mater. 2002, 246: 184–190. 10.1016/S0304-8853(02)00050-1
Baydi ME, Poillerat G, Rehspringer JL, Gautier JL, Koenig JF, Chartier P: A sol–gel route for the preparation of Co 3 O 4 catalyst for oxygen electrocatalysis in alkaline medium. J. Solid State Chem. 1994, 109: 281–288. 10.1006/jssc.1994.1105
Chen Y, Zhang Y, Fu S: Synthesis and characterization of Co 3 O 4 hollow spheres. Mater. Lett. 2007, 61: 701–705. 10.1016/j.matlet.2006.05.046
Li L, Chu Y, Liu Y, Song JL, Wang D, Du XW: A facile hydrothermal route to synthesize novel Co 3 O 4 nanoplates. Mater. Lett. 2008, 62: 1507–1510. 10.1016/j.matlet.2007.09.012
Gu F, Li C, Hu Y, Zhang L: Synthesis and optical characterization of Co 3 O 4 nanocrystals. J. Cryst. Growth 2007, 304: 369–373. 10.1016/j.jcrysgro.2007.03.040
Wang RM, Liu CM, Zhang HZ, Chen CP, Guo L, Xu HB, Yang SH: Porous nanotubes of Co 3 O 4 : synthesis, characterization and magnetic properties. Appl. Phys. Lett. 2004, 85: 2080–2082. 10.1063/1.1789577
Kim DY, Ju SH, Koo HY, Hong SK, Kangf YC: Synthesis of nanosized Co 3 O 4 particles by spray pyrolysis. J. Alloys Compd. 2006, 417: 254–258. 10.1016/j.jallcom.2005.09.013
Mane AU, Shalini K, Wohlfart A, Devi A, Shivashankar SA: Strongly oriented thin films of Co 3 O 4 deposited on single-crystal MgO(100) by low-pressure, low-temperature MOCVD. J. Cryst. Growth 2002, 240: 157–163. 10.1016/S0022-0248(02)00860-6
Xuan Y, Liu R, Jia YQ: Synthesis of a new series of compounds RE 2 Co 2/3 Nb 4/3 O 7 and stability field diagram of RE 2 B 2/3 ′B 4/3 ″O 7 pyrochlore compounds. Mater. Chem. Phys. 1998, 53: 256–261. 10.1016/S0254-0584(98)00002-9
Rumplecker A, Kleitz F, Salabas EL, Schüth F: Hard templating pathways for the synthesis of nanostructured porous Co 3 O 4 . Chem. Mater. 2007, 19: 485–496. 10.1021/cm0610635
Wang WW, Zhu YJ: Microwave-assisted synthesis of cobalt oxalate nanorods and their thermal conversion to Co 3 O 4 rods. Mater. Res. Bull. 2005, 40: 1929–1935. 10.1016/j.materresbull.2005.06.004
Yang LX, Zhu YJ, Li L, Zhang L, Tong H, Wang WW: A facile hydrothermal route to flower-like cobalt hydroxide and oxide. Eur. J. Inorg. Chem. 2006, 23: 4787–4792.
Mohandes F, Davar F, Salavati-Niasari M: Preparation of Co 3 O 4 nanoparticles by nonhydrolytic thermolysis of [Co(Pht)(H 2 O)] n polymers. J. Magn. Magn. Mater. 2010, 322: 872–877. 10.1016/j.jmmm.2009.11.019
Jiang J, Li L: Synthesis of sphere-like Co 3 O 4 nanocrystals via a simple polyol route. Mater. Lett. 2007, 61: 4894–4896. 10.1016/j.matlet.2007.03.067
Ren L, Wang P, Han Y, Hu C, Wei B: Synthesis of CoC 2 O 4 ·2H 2 O nanorods and their thermal decomposition to Co 3 O 4 nanoparticles. Mater. Phys. Lett. 2009, 476: 78–83.
Kumar RV, Diamant Y, Gedanken A: Synthesis and characterization of nanometer-size transition metal oxides from metal acetates. Chem. Mater. 2000, 12: 2301–2305. 10.1021/cm000166z
Oh SW, Bang HJ, Bae YC, Sun Y-K: Effect of calcination temperature on morphology, crystallinity and electrochemical properties of nano-crystalline metal oxides (Co 3 O 4 , CuO, and NiO) prepared via ultrasonic spray pyrolysis. J. Power. Sources 2007, 173: 502–509. 10.1016/j.jpowsour.2007.04.087
Lai T, Lai Y, Lee C, Shu Y, Wang C: Microwave-assisted rapid fabrication of Co 3 O 4 nanorods and application to the degradation of phenol. Catal. Today 2008, 131: 105–110. 10.1016/j.cattod.2007.10.039
Bhatt AS, Bhat DK, Tai C-W, Santosh MS: Microwave-assisted synthesis and magnetic studies of cobalt oxide nanoparticles. Mater. Chem. Phys. 2011, 125: 347–350. 10.1016/j.matchemphys.2010.11.003
Wang X, Chen XY, Gao LS, Zheng HG, Zhang Z, Qian YT: One-dimensional arrays of Co 3 O 4 nanoparticles: synthesis, characterization, and optical and electrochemical properties. J. Phys. Chem. B 2004, 108: 16401–16404. 10.1021/jp048016p
Traversa E, Sakamoto M, Sadaoka Y: A chemical route for the preparation of nanosized rare earth Perovskite-type oxides for electroceramic applications. Part. Sci. Technol. 1998, 16: 185–214. 10.1080/02726359808906794
Salavati-Niasari M, Davar F, Mazaheri M: Preparation of ZnO nanoparticles from [bis(acetylacetonato)zinc(II)]–oleylamine complex by thermal decomposition. Mater. Lett. 2008, 62: 1890–1892. 10.1016/j.matlet.2007.10.032
Salavati-Niasari M, Davar F: Synthesis of copper and copper(I) oxide nanoparticles by thermal decomposition of a new precursor. Mater. Lett. 2009, 63: 441–443. 10.1016/j.matlet.2008.11.023
Salavati-Niasari M, Davar F, Mazaheri M: Synthesis and characterization of ZnS nanoclusters via hydrothermal processing from [bis(salicylidene)zinc(II)]. J. Alloys Compd. 2009, 470: 502–506. 10.1016/j.jallcom.2008.03.048
Davar F, Fereshteh Z, Salavati-Niasari M: Nanoparticles Ni and NiO: synthesis, characterization and magnetic properties. J. Alloys Compd. 2009, 476: 797–801. 10.1016/j.jallcom.2008.09.121
Salavati-Niasari M, Fereshteh Z, Davar F: Synthesis of cobalt nanoparticles from [bis(2-hydroxyacetophenato)cobalt(II)] by thermal decomposition. Polyhedron 2009, 28: 1065–1068. 10.1016/j.poly.2009.01.012
Salavati-Niasari M, Khansari A, Davar F: Synthesis and characterization of cobalt oxide nanoparticles by thermal treatment process. Inorg. Chim. Acta 2009, 362: 4937–4942. 10.1016/j.ica.2009.07.023
Salavati-Niasari M, Mir N, Davar F: Synthesis and characterization of Co 3 O 4 nanorods by thermal decomposition of cobalt oxalate. J. Phys. Chem. Solids 2009, 70: 847–852. 10.1016/j.jpcs.2009.04.006
Farhadi S, Rashidi N: Preparation and characterization of pure single-phase BiFeO 3 nanoparticles through thermal decomposition of the heteronuclear Bi[Fe(CN) 6 ]·5H 2 O complex. Polyhedron 2010, 29: 2959–2965. 10.1016/j.poly.2010.08.019
Farhadi S, Roostaei-Zaniyani Z: Simple and low-temperature synthesis of NiO nanoparticles through solid-state thermal decomposition of the hexa(ammine)Ni(II) nitrate, [Ni(NH 3 ) 6 ](NO 3 ) 2 , complex. Polyhedron 2011, 30: 1244–1249. 10.1016/j.poly.2011.01.028
Farhadi S, Safabakhsh J: Solid-state thermal decomposition of the [Co(NH 3 ) 5 CO 3 ]NO 3 ·0.5H 2 O complex: a simple, rapid and low-temperature synthetic route to Co3O4 nanoparticles. J. Alloys Compd. 2012, 515: 180–185.
Farhadi S, Pourzare K: Simple and low-temperature preparation of Co 3 O 4 sphere-like nanoparticles via solid-state thermolysis of the [Co(NH 3 ) 6 ](NO 3 ) 3 complex. Mater. Res. Bull. 2012, 47: 1550–1556. 10.1016/j.materresbull.2012.02.028
Nakamoto K: Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry. 6th edition. New York: Wiley; 2009.
Pejova B, Isahi A, Najdoski M: Fabrication and characterization of nanocrystalline cobalt oxide thin films. Mater. Res. Bull. 2001, 36: 161–170. 10.1016/S0025-5408(00)00479-7
Klug HP, Alexander LE: X-ray Diffraction Procedures. 2nd edition. New York: Wiley; 1964.
Marinkovic Stanojevic ZV, Romcevic N, Stojanovic B: Spectroscopic study of spinel ZnCr 2 O 4 obtained from mechanically activated ZnO–Cr 2 O 3 mixtures. J. Eur. Ceram. Soc. 2007, 27: 903. 10.1016/j.jeurceramsoc.2006.04.057
Ramana CV, Massot M, Julien CM: XPS and Raman spectroscopic characterization of LiMn 2 O 4 spinels. Surf. Interface Anal. 2005, 37: 412. 10.1002/sia.2022
Hadjiev VG, Iliev MN, Vergilov IV: The Raman spectra of Co 3 O 4 . J. Phys. C. Solid State Phys. 1988, 21: L199. 10.1088/0022-3719/21/7/007
Cao B, Cai W, Duan G, Li Y, Zhao Q, Yu D: Template-free electrochemical deposition route to ZnO nanoneedle arrays and their optical and field emission properties. Nanotechnology 2005, 16: 2567. 10.1088/0957-4484/16/11/017
He T, Chen DR, Jiao XL, Wang YL, Duan YZ: Solubility-controlled synthesis of high-quality Co 3 O 4 nanocrystals. Chem. Mater. 2005, 17: 4023–4030. 10.1021/cm050727s
Gulino A, Dapporto P, Rossi P, Fragala I: A novel self-liquid MOCVD precursor for Co 3 O 4 thin films. Chem. Mater. 2003, 15: 3748–3752. 10.1021/cm034305z
Ichiyanagi Y, Kimishima Y, Yamada S: Magnetic study on Co 3 O 4 nanoparticles. J. Magn. Magn. Mater. 2004, 272–276: e1245-e1246.
Kodama RH, Makhlouf SA, Berkowitz AE: Growth mechanism and magnon excitation in NiO nanowalls. Phys. Rev. Lett. 1997, 79: 1393–1396. 10.1103/PhysRevLett.79.1393
Ozkaya T, Baykal A, Toprak MS, Koseoglu Y, Durmus Z: Reflux synthesis of Co 3 O 4 nanoparticles and its magnetic characterization. J. Magn. Magn. Mater. 2009, 321: 2145–2149. 10.1016/j.jmmm.2009.01.003
Liszka-skoczylas M, Mikuli E, Szklarzewicz J, Hetmanczyk J: Thermal behaviour, phase transition and molecular motions in [Co(NH 3 ) 6 ](NO 3 ) 2 . Thermochim. Acta 2009, 496: 38–44. 10.1016/j.tca.2009.06.017
Acknowledgements
This work was supported by the Lorestan University Research Council and also supported by Iran Nanotechnology Initiative Council (INIC).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SF participated in the idea of the study, the design of the study, interpretation of the results, and writing the manuscript for publication. KP carried out the synthesis and the physicochemical characterization of the Co3O4 nanoparticles and early drafted the manuscript. SS participated in the discussions and interpretation of all characterization results. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Farhadi, S., Pourzare, K. & Sadeghinejad, S. Simple preparation of ferromagnetic Co3O4 nanoparticles by thermal dissociation of the [CoII(NH3)6](NO3)2 complex at low temperature. J Nanostruct Chem 3, 16 (2013). https://doi.org/10.1186/2193-8865-3-16
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2193-8865-3-16