
Abstract
Small cell carcinoma (SmCC) is a distinct clinicopathological entity first described in the lung. It represents approximately 15% of all bronchogenic carcinoma. Extrapulmonary small cell carcinoma (EPSmCC) morphologically indistinguishable from small cell lung cancer (SCLC) was first reported in 1930. Since its first description, EPSmCC has been reported in virtually all anatomical sites, including: gynecologic organs (ovary and cervix); genitourinary organs (urinary bladder and prostate); the gastrointestinal tract (esophagus); skin (Merkel cell carcinoma) and head and neck region. Regardless of the anatomic sites, all SmCCs have similar, if not identical, histo-pathology features and immunohistochemical profile. SmCC is one of the most aggressive malignancies. The molecular mechanisms underlying its development and progression remain poorly understood. Herein, we reviewed the literature in SmCC in respect to its site of occurrence, clinical features, immunohistochemical characteristics. SmCCs have heterogeneous molecular mutations. Dinstinct genetic alterations associated with SmCC from different body sites were reviewed. Some genetic alterations such as RB1, TP53 are commonly seen in different origins of SmCC. Other genes with site specificity were also summarized, such as bladder SmCC with TERT promoter mutations; prostate SmCC with ERG translocations; ovarian SmCC with SMARCA4 mutations; Merkel cell carcinoma (skin) and cervical SmCC with Merkel cell polyomavirus (MCV or MCPyV) and human papillomavirus (HPV). Further studies are needed to employ a genetically oriented approach for the diagnosis and therapy of SmCC.
Similar content being viewed by others

Introduction
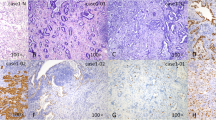
Small-cell carcinoma ("oat-cell carcinoma") is a type of highly malignant cancer that commonly arises in the lung. Uncommonly, small-cell carcinoma arising from outside of the lungs and pleura is referred to as extrapulmonary small-cell carcinoma (EPSmCC). The diagnosis of SmCC is primarily based on path-histologic criteria: sheets, ribbons, clusters, rosettes or peripheral palisading of small to medium sized (2-4x neutrophils) round/oval cells with minimal cytoplasm, salt and pepper chromatin without prominent clumps, hyperchromatic, indistinct nucleoli, nuclear molding, smudging, frequent mitotic figures (Figure 1A). In addition, SmCC cells show neuroendocrine differentiation and are positive for neuroendocrine tissue markers: chromogranin and synaptophysin (Figure 1B and C). The clinical behavior of SmCC from different anatomic sites are quite similar. SmCC cells usually metastasize very early, respond dramatically to chemotherapy (CT) and radiation therapy (RT) [1]. Patients usually have a very poor prognosis and short survival time despite treatment. Treatment of SmCC of lung and EPSmCC is similar. However, recent evidence at molecular and genetic levels suggests that SmCC from different anatomic sites may have distinct genetic biomarkers and is a heterogenous group of diseases. This conceptual change is critical for better understanding of these aggressive malignancies and may lead to a genetically oriented approach for the diagnosis and targeted therapy of SmCC.

Morphology of small cell carcinoma. H&E staining x400 (A); Immunohistochemistry of chromogranin (B) and synaptophysin (C).
I. Small-cell lung cancer
Small-cell lung cancer (SCLC), representing 15% of all bronchogenic carcinoma cases, is a distinct subtype associated with a typical clinical picture of early metastasis. Chemotherapy alone or combined with radiation, but not surgery, is the usual treatment of choice for small cell lung cancer. On this regimen, a large percentage of patients experiences remission. The 5-year survival for small cell lung cancer (6%) is however much lower than that for non-small cell lung cancer (NSCLC) (18%). One major reason is that targeted therapy has been widely used for NSCLC treatment, and mutation analysis is routinely done now for EGFR, KRAS, or ALK. More and more novel agents for targeted therapy of NSCLC are being developed [2–4].
As for SmCC, several tumor suppressor genes are inactivated, including TP53 (80–90% of cases, [5]) RB1 (60–90% of cases [6, 7]) and PTEN (13% of cases [8]). In mice, SCLC is initiated by deletion of two tumor suppressor genes (RB1 and TP53). Deletion of these two genes produces a model that recapitulates the clinical features of human SCLC. Infrequent activating mutations have also been found in PIK3CA, EGFR and KRAS (all 10% or lower). In addition, MYC is amplified in 20% of cases [9]. Mean levels of total PARP1 (a DNA repair protein and E2F1 co-activator) were higher in SCLC cell lines than in NSCLC cell lines, and SCLC growth was inhibited by PARP1 and EZH2 knockdown [10].
II. Small cell carcinoma of genitourinary tract
The genitourinary tract is the most common extrapulmonary site for EPSmCC, with approximately 900 new cases diagnosed every year in the United States [11]. The most common sites for SmCC of the genitourinary tract are the urinary bladder and prostate; however, it is still very rare, accounting for only 0.7% and 0.5% of all bladder and prostate cancer, respectively. SmCC of the genitourinary tract is an aggressive cancer, with a poor prognosis overall. Although there is no standard of care, patients are treated using a multimodality approach analogous to those used in the treatment of small-cell lung cancer [12].
-
1.
Renal SmCC is an extremely rare malignancy and accounts for less than 1% of all renal tumors. Although renal SmCC shares similar morphological and immunohistochemical features with SmCC of other organs, renal SmCC often (60-70%) coexists with conventional urothelial carcinoma, suggesting a potential association between SmCC and urothelial carcinoma in the kidney [13, 14]. Renal SmCC affects patients of various ages and appears to be more common in men (male–female ratio = 2:1) [14]. Its clinical presentations are similar to those of renal cell carcinoma or urothelial carcinoma, and it is histologically and immunohistochemically indistinguishable from its pulmonary or other EPSmCC counterparts. Most patients present at an advanced stage with widespread metastases and a dismal prognosis despite multimodal therapy. However, if the disease is found and treated early, long-term survival may be possible for patients with an organ-confined tumor [13].
-
2.
Small cell carcinoma of the urinary bladder
SmCC of urinary bladder is a malignant neuroendocrine neoplasm derived from the urothelium which histologically mimics its pulmonary counterpart. A recent analysis from the Surveillance, Epidemiology, and End Results of the SmCC of the urinary bladder database indicated a significant rise in the incidence of the SmCC of the urinary bladder in the United States from 0.05 to 0.14 cases per 100,000 people between 1991 and 2005 [15]. This is likely due to increase in the U.S. population’s age. Similar to other bladder cancers, risk factors for SmCC of urinary bladder include smoking, male sex (male-female ratio of 3:1), and advanced age. The average age of incidence was found to be 71.7 ± 11.2 years, with a median age of 73 years [15]. Caucasians were most commonly affected, with a white-nonwhite ratio of 10:1. More than 60% of the SmCC of the lung has metastatic disease at the time of diagnosis. Similar rates have been shown in SmCC of the urinary bladder [11]. Chemotherapy is the mainstay of treatment, with proven survival benefit [16].
One study of immunohistochemical (IHC) stains in SmCC of urinary bladder has shown that nuclear GATA3 expression was encountered in 7 bladder (7/22, 32%), 2 lung (2/15, 13%), and 0 (0/33, 0%) prostate SmCC [17]. TTF-1 expression in SmCC of urinary bladder was found in 40% of the tumors in 2 studies, demonstrating that TTF1 can be expressed in EPSmCC [18, 19]. SmCC of urinary bladder are also stained positive with the epithelial markers: CAM 5.2, CK7, and EMA in 59%, 41%, and 77.7% of the cases, respectively. This supports the urothelial origin of SmCC of urinary bladder [18, 20]. Distingushing SmCC of the prostate and from that of the bladder can be very challenging, if even possible, because of low positivity of GATA3 in SmCC of the bladder and low positivity of PSA and other prostatic markers, such as P501S, in SmCC of the prostate [21, 22].
TERT promoter mutations, originally discovered in ~70% of melanomas, have also been found to be the most common form of genetic mutations in urothelial carcinomas. Interestingly, these mutations have very low incidence in other prevalent carcinomas: lung, prostate and colon cancers. Multiple studies [23–25], including our unpublished results, demonstrated that up to 70-80% of urothelial carcinoma carries the TERT promoter mutations irrespective of grade, stage or location. Our recent study [10] showed that 100% (10 cases) of SmCC of the urinary bladder carry TERT promoter mutation C228T, yet none of SmCC from all other origins including prostate, lung, cervix, esophagus, and skin (Merkel cell carcinoma) contain the TERT promoter mutations. This study indicated that the TERT promoter mutation may be a biomarker to distinguish SmCC of the urinary bladder from SmCC of other origins.
3. Small cell carcinoma of the prostate
SmCC of the prostate was first described by Wenk et al. [26] more than 30 years ago. Since then, it has been reported to occur in 0.5–2% of men with prostate cancer, although autopsy studies of men who have died of castration-resistant prostate cancer have reported the presence of SmCC in up to 10–20% of cases [27, 28]. Neuroendocrine markers such as chromogranin A and synaptophysin are expressed in nearly all cases of conventional prostatic adenocarcinoma, with the proportion of cells that stain positive for these markers increasing during castration [29]. In 24% and 35% of cases, p63 and high-molecular-weight cytokeratin were noted to be positive, which are typically negative in prostatic adenocarcinoma [21]. Studies have demonstrated thyroid transcription factor 1 (TTF-1) expression in over 50% of SmCC of the prostate, limiting its utility in distinguishing primary SmCC of the prostate from metastatic SmCC of the lung [22]. PSA and other prostatic markers, such as P501S, are only positive in about 17–25% cases, often focally [21, 22]. These results demonstrated that IHC stains have very limited value to discriminate SmCC of prostate from SCLC.
Interestingly, both conventional prostatic adenocarcinoma and SmCC of prostate share ERG gene rearrangement which is absent in SmCC from other body sites. This rearrangement occurs between an androgenregulated gene, TMPRSS2 (21q22.3) and an ETS transcription factor family member, most commonly ERG (21q22.2), resulting in a gene fusion product, TMPRSS2–ERG gene fusions. This result not only indicates a common clonal origin between conventional prostatic adenocarcinoma and SmCC of prostate, but also implies the clinical use of ERG gene rearrangement as a biomarker to confirm a prostatic origin for SmCC [30, 31].
III. Small cell carcinoma of gynecologic tract
The SmCC of gynecologic tract is one of the common EPSmCCs, representing up to 2% of all gynecologic malignancies [32, 33]. Reported gynecologic sites include the cervix, endometrium, ovary, fallopian tube, vagina and vulva.
1. Small cell carcinoma of the uterine cervix (SmCCC)
The uterine cervix is the most common gynecologic tract site involved with EPSmCCs. However, SmCCC is a very rare disease, representing only 1% to 3% of all uterine cervical cancers. SmCCC often coexists with conventional squamous cell carcinoma or adenocarcinoma. Depending on the series analyzed and the selection criteria employed, between 11% and 64% of SmCCC cases present admixed histology [34, 35]. Immunohistochemical studies have further revealed that the majority of cases show diffuse nuclear and cytoplasmic p16 positivity [36, 37].
The critical role of human papillomaviruses (HPV) in the carcinogenesis of conventional cervical cancer is well established. The prevalence of the different high-risk HPV types in SmCCC has been preliminarily established and reported to range from 50% to 100% [38, 39]. It has been found that unlike in squamous cell carcinoma of the cervix, HPV 18 may be the most prevalent type of SmCCC [37, 40]. Given this evidence, SmCCC, like other types of cervical cancer, seems to be associated with high-risk HPV infection. For the purpose of differential diagnosis, HPV is specific for SmCC of cervical origin other than ovary or lung, but p16 immunohistochemistry is not useful for this purpose [36].
2. Small-cell carcinoma of the ovary
Small-cell ovarian carcinoma is divided into two categories: pulmonary type (SCCOPT) and hypercalcemic type (SCCOHT). The cellular features and neuroendocrine markers of the pulmonary type resemble small-cell carcinoma of the lung, whereas the immunohistochemical markers and microscopic and ultrastructural examination of SCCOHT do not [41]. Both of these tumors are uncommon, but SCCOPT is extremely rare with only approximately 20 cases reported in the English literatures.
SCCOPT
The mean age of diagnosis of patients with SCCOPT is 51 years (22-85 years) [42]. Bilateral disease is present in about half of the cases and all cases lack hypercalcemia [42]. TTF-1 was found to be diffusely positive in one case but absent in another case [36]. The diagnosis of CSCOPT is exclusion of metastasis of SmCC from other locations, in particular lung. One report showed that malignant transformation of ovarian mature cystic teratoma with a predominant pulmonary type small cell carcinoma component, is CDX2 positive [43].
SCCOHT
SCCOHT represents less than 1% of all ovarian cancer diagnoses, with fewer than 300 cases reported in the literature thus far [44, 45]. The mean age of diagnosis is 23 years, and the majority of affected women present with early-stage disease. Nonetheless, most patients relapse and die within 2 years of diagnosis, regardless of tumor stage, with a long-term survival rate of only 33%, even when disease is confined to the ovary at diagnosis. The tumor appears nearly almost unilaterally, mostly affecting the right ovary [44, 46].
Recently 3 independent studies reported that SCCOHT is a monogenic disease caused by mutations in the SMARCA4 gene. Whole-exome sequencing on DNA obtained from 24 familial or sporadic cases of SCCOHT revealed that 22 of the 24 cases analyzed were due to SMARCA4 mutations; and Immunohistochemical analysis of these cases and additional familial and non-familial cases showed loss of SMARCA4 (BRG1) protein in 38 of 40 tumors [47]. Furthermore, the researchers suggest that SCCOHT tumors are essentially malignant rhabdoid tumors of the ovary, “they are not always comprised of small cells, are not carcinomas, and only two thirds have hypercalcemia”. It is, therefore, possible that chemotherapeutic regimens used to treat rhaboid tumors might help improve the outcome of this disease. Ramos et al. [48] reported that germline and somatic inactivating mutations in the SWI/SNF chromatin-remodeling gene SMARCA4 in 75% (9/12) of SCCOHT cases in addition to SMARCA4 protein loss in 82% (14/17) of SCCOHT tumors but in only 0.4% (2/485) of other primary ovarian tumors. Witkowski et al also reported similar results. These new pieces of evidence demonstrated that alterations in SMARCA4, the major cause of SCCOHT, could lead to improvements in genetic counseling and new treatment approaches [46].
IV. Merkel cell carcinoma (MCC)
MCC is a rare neuroendocrine tumor of the skin with rising incidence and an aggressive behavior. The annual incidence of MCC is 0.6 per 100,000 persons and is increasing (approximately 1,600 new cases per year in the US) [49]. Histologically, MCC shares numerous features with SmCC. Immunohistochemically, MCC stains positive for synaptophysin and chromogranin [50]. cytokeratin CK-20 is positive in 89–100% of Merkel cell tumors with a punctate pattern and may be used to distinguish MCC from other tumor types. However, 33% of small cell lung cancers (SCLCs) and 3–4% of EPSmCCs also stain positively for CK20 [51].
Merkel cell polyomavirus (MCV), a new human polyomavirus, is clonally integrated in 70–80% of Merkel cell carcinoma (MCC) tumors. MCV is part of the normal, healthy skin flora but causes cancer after viral genome mutations eliminate its replication capacity. While similar to known polyomaviruses, MCV oncogenes act in new ways, such as activation of the survivin oncoprotein and PP2A-independent targeting of cap-dependent translation [52, 53]. Survivin inhibition improves survival of mice bearing human MCC xenografts [54].
V. The origin of small cell carcinoma
The identification of the cell type(s) from which small cell carcinoma originates is critical in the development of methods for early diagnosis and treatment.
1. The cell of origin for small cell lung cancer
The usage of cell type-restricted Adeno-Cre vectors to distinct cell populations in the lung of adult mouse showed that loss of TP53 and RB1 can efficiently transform neuroendocrine and Surfactant Protein C (SPC)-expressing cells to SCLC, albeit SPC-expressing cells at a lesser efficiency. In contrast, Clara cells were largely resistant to transformation. These results indicate that although neuroendocrine cells serve as the predominant cell of origin for SCLC, a subset of SPC-expressing cells may also be endowed with this ability [55, 56].
2. The cell of origin for EPSmCCs
The origin of EPSmCCs is controversial. It was assumed that these neoplasms arise from neuroendocrine cells in the Amine Precursor Uptake and Decarboxylation (APUD) system [57, 58]. To date, however, it is thought that the origin of EPSmCCs is either a totipotent stem cell capable of differentiating into a variety of cell types, or that elements of SmCC arise as a late-stage phenomenon in the genetic progression of carcinomas [57]. The presence of mixed carcinomas may have implications for the origin of EPSmCC, which may arise from multipotent stem cells that retain the ability to differentiate into various tissue types. Evidence for this hypothesis comes from an identical pattern of allelic loss in SmCC of urinary bladder mixed with urothelial carcinoma (UC) [59]. For bladder localizations, a malignant transformation of the neuroendocrine cells physiologically located in the urothelium has been proposed [60]. Molecular genetic studies have also suggested a common clonal origin for the coexisting bladder SmCC and conventional UC [59, 61]. X chromosomal inactivation analysis [59] in females illustrated the same nonrandom inactivation in both SmCC of the urinary bladder and UC. Identical point mutations of TP53 were found in invasive bladder SmCC and coexisting UC in situ; additionally, no loss of heterozygosity of 9 microsatellite markers and TP53 was found in either component. This study provided evidence for the development of bladder SmCC out of bladder UC in situ [61].
Other theories of histogenesis include metaplasia from other high-grade malignancies. Neuroendocrine (NE) cells are detected by IHC in some in-situ cervical adenocarcinomas, and these may be the origin of cervical NE carcinomas [62]. Further support for this theory comes from a study where identical loci with loss of heterozygosity were demonstrated in mixed adenocarcinoma and EPSmCC of the appendix, and an additional locus was found in the EPSmCC component [63]. Similarly, additional loci with loss of heterozygosity were noted in breast EPSmCC compared with adjacent ductal carcinoma in situ.
There is a high concordance rate of ERG rearrangement between the SmCC of prostate and prostatic acinar components in a given patient; however, the absence of ERG rearrangement in bladder or lung small cell carcinomas supports a common origin for these two subtypes of prostate cancer.
In one report, malignant transformation of ovarian mature cystic teratoma consists of a predominant pulmonary type SmCC component (65%), as well as minor components including adenocarcinoma (25%), squamous cell carcinoma (5%), and transitional cell carcinoma (5%). CDX2 positivity was retained in all of the carcinomas. It may suggest that the adenocarcinoma had arisen from an intestinal epithelium in the mature cystic teratoma and then differentiated into the diverse histological types mentioned above [43].
Recent reports [46, 48] also revealed that small cell carcinoma of the ovary, hypercalcemic type, display frequent inactivating germline and somatic mutations in SMARCA4, a mutation rare in other common tumors. It has been well established that Merkel cell virus is strongly associated with only Merkel cell carcinoma, but not SmCC of any other origins [52].
Loss of RB1 function is associated with the development of neuroendocrine tumors, including prostate, pituitary, thyroid and adrenal gland tumors [64–67]. This mutation may give this type of cancer mutual morphologic features. Loss of RB1 by deletion is a common event in prostatic small cell carcinoma (90, 26/29), but rarely occurs in high-grade acinar tumors (7%, 10/150) and primary acinar carcinomas with neuroendocrine differentiation (11%, 4/35) [68]. General inactivation of the RB1 pathway and deregulation of the cell cycle was a common early event in human cancers [69, 70], however homozygous deletion of RB1 is a relatively late-stage genomic alteration in acinar prostate cancer progression [68].
These results suggested that at least some EPSmCC has the same cell origin of conventional carcinoma, such as urothelial carcinoma, prostatic adenocarcinoma, et al.
Conclusion
Current evidences strongly suggest that despite significant overlap of morphology and immunophenotype among SmCCs from different anatomic sites, SmCCs have heterogeneous molecular mutations (Table 1). A genetically oriented approach for the diagnosis and therapy of SmCCs becomes necessary for significant clinical impact on the prognosis and therapy outcome of this heterogeneous group of aggressive malignancies.
References
Mountain C: Clinical biology of small cell carcinoma: relationship to surgical therapy. Semi Oncol 1978, 5:272–9.
Molife L, Yan L, Vitfell-Rasmussen J, Zernhelt A, Sullivan D, Cassier P, et al.: Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J Hematol Oncol 2014, 7:1. 10.1186/1756-8722-7-1
Zhang J, Cao J, Li J, Zhang Y, Chen Z, Peng W, et al.: A phase I study of AST1306, a novel irreversible EGFR and HER2 kinase inhibitor, in patients with advanced solid tumors. J Hematol Oncol 2014, 7:22. 10.1186/1756-8722-7-22
Niu F-Y, Wu Y-L: Novel agents and strategies for overcoming EGFR TKIs resistance. Experiment Hematol Oncol 2014, 3:2. 10.1186/2162-3619-3-2
Wistuba II, Gazdar AF, Minna JD: Molecular genetics of small cell lung carcinoma. Semin Oncol 2001, 28:3–13. 10.1053/sonc.2001.20744
Horowitz JM, Park SH, Bogenmann E, Cheng JC, Yandell DW, Kaye FJ, et al.: Frequent inactivation of the retinoblastoma anti-oncogene is restricted to a subset of human tumor cells. Proc Natl Acad Sci U S A 1990, 87:2775–9. 10.1073/pnas.87.7.2775
Mori N, Yokota J, Akiyama T, Sameshima Y, Okamoto A, Mizoguchi H, et al.: Variable mutations of the RB gene in small-cell lung carcinoma. Oncogene 1990, 5:1713–7.
Yokomizo A, Tindall DJ, Drabkin H, Gemmill R, Franklin W, Yang P, et al.: PTEN/MMAC1 mutations identified in small cell, but not in non-small cell lung cancers. Oncogene 1998, 17:475–9. 10.1038/sj.onc.1201956
Pleasance ED, Stephens PJ, O'Meara S, McBride DJ, Meynert A, Jones D, et al.: A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010, 463:184–90. 10.1038/nature08629
Zheng X, Zhuge J, Bezerra SM, Faraj SF, Munari E, Fallon JT, et al.: High frequency of TERT promoter mutation in small cell carcinoma of bladder, but not in small cell carcinoma of other origins. J Hematol Oncol 2014, 7:47. 10.1186/s13045-014-0047-7
Eble JN, Organization WH, Cancer IAR: Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. WHO: IARC Press; 2004.
Pervez N, El-Gehani F, Joseph K, Dechaphunkul A, Kamal M, Pertschy D, et al.: Genitourinary small-cell carcinoma: a single-institution experience. Curr Oncol 2013, 20:258–64. 10.3747/co.20.1338
Si Q, Dancer J, Stanton ML, Tamboli P, Ro JY, Czerniak BA, et al.: Small cell carcinoma of the kidney: a clinicopathologic study of 14 cases. Hum Pathol 2011, 42:1792–8. 10.1016/j.humpath.2011.03.005
Ouzzane A, Ghoneim TP, Udo K, Verhasselt-Crinquette M, Puech P, Betrouni N, et al.: Small cell carcinoma of the upper urinary tract (UUT-SCC): report of a rare entity and systematic review of the literature. Cancer Treat Rev 2011, 37:366–72. 10.1016/j.ctrv.2010.12.005
Koay EJ, Teh BS, Paulino AC, Butler EB: A surveillance, epidemiology, and end results analysis of small cell carcinoma of the bladder. Cancer 2011, 117:5325–33. 10.1002/cncr.26197
Mackey JR, Au HJ, Hugh J, Venner P: Genitourinary small cell carcinoma: determination of clinical and therapeutic factors associated with survival. J Urol 1998, 159:1624–9. 10.1097/00005392-199805000-00058
Bezerra SM, Lotan TL, Faraj SF, Karram S, Sharma R, Schoenberg M, et al.: GATA3 expression in small cell carcinoma of bladder and Prostate and its Potential Role in Determining Primary Tumor Origin. Hum Pathol 2014. doi: http://dx.doi.org/10.1016/j.humpath.2014.04.011
Jones TD, Kernek KM, Yang XJ, Lopez-Beltran A, MacLennan GT, Eble JN, et al.: Thyroid transcription factor 1 expression in small cell carcinoma of the urinary bladder: an immunohistochemical profile of 44 cases. Hum Pathol 2005, 36:718–23. 10.1016/j.humpath.2005.04.007
Agoff SN, Lamps LW, Philip AT, Amin MB, Schmidt RA, True LD, et al.: Thyroid transcription factor-1 is expressed in extrapulmonary small cell carcinomas but not in other extrapulmonary neuroendocrine tumors. Modern Pathol 2000, 13:238–42. 10.1038/modpathol.3880044
Ismaili N: A rare bladder cancer–small cell carcinoma: review and update. Orphanet J Rare Dis 2011, 6:75. 10.1186/1750-1172-6-75
Yao JL, Madeb R, Bourne P, Lei J, Yang X, Tickoo S, et al.: Small cell carcinoma of the prostate: an immunohistochemical study. Am J Surg Pathol 2006, 30:705–12. 10.1097/00000478-200606000-00005
Wang W, Epstein JI: Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am J Surg Pathol 2008, 32:65–71. 10.1097/PAS.0b013e318058a96b
Liu X, Wu G, Shan Y, Hartmann C, von Deimling A, Xing M: Highly prevalent TERT promoter mutations in bladder cancer and glioblastoma. Cell Cycle 2013, 12:1637–8. 10.4161/cc.24662
Allory Y, Beukers W, Sagrera A, Flandez M, Marques M, Marquez M, et al.: Telomerase reverse transcriptase promoter mutations in bladder cancer: high frequency across stages, detection in urine, and lack of association with outcome. Eur Urol 2014, 65:360–6. 10.1016/j.eururo.2013.08.052
Kinde I, Munari E, Faraj SF, Hruban RH, Schoenberg M, Bivalacqua T, et al.: TERT promoter mutations occur early in urothelial neoplasia and are biomarkers of early disease and disease recurrence in urine. Cancer Res 2013, 73:7162–7. 10.1158/0008-5472.CAN-13-2498
Wenk RE, Bhagavan BS, Levy R, Miller D, Weisburger W: Ectopic ACTH, prostatic oat cell carcinoma, and marked hypernatremia. Cancer 1977, 40:773–8. Publisher Full Text 10.1002/1097-0142(197708)40:2<773::AID-CNCR2820400226>3.0.CO;2-I
Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, et al.: Androgen-independent prostate cancer is a heterogeneous group of diseases lessons from a rapid autopsy program. Cancer Res 2004, 64:9209–16. 10.1158/0008-5472.CAN-04-2442
Turbat-Herrera E, Herrera G, Gore I, Lott R, Grizzle W, Bonnin J: Neuroendocrine differentiation in prostatic carcinomas. A retrospective autopsy study. Archiv Pathol Lab Med 1988, 112:1100–5.
di Sant'Agnese PA: Neuroendocrine differentiation in prostatic carcinoma: an update on recent developments. Ann Oncol 2001,12(Suppl 2):S135–40. 10.1093/annonc/12.suppl_2.S135
Guo CC, Dancer JY, Wang Y, Aparicio A, Navone NM, Troncoso P, et al.: TMPRSS2-ERG gene fusion in small cell carcinoma of the prostate. Hum Pathol 2011, 42:11–7. 10.1016/j.humpath.2010.05.026
Lotan TL, Gupta NS, Wang W, Toubaji A, Haffner MC, Chaux A, et al.: ERG gene rearrangements are common in prostatic small cell carcinomas. Mod Pathol 2011, 24:820–8. 10.1038/modpathol.2011.7
Walenkamp AM, Sonke GS, Sleijfer DT: Clinical and therapeutic aspects of extrapulmonary small cell carcinoma. Cancer Treat Rev 2009, 35:228–36. 10.1016/j.ctrv.2008.10.007
Brennan SM, Gregory DL, Stillie A, Herschtal A, Mac Manus M, Ball DL: Should extrapulmonary small cell cancer be managed like small cell lung cancer? Cancer 2010, 116:888–95. 10.1002/cncr.24858
Mannion C, Park WS, Man YG, Zhuang Z, Albores-Saavedra J, Tavassoli FA: Endocrine tumors of the cervix: morphologic assessment, expression of human papillomavirus, and evaluation for loss of heterozygosity on 1p,3p, 11q, and 17p. Cancer 1998, 83:1391–400. Publisher Full Text 10.1002/(SICI)1097-0142(19981001)83:7<1391::AID-CNCR17>3.0.CO;2-#
Hoskins P, Wong F, Swenerton K, Pike J, Manji M, McMurtrie E, et al.: Small cell carcinoma of the cervix treated with concurrent radiotherapy, cisplatin, and etoposide. Gynecol Oncol 1995, 56:218–25. 10.1006/gyno.1995.1035
Carlson JW, Nucci MR, Brodsky J, Crum CP, Hirsch MS: Biomarker-assisted diagnosis of ovarian, cervical and pulmonary small cell carcinomas: the role of TTF-1, WT-1 and HPV analysis. Histopathology 2007, 51:305–12. 10.1111/j.1365-2559.2007.02790.x
Atienza-Amores M, Guerini-Rocco E, Soslow RA, Park KJ, Weigelt B: Small cell carcinoma of the gynecologic tract: a multifaceted spectrum of lesions. Gynecol Oncol 2014,134(2):410–8. 10.1016/j.ygyno.2014.05.017
Masumoto N, Fujii T, Ishikawa M, Saito M, Iwata T, Fukuchi T, et al.: P16 overexpression and human papillomavirus infection in small cell carcinoma of the uterine cervix. Hum Pathol 2003, 34:778–83. 10.1016/S0046-8177(03)00284-3
Wistuba II, Thomas B, Behrens C, Onuki N, Lindberg G, Albores-Saavedra J, et al.: Molecular abnormalities associated with endocrine tumors of the uterine cervix. Gynecol Oncol 1999, 72:3–9. 10.1006/gyno.1998.5248
Ishida GM, Kato N, Hayasaka T, Saito M, Kobayashi H, Katayama Y, et al.: Small cell neuroendocrine carcinomas of the uterine cervix: a histological, immunohistochemical, and molecular genetic study. Int J Gynecol Pathol 2004, 23:366–72. 10.1097/01.pgp.0000139637.01977.61
Clement PB: Selected miscellaneous ovarian lesions: small cell carcinomas, mesothelial lesions, mesenchymal and mixed neoplasms, and non-neoplastic lesions. Mod Pathol 2005,18(Suppl 2):S113–29.
Eichhorn JH, Young RH, Scully RE: Primary ovarian small cell carcinoma of pulmonary type. A clinicopathologic, immunohistologic, and flow cytometric analysis of 11 cases. Am J Surg Pathol 1992, 16:926–38. 10.1097/00000478-199210000-00002
Ikota H, Kaneko K, Takahashi S, Kawarai M, Tanaka Y, Yokoo H, et al.: Malignant transformation of ovarian mature cystic teratoma with a predominant pulmonary type small cell carcinoma component. Pathol Int 2012, 62:276–80. 10.1111/j.1440-1827.2012.02804.x
Estel R, Hackethal A, Kalder M, Munstedt K: Small cell carcinoma of the ovary of the hypercalcaemic type: an analysis of clinical and prognostic aspects of a rare disease on the basis of cases published in the literature. Arch Gynecol Obstet 2011, 284:1277–82. 10.1007/s00404-011-1846-5
Young RH, Oliva E, Scully RE: Small cell carcinoma of the ovary, hypercalcemic type. A clinicopathological analysis of 150 cases. Am J Surg Pathol 1994, 18:1102–16. 10.1097/00000478-199411000-00004
Jelinic P, Mueller JJ, Olvera N, Dao F, Scott SN, Shah R, et al.: Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet 2014, 46:424–6. 10.1038/ng.2922
Witkowski L, Carrot-Zhang J, Albrecht S, Fahiminiya S, Hamel N, Tomiak E, et al.: Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet 2014, 46:438–43. 10.1038/ng.2931
Ramos P, Karnezis AN, Craig DW, Sekulic A, Russell ML, Hendricks WP, et al.: Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet 2014, 46:427–9. 10.1038/ng.2928
Afanasiev OK, Yelistratova L, Miller N, Nagase K, Paulson K, Iyer JG, et al.: Merkel polyomavirus-specific T cells fluctuate with merkel cell carcinoma burden and express therapeutically targetable PD-1 and Tim-3 exhaustion markers. Clin Cancer Res 2013, 19:5351–60. 10.1158/1078-0432.CCR-13-0035
Mott RT, Smoller BR, Morgan MB: Merkel cell carcinoma: a clinicopathologic study with prognostic implications. J Cutan Pathol 2004, 31:217–23. 10.1111/j.0303-6987.2004.00149.x
Bobos M, Hytiroglou P, Kostopoulos I, Karkavelas G, Papadimitriou CS: Immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol 2006, 28:99–104. 10.1097/01.dad.0000183701.67366.c7
Feng H, Shuda M, Chang Y, Moore PS: Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319:1096–100. 10.1126/science.1152586
Arora R, Chang Y, Moore PS: MCV and Merkel cell carcinoma: a molecular success story. Curr Opin Virol 2012, 2:489–98. 10.1016/j.coviro.2012.05.007
Arora R, Shuda M, Guastafierro A, Feng H, Toptan T, Tolstov Y, et al.: Survivin is a therapeutic target in Merkel cell carcinoma. Sci Trans Med 2012, 4:133ra156–6.
Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A: Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011, 19:754–64. 10.1016/j.ccr.2011.04.019
Park KS, Liang MC, Raiser DM, Zamponi R, Roach RR, Curtis SJ, et al.: Characterization of the cell of origin for small cell lung cancer. Cell Cycle 2011, 10:2806–15. 10.4161/cc.10.16.17012
Frazier SR, Kaplan PA, Loy TS: The pathology of extrapulmonary small cell carcinoma. Semin Oncol 2007, 34:30–8. 10.1053/j.seminoncol.2006.11.017
Pearse AG: The diffuse endocrine system and the implications of the APUD concept. Int Surg 1979, 64:5–7.
Cheng L, Jones TD, McCarthy RP, Eble JN, Wang M, MacLennan GT, et al.: Molecular genetic evidence for a common clonal origin of urinary bladder small cell carcinoma and coexisting urothelial carcinoma. Am J Pathol 2005, 166:1533–9. 10.1016/S0002-9440(10)62369-3
Ali SZ, Reuter VE, Zakowski MF: Small cell neuroendocrine carcinoma of the urinary bladder. A clinicopathologic study with emphasis on cytologic features. Cancer 1997, 79:356–61. Publisher Full Text 10.1002/(SICI)1097-0142(19970115)79:2<356::AID-CNCR19>3.0.CO;2-#
Gaisa NT, Tilki D, Losen I, Dahl E, Stoehr R, Stief CG, et al.: Insights from a whole cystectomy specimen–association of primary small cell carcinoma of the bladder with transitional cell carcinoma in situ. Hum Pathol 2008, 39:1258–62. 10.1016/j.humpath.2007.12.017
McCluggage WG, Kennedy K, Busam KJ: An immunohistochemical study of cervical neuroendocrine carcinomas: Neoplasms that are commonly TTF1 positive and which may express CK20 and P63. Am J Surg Pathol 2010, 34:525–32. 10.1097/PAS.0b013e3181d1d457
Rossi G, Bertolini F, Sartori G, Bigiani N, Cavazza A, Foroni M, et al.: Primary mixed adenocarcinoma and small cell carcinoma of the appendix: a clinicopathologic, immunohistochemical, and molecular study of a hitherto unreported tumor. Am J Surg Pathol 2004, 28:1233–9. 10.1097/01.pas.0000128666.89191.48
Bezerra SM, Lotan TL, Faraj SF, Karram S, Sharma R, Schoenberg M, et al.: GATA3 expression in small cell carcinoma of bladder and prostate and its potential role in determining primary tumor origin. Hum Pathol 2014,45(8):1682–7. 10.1016/j.humpath.2014.04.011
Cecen K, Karadag MA, Demir A, Kocaaslan R: Small cell carcinoma of the prostate presenting with skin metastasis: a case report. J Med Case Rep 2014, 8:146. 10.1186/1752-1947-8-146
Scott AF, Mohr DW, Ling H, Scharpf RB, Zhang P, Liptak GS: Characterization of the genomic architecture and mutational spectrum of a small cell prostate carcinoma. Genes (Basel) 2014, 5:366–84. 10.3390/genes5020366
Kimura H, Uegaki M, Aoyama T, Kawai J, Hamano T, Hashimura T: Carboplatin plus irinotecan induced partial response in a patient with small cell carcinoma of the prostate; a case report. Hinyokika Kiyo 2014, 60:39–43.
Tan H-L, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, et al.: Rb Loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res 2013. doi:10.1158/1078–0432.CCR-13–1982
Burkhart DL, Sage J: Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer 2008, 8:671–82. 10.1038/nrc2399
Williams BO, Schmitt EM, Remington L, Bronson RT, Albert DM, Weinberg RA, et al.: Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J 1994, 13:4251–9.
Tan H-L, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, et al.: Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res 2014, 20:890–903. 10.1158/1078-0432.CCR-13-1982
Wang X, Jones TD, Maclennan GT, Yang XJ, Lopez-Beltran A, Eble JN, et al.: P53 expression in small cell carcinoma of the urinary bladder: biological and prognostic implications. Anticancer Res 2005, 25:2001–4.
Salagierski M, Schalken JA: Molecular diagnosis of prostate cancer: PCA3 and TMPRSS2:ERG gene fusion. J Urol 2012, 187:795–801. 10.1016/j.juro.2011.10.133
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Dr. XZ and Dr. MZ drafted the manuscript. Dr. JTF and Dr. DL partly drafted and revised the manuscript. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Zheng, X., Liu, D., Fallon, J.T. et al. Distinct genetic alterations in small cell carcinoma from different anatomic sites. Exp Hematol Oncol 4, 2 (2015). https://doi.org/10.1186/2162-3619-4-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2162-3619-4-2