
Abstract
Since the recognition of epidermal growth factor receptor (EGFR) as a therapeutic target, EGFR tyrosine kinase inhibitors (TKIs) have been used in lung cancer patients with EGFR mutations, which has been a major breakthrough for lung cancer treatment.. The progression-free survival (PFS) of patients with EGFR mutations treated with EGFR TKIs is significantly prolonged compared with that of patients who underwent standard chemotherapy. However, all patients who initially respond to EGFR TKIs eventually develop acquired resistance (AR). Many small molecule agents and monoclonal antibodies (McAb) targeting signaling pathways are potential therapeutic regimens for overcoming resistance, and various therapeutic strategies are used in clinical practice. Here we review the novel agents and therapeutic strategies for overcoming AR to EGFR TKIs.
Similar content being viewed by others
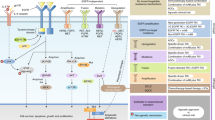

Introduction
Non-small cell lung cancer (NSCLC) accounts for approximately 80% of all lung cancer cases [1]. The traditional treatment for NSCLC is platinum-based chemotherapy, which inhibits normal and cancer cells, but it has reached a therapeutic plateau [2]. In contrast, personalized therapy has numerous advantages [3]. The ERBB family includes EGFR/HER1, ERBB2/HER2, ERBB3/HER3 and ERBB4/HER4. Somatic EGFR mutations result in conformational changes and the activation of downstream signaling. Gefitinib and erlotinib are first-generation EGFR TKIs that reversibly compete with the ATP-binding site in the EGFR. Gefitinib/erlotinib (G/E) prolongs progression-free survival (PFS) and improves quality of life compared with standard chemotherapy, while almost all patients develop AR to G/E in 9 to 11 months [4].
The mechanisms responsible for AR to G/E include the following: first, secondary EGFR mutation, which is predominantly T790M; second, activation of parallel signaling pathways; third, activation of downstream signaling pathways; fourth, phenotypic transformation, such as epithelial to mesenchymal transition (EMT) and small cell lung cancer (SCLC) transformation. We will discuss the mechanisms of AR to G/E and corresponding novel targeting agents (Figure 1). The potential therapeutic options for patients with AR to EGFR TKIs are also reviewed.

New agents for overcoming AR. This figure shows the parallel and downstream signaling pathways of the EGFR. The gray boxes depict new agents available for treatment.
New agents for overcoming AR
Secondary EGFR mutations
The most common type of AR is mutation in codon 790 (T790M) in EGFR exon 20, which accounts for 50 ~ 60% of cases with AR to G/E [5]. EGFR T790M leads to a higher affinity of ATP for EGFR, thus affecting G/E binding [6]. Second generation EGFR TKIs e.g., BIBW 2992 (afatinib) and PF00299804 (dacomitinib), and third generation EGFR TKIs e.g., CO-1686 and AZD9291 are irreversible inhibitors that could overcome the AR caused by T790M. Afatinib has been approved by the FDA. Dacomitinib, CO-1686 and AZD9291 are currently being tested in ongoing trials.
Afatinib
The safety and efficacy of afatinib have been characterized in many clinical trials. The most classic trial is the LUX-Lung program. This program evaluated the efficacy of afatinib for first-line treatment and for patients who have been treated with G/E. The efficacy of afatinib in overcoming AR to G/E was confirmed in the LUX-Lung 1/4/5 trials.
A total of 585 patients who had received G/E and chemotherapy were enrolled in LUX-Lung 1 [7], and these patients were randomized to receive afatinib or placebo. The primary endpoint, overall survival (OS), was 10.8 months in the afatinib group, while the outcome was 12.0 months in placebo group (HR = 1.077, P = 0.74). However, PFS was significantly prolonged in the afatinib group (3.3 vs. 1.1 months, HR = 0.38; p < 0.0001). Moreover, symptoms such as cough and dyspnea were significantly improved in the afatinib group.
LUX-Lung 4 [8] also confirmed the efficacy of afatinib in patients who progressed on G/E and chemotherapy. In the 61 evaluable patients, partial response (PR) was achieved in 4 patients, and the median PFS and OS was 4.4 and 19.0 months, respectively.
Afatinib was investigated in patients who failed chemotherapy and G/E in LUX-Lung 5 [9]. The patients were randomized into two groups after afatinib resistance i.e., afatinib alone or afatinib combined with paclitaxol. In part A of the trial, 63% of the patients achieved complete response (CR)/PR/stable disease (SD), and the median PFS was 3.3 months. The results of part B are pending.
Dacomitinib
In a phase II trial, dacomitinib was given to patients with advanced NSCLC after chemotherapy (CT) and erlotinib (E) failure. In 62 evaluable cases, 3 had a partial response, and 35 had stable disease (SD) for more than 6 weeks. The side effects included diarrhea (86%), fatigue (40%), rash (45%), and stomatitis/mucosal inflammation, which was mainly grade 1/2 and manageable [10]. One patient with a T790M mutation caused by prior gefitinib treatment achieved SD after exposure to dacomitinib in the NCT00783328 trial [11]. Four patients who have failed G/E achieved PR in a phase I trial (NCT00225121) [12]. NCT01000025 is an ongoing phase III trial that investigates the efficacy of dacomitinib in patients with NSCLC who have failed chemotherapy and G/E.
CO-1686 and AZD9291
CO-1686 is a third-generation EGFR TKI, which may be more effective in patients with T790M. NCT01526928 is an ongoing phase I/II trial that has enrolled patients who have failed prior chemotherapy and EGFR-targeted therapy including erlotinib, gefitinib, neratinib, afatinib, or dacomitinib. Three cases treated with CO-1686 demonstrated clinical benefit or tumor shrinkage, and the tolerance was acceptable [13].
AZD9291 is a potent oral, irreversible inhibitor of the EGFR with EGFR-TKI-sensitizing (EGFR+) and resistance mutations (T790M). The AZD9291 IC50 for the EGFR+/T790M H1975 cells is 15 nM. In a phase I open-label multicenter study, AZD9291 was shown to have mostly mild AEs, and no grade 3-4 SAEs were observed. Moreover, 89 patients received at least one dose, and no DLTs were observed. Good evidence for efficacy has been observed at all doses studied thus far, including 9/18 patients with T790M who had confirmed or unconfirmed partial responses [14].
Parallel pathway activation
MET amplication/overexpression
The incidence of MET gene amplication or protein overexpression is 5 ~ 22% in AR patients [15]. MET activates ERBB3 and the PI3K/AKT pathway independent of EGFR [16]. MetMAb [17] and ARQ197 (tivantinib) [18] are the most prominent MET inhibitors, but they have not been investigated for overcoming TKI resistance.
NCT01610336 and NCT01911507 are ongoing trials that test the safety and efficacy of INC280 in NSCLC patients with AR to G/E. INC280 and gefitinib are simultaneously given to patients with MET-amplification in the NCT01610336 trial. Patients with MET overexpression are treated with INC280 and erlotinib in the NCT01911507 trial. The results of these trials are not yet available.
XL184 is a tyrosine kinase inhibitor of multiple receptors, including VEGFR2, MET, and RET. NCT00596648 is a phase Ib/II trial evaluating the safety and efficacy of XL184 with or without erlotinib in patients resistant to EGFR TKIs, and its results are pending.
HGF overexpression
Approximately 60% patients with AR to G/E have HGF overexpression [19]. The overexpression results in MET phosphorylation and the activation of GAB1 and PI3K/AKT [20]. BEZ235 is PI3K-mTOR inhibitor that has the potential to overcome AR in vitro [21]. However, the efficacy of BEZ235 has not been tested in vivo. Heat shock protein 90 (HSP90) is a molecular chaperone for several proteins, including EGFR and MET. 17-DMAG is an HSP90 inhibitor that has efficacy for HGF-triggered erlotinib resistance in cell lines and animal models [22]. AUY922 is also an HSP90 inhibitor that is currently being tested in the phase II trial NCT01259089. This trial enrolled patients with AR to G/E. The ORR was 13%, and the two patients with a PR had an EGFR T790M mutation [23].
Downstream signaling pathway activation
The downstream signaling pathways of EGFR include the RAS/RAF/MEK/ERK and PI3K/AKT/mTOR pathways. The former is associated with proliferation, and the latter is related to survival. Mutations in EGFR result in activation of the PI3K/AKT pathway and the survival of tumor cells without affecting tumor cell proliferation. The mutation of key genes in these two pathways leads to G/E resistance.
PIK3CA mutation
PIK3CA mutations were detected in 5% of patients with AR to G/E [24]. The most promising PI3K inhibitor is BKM120, which is a pan-PI3K inhibitor. The antitumor activation of gefitinib plus BKM120 was observed in patients with AR to G/E in the phase Ib trial NCT01570296 [25]. A reduction in SUVmax (>25%) was observed in 4/10 patients, and the median PFS was 2.8 months. Given the favorable central nervous system (CNS) penetration of BKM120, patients with brain metastases were included, and two patients with CNS penetration had a PFS of 2.8 and 10.7 months. However, molecular analysis revealed that no patient harbored a PIK3CA mutation. NCT01487265 is an ongoing phase I/II trial examines the efficacy of BKM120 combined with erlotinib in patients with AR to erlotinib, and the results of this trial are pending.
AKT
AKT mutations were not detected in patients with AR to G/E [26], but AKT might be activated in tumors resistant to G/E. MK2206 is one of the most potent AKT inhibitors. Growth inhibition was greatly enhanced with the combination of MK2206 and erlotinib in TKI-sensitive and TKI-resistant NSCLC cell lines, and MK-2206 restored erlotinib sensitivity in HGF-induced AR cells [27]. There are two ongoing trials, NCT01294306 and NCT01147211, which examine the efficacy of MK2206 in NSCLC patients with AR to erlotinib and gefitinib, respectively, but their results are unavailable.
mTOR
mTOR regulates cell growth and metabolism by controlling several catabolic and anabolic processes. mTOR inhibitors include temsirolimus, everolimus (RAD001), Ku-0063794, and sirolimus.
Temsirolimus and everolimus could overcome HGF-induced EGFR TKI AR in vitro [28]. Everolimus enhanced the antitumor activation of gefitinib in sensitive and resistant cell lines [29] but failed to meet the primary endpoint in a phase II trial [30]. Ku-0063794 could effectively inhibit gefitinib-resistant cells [31]. NCT00993499 is an ongoing phase Ib trial in which the efficacy of the combination of afatinib and sirolimus is being tested in patients with AR to gefitinib or erlotinib, and the results are pending.
ERK2 amplification
ERK2 (MAPK1) amplification has been reported in 5% of patients with AR to G/E [32]. AZD6244 and CI1040 could reverse resistance in cells with AR to gefitinib [33]; however, there is no clinical trial that has evaluated the efficacy of overcoming AR to AZD6244 and CI1040.
Strategies for overcoming AR
IMPRESS model
Some patients may have accelerated disease progression after discontinuing G/E during washout periods. This phenomenon is called disease flare, which was observed in 23% of patients with AR [34]. To avoid disease flare, some clinicians suggest G/E continuation plus chemotherapy. There was a retrospective study demonstrating that erlotinib continuation in addition to chemotherapy significantly improved the respond rate (RR) compared with chemotherapy alone after erlotinib failure [35]. IMPRESS is an ongoing phase III trial enrolling patients with EGFR mutations who failed first-line gefitinib. The patients are randomized to receive cisplatin and pemetrexed plus gefitinib or placebo. The primary endpoint of this study is PFS, and the results are pending.
ASPIRATION model
As EGFR TKIs greatly improve the PFS of patients with EGFR positive mutations, the RECIST definition of progression now appears to be challenged. Recently Yang et al. reported a retrospective study that divided patients with AR into three groups: dramatic progression, local progression and gradual progression. The authors recommended chemotherapy for patients with dramatic progression, the continuation of TKIs plus local intervention for those with local progression and the continuation of TKIs for those with gradual progression [36]. ASPIRATION is an ongoing phase II study evaluating the continuation of erlotinib beyond RECIST PD in selected patients who have slowed PD (>6 months of partial response/stable disease), asymptomatic minimal PD, or new, locally controlled brain metastases.
Switching to or combining with novel compounds
With the unveiling of the underlying mechanisms of AR, novel small molecule inhibitors alone or in combination will be effective in overcoming AR e.g., INC280 plus gefitinib or erlotinib as mentioned above. Therefore, we should recommend that patients receive rebiopsies after progression on G/E to identify the underlying mechanisms of AR. Based on the understanding of resistance mechanisms, clinicians should encourage patients to enter clinical trials.
Conclusion
EGFR TKIs are increasingly used in the clinic. The standard treatment for patients with AR to EGFR TKIs is changing to chemotherapy at RECIST progression. However, this standard treatment is now being challenged due to a growing number of effective novel compounds and clinical trial strategies.
Abbreviations
- EGFR:
-
Epidermal growth factor receptor
- TKIs:
-
Tyrosine kinase inhibitors
- PFS:
-
Progression-free survival
- AR:
-
Acquired resistance
- McAb:
-
Monoclonal antibodies
- G/E:
-
Gefitinib/erlotinib
- EMT:
-
Epithelial mesenchymal transition
- SCLC:
-
Small cell lung cancer
- OS:
-
Overall survival
- PR:
-
(Partial response)
- CR:
-
Complete response
- SD:
-
Stable disease
- HSP90:
-
Heat shock protein 90
- RR:
-
Response rate.
References
Hung JJ, Hsueh CT, Chen KH, Hsu WH, Wu YC: Clinical significance of E2F1 protein expression in non-small cell lung cancer. Exp Hematol Oncol 2012,1(1):18. 10.1186/2162-3619-1-18
Cheng H, An SJ, Dong S, Zhang YF, Zhang XC, Chen ZH, Jian-Su , Wu YL: Molecular mechanism of the schedule-dependent synergistic interaction in EGFR-mutant non-small cell lung cancer cell lines treated with paclitaxel and gefitinib. J Hematol Oncol 2011, 4–5.
Yuan Y, Liao YM, Hsueh CT, Mirshahidi HR: Novel targeted therapeutics: inhibitors of MDM2, ALK and PARP. J Hematol Oncol 2011, 4–16.
Xu C, Zhou Q, Wu YL: Can EGFR-TKIs be used in first line treatment for advanced non-small cell lung cancer based on selection according to clinical factors? - A literature-based meta-analysis. J Hematol Oncol 2012, 5–62.
Gazdar AF: Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors[J]. Oncogene 2009,28(Suppl 1):S24-S31.
Katzel JA, Fanucchi MP, Li Z: Recent advances of novel targeted therapy in non-small cell lung cancer. J Hematol Oncol 2009, 2–2.
Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, Zhou C, Su WC, Wang M, Sun Y, et al.: Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial[J]. Lancet Oncol 2012,13(5):528–538. 10.1016/S1470-2045(12)70087-6
Katakami N, Atagi S, Goto K, Hida T, Horai T, Inoue A, Ichinose Y, Koboyashi K, Takeda K, Kiura K, et al.: LUX-lung 4: a phase II trial of afatinib in patients with advanced Non-small-cell lung cancer Who progressed during prior treatment with Erlotinib, Gefitinib, or both[J]. J Clin Oncol 2013,31(27):3335–41. 10.1200/JCO.2012.45.0981
Martin H, Schuler DP, Yang JCH, Kim J, De Marinis F, Chen Y, Zhou C, Bennouna J, Liu X, Feng JF, et al.: Interimanalysis of afatinib monotherapy in patients with metastatic NSCLC progressing after chemotherapy and erlotinib/gefitinib (E/G) in a trial of afatinib plus paclitaxel versus investigator’s choice chemotherapy following progression on afatinib monotherapy[J]. J Clin Oncol 2012., 30: abstr 7557
Campbell A, Reckamp KL, Camidge DR, Giaccone G, Gadgeel SM, Khuri FR, Engelman JA, Denis LJ, O'Connell JP, Janne PA: PF-00299804 (PF299) patient (pt)-reported outcomes (PROs) and efficacy in adenocarcinoma (adeno) and nonadeno non-small cell lung cancer (NSCLC): a phase (P) II trial in advanced NSCLC after failure of chemotherapy (CT) and erlotinib (E). J Clin Oncol 2010,28(5s):7596. suppl; abstr
Takahashi T, Boku N, Murakami H, Naito T, Tsuya A, Nakamura Y, Ono A, Machida N, Yamazaki K, Watanabe J, et al.: Phase I and pharmacokinetic study of dacomitinib (PF-00299804), an oral irreversible, small molecule inhibitor of human epidermal growth factor receptor-1, -2, and -4 tyrosine kinases, in Japanese patients with advanced solid tumors. Invest New Drugs 2012,30(6):2352–2363. 10.1007/s10637-011-9789-z
Janne PA, Boss DS, Camidge DR, Britten CD, Engelman JA, Garon EB, Guo F, Wong S, Liang J, Letrent S, et al.: Phase I dose-escalation study of the pan-HER inhibitor, PF299804, in patients with advanced malignant solid tumors. Clin Cancer Res 2011,17(5):1131–1139. 10.1158/1078-0432.CCR-10-1220
Sequist LV, Soria JC, Gadgeel SM, Wakelee HA, Camidge DR: First-in-human evaluation of CO-1686, an irreversible, selective, and potent tyrosine kinase inhibitor of EGFR T790M[J]. J Clin Oncol 2013, 31: 2524. suppl; abstr
Ranson M, Pao W, Kim DW: AZD9291: an irreversible, potent and selective tyrosine kinase inhibitor (TKI) of activating (EGFR+) and resistance (T790M) mutations in advanced NSCLC The AURA study. J Thorac Oncol 2013, 11–034. suppl; abstr P1.11
Robinson KW, Sandler AB: The role of MET receptor tyrosine kinase in non-small cell lung cancer and clinical development of targeted anti-MET agents. Oncologist 2013,18(2):115–122. 10.1634/theoncologist.2012-0262
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al.: MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007,316(5827):1039–1043. 10.1126/science.1141478
Vashishtha A, Patel PH, Yu W, Bothos JG, Simpson J, Maneatis T, Doessegger L, Peterson AC, Clement-Duchene C, Robinet G, et al.: Safety data and patterns of progression in met diagnostic subgroups in OAM4558g; A phase II trial evaluating MetMAb in combination with erlotinib in advanced NSCLC. J Clin Oncol 2011, 29: 7604. suppl; abstr
Sequist LV, Von Pawel J, Garmey EG, Akerley WL, Brugger W, Ferrari D, Chen Y, Costa DB, Gerber DE, Orlov S, et al.: Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol 2011,29(24):3307–3315. 10.1200/JCO.2010.34.0570
Yano S, Yamada T, Takeuchi S, Tachibana K, Minami Y, Yatabe Y, Mitsudomi T, Tanaka H, Kimura T, Kudoh S, et al.: Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol 2011,6(2):2011–2017.
Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka Y, et al.: Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res 2008,68(22):9479–9487. 10.1158/0008-5472.CAN-08-1643
Sano T, Takeuchi S, Nakagawa T, Ishikawa D, Nanjo S, Yamada T, Nakamura T, Matsumoto K, Yano S: The novel phosphoinositide 3-kinase-mammalian target of rapamycin inhibitor, BEZ235, circumvents erlotinib resistance of epidermal growth factor receptor mutant lung cancer cells triggered by hepatocyte growth factor. Int J Cancer 2013,133(2):505–513. 10.1002/ijc.28034
Koizumi H, Yamada T, Takeuchi S, Nakagawa T, Kita K, Nakamura T, Matsumoto K, Suda K, Mitsudomi T, Yano S: Hsp90 inhibition overcomes HGF-triggering resistance to EGFR-TKIs in EGFR-mutant lung cancer by decreasing client protein expression and angiogenesis. J Thorac Oncol 2012,7(7):1078–1085. 10.1097/JTO.0b013e3182519a2c
Johnson ML, Hart EM, Rademaker A, Weitner BB, Urman A, Simm HD, Fountas LM, Worden R, Patel JD, Miller VA, et al.: A phase II study of HSP90 inhibitor AUY922 and erlotinib (E) for patients (pts) with EGFR-mutant lung cancer and acquired resistance (AR) to EGFR tyrosine kinase inhibitors (EGFR TKIs). J Clin Oncol 2013, 8036. suppl; abstr
Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, et al.: Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011,3(75):26r-75r.
Tan DS, Lim KH, Tai WM, Ahmad A, Pan S, Ng QS, Ang M, Gogna A, Ng YL, Tan BS, et al.: A phase Ib safety and tolerability study of a pan class I PI3K inhibitor buparlisib (BKM120) and gefitinib (gef) in EGFR TKI-resistant NSCLC. J Clin Oncol 2013, 8107. suppl; abstr
Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, Riely GJ: Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013,19(8):2240–2247. 10.1158/1078-0432.CCR-12-2246
Mack PC, Farneth N, Mahaffey C, Lara P, Gandara DR: Impact of AKT inhibitor MK-2206 on erlotinib resistance in non-small cell lung cancer (NSCLC). J Clin Oncol 2011, 7573. suppl; abstr
Ishikawa D, Takeuchi S, Nakagawa T, Sano T, Nakade J, Nanjo S, Yamada T, Ebi H, Zhao L, Yasumoto K, et al.: mTOR inhibitors control the growth of EGFR mutant lung cancer even after acquiring resistance by HGF. PLoS One 2013,8(5):e62104. 10.1371/journal.pone.0062104
Dong S, Zhang X, Cheng H, Guo A, Zhu J, Yang S, Wu YL: Effect of everolimus and gefitinib on PI3K/akt/mTOR and raf/MEK/ERK pathways in NSCLC cells. J Clin Oncol 2010, e13654. suppl; abstr
Price KA, Azzoli CG, Krug LM, Pietanza MC, Rizvi NA, Pao W, Kris MG, Riely GJ, Heelan RT, Arcila ME, et al.: Phase II trial of gefitinib and everolimus in advanced non-small cell lung cancer. J Thorac Oncol 2010,5(10):1623–1629. 10.1097/JTO.0b013e3181ec1531
Fei SJ, Zhang XC, Dong S, Cheng H, Zhang YF, Huang L, Zhou HY, Xie Z, Chen ZH, Wu YL: Targeting mTOR to overcome epidermal growth factor receptor tyrosine kinase inhibitor resistance in Non-small cell lung cancer cells. PLoS One 2013,8(7):e69104. 10.1371/journal.pone.0069104
Remon J, Moran T, Majem M, Reguart N, Dalmau E, Marquez-Medina D, Lianes P: Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: A new era begins. Cancer Treat Rev 2014,40(1):93–101. 10.1016/j.ctrv.2013.06.002
Huang MH, Lee JH, Chang YJ, Tsai HH, Lin YL, Lin AM, Yang JC: MEK inhibitors reverse resistance in epidermal growth factor receptor mutation lung cancer cells with acquired resistance to gefitinib. Mol Oncol 2013,7(1):112–120. 10.1016/j.molonc.2012.09.002
Chaft JE, Oxnard GR, Sima CS, Kris MG, Miller VA, Riely GJ: Disease flare after tyrosine kinase inhibitor discontinuation in patients with EGFR-mutant lung cancer and acquired resistance to erlotinib or gefitinib: implications for clinical trial design. Clin Cancer Res 2011,17(19):6298–6303. 10.1158/1078-0432.CCR-11-1468
Goldberg SB, Oxnard GR, Digumarthy S, Muzikansky A, Jackman DM, Lennes IT, Sequist LV: Chemotherapy with erlotinib or chemotherapy alone in advanced NSCLC with acquired resistance to EGFR tyrosine kinase inhibitors (TKI). J Clin Oncol 2012, 7524. suppl; abstr
Yang JJ, Chen HJ, Yan HH, Zhang XC, Zhou Q, Su J, Wang Z, Xu CR, Huang YS, Wang BC, et al.: Clinical modes of EGFR tyrosine kinase inhibitor failure and subsequent management in advanced non-small cell lung cancer. Lung Cancer-J Iaslc 2013,79(1):33–39. 10.1016/j.lungcan.2012.09.016
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors have no relevant conflicts of interests.
Authors’ contributions
Both authors were involved in preparing, drafting and revising the manuscript, and both have read and approved the final version.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Niu, FY., Wu, YL. Novel agents and strategies for overcoming EGFR TKIs resistance. Exp Hematol Oncol 3, 2 (2014). https://doi.org/10.1186/2162-3619-3-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2162-3619-3-2