
Abstract
Increased blood lactate levels (hyperlactataemia) are common in critically ill patients. Although frequently used to diagnose inadequate tissue oxygenation, other processes not related to tissue oxygenation may increase lactate levels. Especially in critically ill patients, increased glycolysis may be an important cause of hyperlactataemia. Nevertheless, the presence of increased lactate levels has important implications for the morbidity and mortality of the hyperlactataemic patients. Although the term lactic acidosis is frequently used, a significant relationship between lactate and pH only exists at higher lactate levels. The term lactate associated acidosis is therefore more appropriate. Two recent studies have underscored the importance of monitoring lactate levels and adjust treatment to the change in lactate levels in early resuscitation. As lactate levels can be measured rapidly at the bedside from various sources, structured lactate measurements should be incorporated in resuscitation protocols.
Similar content being viewed by others

Review
Introduction
Many variables measured in critically ill patients have been used to estimate severity of disease, prognosticate morbidity and mortality, evaluate costs of treatment, and finally indicate specific treatment and monitor the adequacy of treatment and its timing. It is unlikely that one measurement can replace all of these, but in the remainder of this manuscript we will show that lactate levels may come close. Although in our mind strongly linked to tissue hypoxia, lactate levels follow many more metabolic processes not related to tissue hypoxia and, therefore, subject to many disturbances found in various clinical situations.
History of lactate
The first description of lactate originates from 1780 when Karl Scheele found lactate in sour milk. It took almost 70 years before the German physician-chemist Joseph Scherer demonstrated the presence of lactate in human blood. Where Scherer analysed blood drawn from a young woman who had just died from what we now call septic shock, it was Carl Folwarczny in 1858 who demonstrated the presence of lactate in the blood of a living patient [1]. Araki and Zillessen made an important observation that has shaped our association of increased lactate levels and tissue hypoxia. These authors observed that when they interrupted oxygen supply to muscles in mammals and birds, lactic acid was formed and increased [2]. In current practice, lactate is frequently measured in many kinds of patients, usually with the goal of detecting tissue hypoxia. However, given the metabolism of lactate and the effect of acute illness on glucose metabolism, increased lactate levels can reflect more than only tissue hypoxia.
Metabolism of lactate
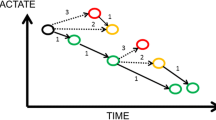
Lactate is a crucial metabolite in the two main energy (ATP)-producing processes that power life: glycolysis and oxidative phosphorylation (OxPhos). Glycolysis, a process that occurred very early in evolution (approximately 3 billion years ago), converts glucose into two molecules of pyruvate with the concomitant generation of 2 ATP. When atmospheric oxygen levels rose (1 billion years ago), mitochondria developed to generate far more energy from glucose (36 ATP molecules for 1 glucose molecule), although following a much more complicated process (Krebs cycle and OxPhos). Glycolysis and OxPhos steadily metabolize glucose when conditions are stable (Figure 1a). Pyruvate is the molecule that links these two reactions. Because the rate of glycolysis can increase two to three orders of a magnitude faster than OxPhos, glycolysis can briefly provide far more ATP. Excess pyruvate will rapidly accumulate and is diverted to lactate in order for glycolysis to proceed (Figure 1b). During recovery (Figure 1c) lactate is converted into pyruvate. In both directions this is catalyzed by the ubiquitous enzyme lactate dehydrogenase (LDH). Thus, when rapidly large amounts of energy are required, such as under circumstances of cellular stress, lactate serves as a critical buffer that allows glycolysis to accelerate. Also, at the level of the organism (Figure 2), lactate has a similar role as an intermediate fuel that is readily exchanged between various tissues, facilitated by a family of membrane-bound mono-carboxylate transporters (MCT). Over the past two decades, lactate shuttles between astrocytes, neurons, striated muscle, cardiac muscle, as well as the liver and kidneys have been demonstrated [3, 4]. The long-known Cori cycle can now be considered one of many lactate shuttles (Figure 2). It should be noted that whereas the Cori cycle involves energy-consuming hepatic or renal gluconeogenesis to convert lactate into glucose, direct interorgan exchange of lactate itself does not carry an energy penalty and even exogenous lactate may serve as a suitable substrate [3].

Lactate at the cellular level. Usually not oxygen shortage per se, but acute energy requirements is a key determinant of lactate levels. a Under stable conditions, glucose is converted to pyruvate, generating 2 ATP, and pyruvate is then subsequently fully oxidized to CO2 generating ~36 ATP. b Under stress, glycolysis can increase by a factor 100 to 1,000, provided that glucose is present and pyruvate is converted to lactate. Irrespective of optimal mitochondrial function and oxygenation, such a rate of pyruvate production will saturate the mitochondrial tricarboxylic acid cycle and oxidative phosphorylation (OxPhos). c During recovery, lactate is converted back to pyruvate and fully oxidized.

Lactate at the physiological level. The flexible use of glucose and lactate as fuels on the cellular level is mirrored at the organism level. All living tissues can consume glucose. From the glucose/lactate point of view, three sorts of tissues/cells exist: 1) cells that must produce lactate because they lack mitochondria, e.g., red blood cells; 2) tissues or cells that either produce or consume lactate depending on circumstances, i.e., all mitochondria-containing cells; 3) tissues that can perform gluconeogenesis and export glucose that is resynthesized from lactate. The liver and the kidneys can only perform gluconeogenesis and export glucose. Only this so-called Cori cycle (denoted by *) carries an energy penalty, whereas the other shuttles do not lead to “waste” of energy.
Lactate and acidosis
The metabolism of glucose during tissue hypoxia results in the production of [lactate]-, ATP, and water [5]. The production of H+ originates from the hydrolysis of ATP to ADP. In the presence of oxygen and provided that OxPhos can keep up with glycolysis, these H+ ions can be used together with lactate in the OxPhos in the mitochondria and acidosis is thus less likely to develop. Stewart challenged the classic Henderson-Hasselbalch approach where the acidosis in his approach is the result of the dissociation of water to maintain acid–base equilibrium by the addition of the strong ion lactate- to the circulation [6]. There is however not a strong relationship between arterial pH and lactate levels. Even at higher lactate levels, only a weak, although significant, correlation exists (Figure 3). When evaluating the significance for patient outcome and the origin of the metabolic acidosis, it is probably more realistic to use the term: lactate-associated metabolic acidosis, a combination that also carries the highest risk of mortality [7].

1,745 combined measurements of arterial pH and arterial lactate in 171 critically ill patients. Horizontal and vertical lines represent suggested definition of lactic acidosis [8]. For lactate levels ≥ 5.0 mmol/L, a significant linear regression analysis reveals a R2 = 0.28 (p < 0.001).
Lactate and tissue hypoxia
Many experimental studies have confirmed the relationship between tissue hypoxia and the generation of lactate by reducing the components of systemic oxygen delivery (haemoglobin level, oxygen saturation, and cardiac output) until the extraction of oxygen can no longer maintain oxygen availability to the cells to meet their demands [9, 10]. At a critical level of oxygen delivery, oxygen consumption becomes limited by oxygen delivery, and this coincides with a sharp increase in lactate levels. Also, clinical data indicate the relationship between the presence of this supply dependent state of oxygen consumption and increased lactate levels similar to animal studies [11]. In a landmark study, Ronco et al. showed that this phenomenon also was present in patients when oxygen delivery decreased until circulatory arrest during end-of-life care [12]. In addition, Friedman et al. [13] showed that this phenomenon is present in the early resuscitation phase of critical illness, suggesting that resuscitation resolves a state of supply dependent oxygen consumption and thereby the hyperlactataemia. This has been confirmed in an experimental study of cardiac tamponade by Zhang et al. [10], which showed that resolution of the supply-dependent state of oxygen consumption by resolving the tamponade was associated with an increase in oxygen consumption to baseline levels and normalisation of lactate levels.
Because the exchange of oxygen takes place in the microcirculation, alterations in microcirculatory perfusion also can result in limited oxygen availability. Particularly in sepsis, microcirculatory derangement or shunting may lead to insufficient oxygen that is delivered to the cell, thereby increasing lactate levels [10]. This is indirectly illustrated by the observation that improving capillary perfusion has been correlated to a reduction in lactate levels in patients with septic shock, independent of changes in systemic haemodynamic variables [14].
Given the near equilibrium reaction between lactate and pyruvate and its connection with the cellular oxido-reduction state, the lactate-to-pyruvate ratio (L:P) provides additional information as L:P is coupled to the cytoplasmic NADH:NAD+ [15–17]. However, it must be noted that in contrast to lactate, pyruvate is far from trivial to reliably measure in clinical practice and therefore its use is limited in critically ill patients [18].
Lactate production in aerobic metabolism
As discussed earlier, aerobic glucose metabolism to lactate may be a preferred way to rapidly produce significant energy amounts. Therefore, stimulating increased aerobic glucose metabolism has been shown to increase lactate levels in the absence of tissue hypoxia. Most notably, the administration of epinephrine has long been shown to result in a dose-dependent increase in lactate levels [19]. Also, stimulation of the phosphofructokinase enzyme by alkalosis (respiratory and metabolic) has been shown to increase lactate levels [20]. Clinically often-used therapeutic interventions also have been shown to increase aerobic lactate production [21]. The aerobic production of lactate as an energy source is related to the very high lactate levels found in patients with lymphoma, a phenomenon referred to as the Warburg effect [22]. When treating the lymphoma, both lactate levels and LDH respond to chemotherapy (Figure 4). Recently, it has been shown that the activity of the Na+/K+ pump system, which requires significant amounts of ATP for its function, is related to increased lactate levels in both experimental and clinical conditions [23, 24], unrelated to the presence of tissue hypoxia. Such enhanced glycolysis can be triggered by cytokine-mediated uptake of glucose [25] or catecholamine-stimulated increased Na-K-pump activity [26, 27], supported by both experimental and clinical studies [23, 28]. A recent, still unresolved discussion, has focused on the presence of mitochondrial dysfunction in critically ill that could limit pyruvate metabolism (and thus increase lactate levels) in the absence of limited oxygen availability [29, 30].

Lactate levels and LDH levels in a patient with a lymphoma admitted to the ICU because of respiratory failure. Following diagnosis, treatment with chemotherapy was started. The effect of the first and second chemotherapy on lactate and LDH is shown.
Infusion of Ringer’s lactate does not hamper the accuracy of lactate measurement [31]. Finally, renal replacement therapy eliminates only negligible amounts of lactate [32], but using lactate-containing buffer solutions can induce transient hyperlactataemia [33, 34]. Other causes of increased lactate levels (probably) not related to the presence of tissue hypoxia have been described [35–39]. In the case of ethylene glycol intoxication, falsely increased measured lactate levels may result from an adverse reaction to the lactate electrode [40]. Thus, a normal lactate level in the laboratory in contrast to a high level in a point of care device may even be diagnostic in these cases [41].
Clearance of lactate
Corresponding with its versatile functions, the body is able to clear large lactate loads as shown following the rapid decrease in lactate levels following exercise or return of circulation in cardiac arrest. Likewise, the body is equally adept in clearing very large exogenous loads of lactate during high-volume continuous veno-venous hemofiltration [3, 33, 42].
Several clinical conditions have been associated with impaired clearance of lactate. First liver dysfunction has been shown to impair lactate clearance [37, 43]. Second, in patients following cardiac surgery lactate clearance also may be impaired [44]. Third, in addition to increased glucose metabolism and thus lactate production, sepsis may impair lactate clearance by the inhibition of the rate limiting enzyme pyruvate dehydrogenase [45]. Although this enzyme can be stimulated by dichloroacetate, thus forcing lactate and pyruvate into mitochondrial oxidation, clinical studies have not shown benefit of this increased lactate clearance [46].
How and where to measure lactate levels
Blood lactate levels can be measured using various devices (central laboratory, point-of-care blood gas analysers, hand-held devices) and generally most devices used at the bedside have acceptable limits of agreement compared to the laboratory devices [47, 48]. In addition, the sampling site of the blood (arterial, venous, capillary, etc.) also does not seem to affect the results much [49–51]. However, especially when targeting changes in lactate levels in relatively short intervals, it is not appropriate to use devices and sampling site interchangeably. To prevent an in vitro rise in blood lactate levels, especially when leucocytosis or a high haematocrit (red blood cells do not have mitochondria) are present, a maximum turnaround time of 15 min or storage of the sample on ice is advised [52–54]. Alternatively, tubes containing fluoride, a potent inhibitor of in vitro glycolysis, are widely used.
Prognosis
Since its first description in humans, increased blood lactate levels have been related to morbidity and mortality. In a recent health technology assessment on the use of lactate levels in critically ill patients, we showed that both in the emergency department and in the ICU blood lactate levels have a place in risk-stratification [55]. Not only one point in time measurements are related to outcome but also the duration and area under the curve of increased lactate levels are related to both morbidity (organ failure) and mortality in different patient groups [56, 57]. In the early phase of resuscitation, lactate levels seem to be more closely related to outcome than frequently used haemodynamics, including oxygen delivery and oxygen consumption [58–61]. Moreover, a holistic view incorporating many parameters may be more appropriate in early resuscitation [62].
Lactate as a goal of therapy
The latter observations stress the importance to define adequate resuscitation goals. Although generally believed to be inadequate, mean arterial pressure is frequently used as an important diagnostic and goal of therapy in patients with haemodynamic instability [63]. Given its strong relationship to the occurrence of inadequate tissue oxygenation and its long-time established relationship with morbidity and mortality, lactate levels could represent a useful goal of initial resuscitation in many clinical conditions. Until recently, the only known single-centre clinical trial advocating such lactate-directed therapy was performed in postcardiac surgery patients [64]. This study showed a reduction in morbidity but was not powered to study the effect on mortality. In addition, translating these findings to a more general and frequently much sicker population is difficult. However, in 2010, two multicentre clinical trials were published on the clinical value of lactate-directed therapy studying a specific group of patients (sepsis) in the Emergency Department [65] and a heterogeneous group of patients with increased lactate levels, not likely to be associated with confounding factors in lactate metabolism, in the ICU [66].
Lactate-guided therapy: the United States
In a multicentre, open-label, randomized controlled study, 300 patients were randomized to test the noninferiority between lactate clearance (≥10%) and central venous oxygen saturation (ScvO2 ≥70%) as goals of early resuscitation in patients presenting to the ED with severe sepsis or septic shock [65]. The intervention lasted until either all goals were achieved or 6 hours after start of the study. There were no differences in treatments administered during the initial 72 hours of hospitalization. In-hospital mortality in the lactate group was noninferior to the ScvO2 group. Although one might conclude that thus both lactate levels and ScvO2 are equally effective as a goal of therapy, the study has some limitations that prohibit this. First, venous oxygen saturation might help to differentiate anaerobic from aerobic hyperlactataemia probably resulting in a different treatment [67]. Second, it is questionable whether a 10% reduction in lactate in 6 hours represents an effective resuscitation. The study would implicate that a patient is adequately treated when the initial lactate level of 5.0 mmol/L decreases to 4.5 mmol/L after 6 hours of treatment. Also, a decrease of this magnitude even in the first hour of treatment is not likely to be associated with survival as early resuscitation studies have shown that survivors show an almost 30% decrease in lactate levels in the first hour [68]. Probably the failure to decrease lactate levels at all in response to treatment has more implications for both treatment and prognosis [59, 68]. Finally, as only 10% of the patients received either dobutamine or red blood cell transfusion and fluids and vasopressors were guided by CVP and MAP in both groups, the potential difference in protocol actions directly attributable to either lactate or ScvO2 was very small. Therefore, it seems unlikely that a change in this resuscitation target could increase mortality by 10%, the noninferiority margin selected for the trial [69].
Lactate-guided therapy: the Netherlands
In a multicentre, open-label, randomized, controlled trial, 348 patients were randomly allocated to either lactate-guided treatment (lactate group) or nonlactate-guided treatment (control group) during the first 8 hours of ICU stay [66]. In the lactate group, the treatment goals were a 20% or more decrease in lactate levels per 2 hours and the normalization of ScvO2 (>70%). In the control group, lactate levels were not available to the treating physicians during the first 8 hours except for the admission level required for randomization. An important addition to the protocol treatment was the administration of a vasodilator when ScvO2 levels where normal but lactate levels did not decrease sufficiently. This is the first study to address this important problem in the resuscitation of critically ill patients, because normalisation of ScvO2 is generally regarded as a restoration of the balance between oxygen delivery and oxygen demand that should result in normalization of lactate levels as demonstrated by Zhang et al. [70]. Assuming abnormal microcirculatory perfusion in a state like this that could be improved by the administration of nitroglycerine [71] led to the addition of this intervention to the protocol treatment. We recently showed that nitroglycerine improves abnormal tissue oxygenation in critically ill patients supporting this concept [72]. For a recent, double-blinded, randomized study by Boerma et al., [73] nitroglycerin administration was used in addition to a standard resuscitation protocol in all patients in the protocol group. This study showed no differences in the microcirculation between the two groups and a trend towards an increased mortality in the protocol group. The use of nitroglycerin this study is very different from its use in the study by Jansen et al. [66]. In the latter study, the use of nitroglycerin was guided by a clinical problem (no adequate decrease in lactate levels despite optimal balance between oxygen delivery and oxygen demand), whereas in the study by Boerma et al. nitroglycerin also was used in patients with normal microcirculation, decreasing, or even normal lactate levels, etc. We therefore do not advocate adding nitroglycerin to the standard resuscitation protocols.
Just like in the landmark study on early goal directed therapy [74], protocol patients received significantly more fluids during the intervention period, whereas these patients received less fluid during the subsequent observation period. In addition, significantly more patients were treated with vasodilators (predominantly nitroglycerine) in the protocol group. These differences in treatment were associated with an almost statistically significant (p = 0.067), 20% relative reduction in mortality in addition to a strong statistically significant reduction in morbidity (duration of mechanical ventilation and ICU stay, p = 0.006).
However, despite the outcome benefit, the course of lactate levels in the two groups was similar. This in fact suggests no causal relationship between the resuscitation therapy and hyperlactataemia. Instead, lactate might be an epiphenomenon of severity of disease. By acting as a warning signal, clinicians might have interpreted hyperlactataemia as a warning that their patients did not clinically improve or even deteriorate in the presence of stable haemodynamic parameters. This could have triggered additional diagnostic and therapeutic interventions.
Summarizing, these two recent studies show that lactate-directed resuscitation therapy has clinical benefit for critically ill patients, although the exact mechanism behind it remains uncertain. Lactate measurement probably should be accompanied by venous saturations monitoring to guide decision-making and therapy.
Conclusions
To understand the importance of an increased lactate level, it is important not only to consider anaerobic production but also aerobic mechanisms and changes in lactate clearance. Despite this complex evaluation, increased lactate levels usually reflect increased morbidity and high mortality. In addition, two recent multicentre trials suggest that the use of lactate levels in goal-directed therapy may improve clinical outcome. These findings confirm that lactate monitoring is a valuable parameter in the early resuscitation of critically ill patients.
References
Kompanje EJ, Jansen TC, van der Hoven B, Bakker J: The first demonstration of lactic acid in human blood in shock by Johann Joseph Scherer (1814–1869) in January 1843. Intensive Care Med 2007,33(11):1967–1971. 10.1007/s00134-007-0788-7
Araki T: Ueber die Bildung von Milchsäure und Glycose im Organismus bei Sauerstoffmangel. Z Physiol Chem 1891, 15: 335–370.
Leverve XM: Energy metabolism in critically ill patients: lactate is a major oxidizable substrate. Curr Opin Clin Nutr Metab Care 1999,2(2):165–169. 10.1097/00075197-199903000-00013
Brooks GA: Lactate shuttles in nature. Biochem Soc Trans 2002,30(2):258–264.
Zilva JF: The origin of the acidosis in hyperlactataemia. Ann Clin Biochem 1978,15(1):40–43.
Stewart PA: Modern quantitative acid–base chemistry. Can J Physiol Pharmacol 1983,61(12):1444–1461. 10.1139/y83-207
Gunnerson KJ, Saul M, He S, Kellum JA: Lactate versus non-lactate metabolic acidosis: a retrospective outcome evaluation of critically ill patients. Crit Care 2006,10(1):R22. 10.1186/cc3987
Fall PJ, Szerlip HM: Lactic acidosis: from sour milk to septic shock. J Intensive Care Med 2005,20(5):255–271. 10.1177/0885066605278644
Cain SM: Appearance of excess lactate in anesthetized dogs during anemic and hypoxic hypoxia. Am J Physiol 1965, 209: 604–608.
Zhang H, Vincent JL: Oxygen extraction is altered by endotoxin during tamponade-induced stagnant hypoxia in the dog. Circ Shock J1 - CS 1993,40(3):168–176.
Bakker J, Vincent J-L: The oxygen supply dependency phenomenon is associated with increased blood lactate levels. J Crit Care 1991, 6: 152–159. 10.1016/0883-9441(91)90006-F
Ronco JJ, Fenwick JC, Tweeddale MG, Wiggs BR, Phang PT, Cooper DJ, Cunningham KF, Russell JA, Walley KR: Identification of the critical oxygen delivery for anaerobic metabolism in critically ill septic and nonseptic humans. JAMA 1993,270(14):1724–1730. 10.1001/jama.1993.03510140084034
Friedman G, De Backer D, Shahla M, Vincent JL: Oxygen supply dependency can characterize septic shock. Intensive Care Med 1998,24(2):118–123. 10.1007/s001340050531
De Backer D, Creteur J, Dubois MJ, Sakr Y, Koch M, Verdant C, Vincent JL: The effects of dobutamine on microcirculatory alterations in patients with septic shock are independent of its systemic effects. Crit Care Med 2006,34(2):403–408. 10.1097/01.CCM.0000198107.61493.5A
Leverve XM: From tissue perfusion to metabolic marker: assessing organ competition and co-operation in critically ill patients? Intensive Care Med 1999,25(9):890–892. 10.1007/s001340050976
Levy B, Sadoune LO, Gelot AM, Bollaert PE, Nabet P, Larcan A: Evolution of lactate/pyruvate and arterial ketone body ratios in the early course of catecholamine-treated septic shock [see comments]. Crit Care Med 2000,28(1):114–119. 10.1097/00003246-200001000-00019
Levy B, Bollaert PE, Charpentier C, Nace L, Audibert G, Bauer P, Nabet P, Larcan A: Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997,23(3):282–287. 10.1007/s001340050329
Weil MH, Tang W: Forty-five-year evolution of stat blood and plasma lactate measurement to guide critical care. Clin Chem 2009,55(11):2053–2054. 10.1373/clinchem.2009.133553
Griffith FR Jr, Lockwood JE, Emery FE: Adrenalin lactacidemia: proportionality with dose. Am J Physiol 1939,127(3):415–421.
Zborowska-Sluis DT, Dossetor JB: Hyperlactatemia of hyperventilation. J Appl Physiol J1 - JAP 1967,22(4):746–755.
McMahon M, Gerich J, Rizza R: Effects of glucocorticoids on carbohydrate metabolism. Diabetes Metab Rev 1988,4(1):17–30. 10.1002/dmr.5610040105
Warburg O: On respiratory impairment in cancer cells. Science 1956,124(3215):269–270.
Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE: Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005,365(9462):871–875. 10.1016/S0140-6736(05)71045-X
Levy B, Desebbe O, Montemont C, Gibot S: Increased aerobic glycolysis through beta-2 stimulation is a common mechanism involved in lactate formation during shock states. Shock 2008,30(4):417–421. 10.1097/SHK.0b013e318167378f
Taylor DJ, Faragher EB, Evanson JM: Inflammatory cytokines stimulate glucose uptake and glycolysis but reduce glucose oxidation in human dermal fibroblasts in vitro. Circ Shock 1992,37(2):105–110.
McCarter FD, Evans JA, Luchette FA, Friend LA, James JH, Davis K, Frame SB: Concurrent reduction of glycogenolysis, glycolysis, and Na(+)/K(+) pump activity after hemorrhagic shock. Curr Surg 2000,57(6):639.
Luchette FA, Friend LA, Brown CC, Upputuri RK, James JH: Increased skeletal muscle Na+, K+-ATPase activity as a cause of increased lactate production after hemorrhagic shock. J Trauma 1998,44(5):796–801. discussion 801–793 10.1097/00005373-199805000-00010
Haji-Michael PG, Ladriere L, Sener A, Vincent JL, Malaisse WJ: Leukocyte glycolysis and lactate output in animal sepsis and ex vivo human blood. Metabolism 1999,48(6):779–785. 10.1016/S0026-0495(99)90179-8
Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M: Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002,360(9328):219–223. 10.1016/S0140-6736(02)09459-X
Crouser ED, Julian MW, Blaho DV, Pfeiffer DR: Endotoxin-induced mitochondrial damage correlates with impaired respiratory activity. Crit Care Med 2002,30(2):276–284. 10.1097/00003246-200202000-00002
Didwania A, Miller J, Kassel D, Jackson EVJ, Chernow B: Effect of intravenous lactated Ringer’s solution infusion on the circulating lactate concentration: Part 3. Results of a prospective, randomized, double-blind, placebo-controlled trial. Crit Care Med 1997,25(11):1851–1854. 10.1097/00003246-199711000-00024
Levraut J, Ciebiera JP, Jambou P, Ichai C, Labib Y, Grimaud D: Effect of continuous venovenous hemofiltration with dialysis on lactate clearance in critically ill patients. Crit Care Med 1997,25(1):58–62. 10.1097/00003246-199701000-00013
Cole L, Bellomo R, Baldwin I, Hayhoe M, Ronco C: The impact of lactate-buffered high-volume hemofiltration on acid–base balance. Intensive Care Med 2003,29(7):1113–1120. 10.1007/s00134-003-1812-1
Bollmann MD, Revelly JP, Tappy L, Berger MM, Schaller MD, Cayeux MC, Martinez A, Chiolero RL: Effect of bicarbonate and lactate buffer on glucose and lactate metabolism during hemodiafiltration in patients with multiple organ failure. Intensive Care Med 2004,30(6):1103–1110. 10.1007/s00134-004-2251-3
Lalau JD, Lacroix C, Compagnon P, de Cagny B, Rigaud JP, Bleichner G, Chauveau P, Dulbecco P, Guerin C, Haegy JM: Role of metformin accumulation in metformin-associated lactic acidosis. Diabetes Care 1995,18(6):779–784. 10.2337/diacare.18.6.779
Marinella MA: Lactic acidosis associated with propofol. Chest 1996,109(1):292. 10.1378/chest.109.1.292
Lonergan JT, Behling C, Pfander H, Hassanein TI, Mathews WC: Hyperlactatemia and hepatic abnormalities in 10 human immunodeficiency virus-infected patients receiving nucleoside analogue combination regimens. Clin Infect Dis 2000,31(1):162–166. 10.1086/313912
Claessens YE, Cariou A, Monchi M, Soufir L, Azoulay E, Rouges P, Goldgran-Toledano D, Branche F, Dhainaut JF: Detecting life-threatening lactic acidosis related to nucleoside-analog treatment of human immunodeficiency virus-infected patients, and treatment with L-carnitine. Crit Care Med 2003,31(4):1042–1047. 10.1097/01.CCM.0000053649.69377.08
Naidoo DP, Gathiram V, Sadhabiriss A, Hassen F: Clinical diagnosis of cardiac beriberi. S Afr Med J 1990,77(3):125–127.
Morgan TJ, Clark C, Clague A: Artifactual elevation of measured plasma L-lactate concentration in the presence of glycolate. Crit Care Med 1999,27(10):2177–2179. 10.1097/00003246-199910000-00017
Brindley PG, Butler MS, Cembrowski G, Brindley DN: Falsely elevated point-of-care lactate measurement after ingestion of ethylene glycol. CMAJ 2007,176(8):1097–1099.
Leverve XM, Boon C, Hakim T, Anwar M, Siregar E, Mustafa I: Half-molar sodium-lactate solution has a beneficial effect in patients after coronary artery bypass grafting. Intensive Care Med 2008,34(10):1796–1803. 10.1007/s00134-008-1165-x
Almenoff PL, Leavy J, Weil MH, Goldberg NB, Vega D, Rackow EC: Prolongation of the half-life of lactate after maximal exercise in patients with hepatic dysfunction. Crit Care Med 1989,17(9):870–873. 10.1097/00003246-198909000-00004
Mustafa I, Roth H, Hanafiah A, Hakim T, Anwar M, Siregar E, Leverve XM: Effect of cardiopulmonary bypass on lactate metabolism. Intensive Care Med 2003,29(8):1279–1285. 10.1007/s00134-003-1860-6
Vary TC: Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996,6(2):89–94. 10.1097/00024382-199608000-00002
Stacpoole PW, Wright EC, Baumgartner TG, Bersin RM, Buchalter S, Curry SH, Duncan CA, Harman EM, Henderson GN, Jenkinson S: A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. The Dichloroacetate-Lactic Acidosis Study Group. N Engl J Med 1992,327(22):1564–1569. 10.1056/NEJM199211263272204
Aduen J, Bernstein WK, Khastgir T, Miller J, Kerzner R, Bhatiani A, Lustgarten J, Bassin AS, Davison L, Chernow B: The use and clinical importance of a substrate-specific electrode for rapid determination of blood lactate concentrations. JAMA 1994,272(21):1678–1685. 10.1001/jama.1994.03520210062033
Brinkert W, Rommes JH, Bakker J: Lactate measurements in critically ill patients with a hand-held analyser. Intensive Care Med 1999,25(9):966–969. 10.1007/s001340050990
Weil MH, Michaels S, Rackow EC: Comparison of blood lactate concentrations in central venous, pulmonary artery, and arterial blood. Crit Care Med 1987,15(5):489–490.
Younger JG, Falk JL, Rothrock SG: Relationship between arterial and peripheral venous lactate levels. Acad Emerg Med 1996,3(7):730–734. 10.1111/j.1553-2712.1996.tb03502.x
Fauchere JC, Bauschatz AS, Arlettaz R, Zimmermann-Bar U, Bucher HU: Agreement between capillary and arterial lactate in the newborn. Acta Paediatr 2002,91(1):78–81. 10.1111/j.1651-2227.2002.tb01645.x
Noordally O, Vincent JL: Evaluation of a new, rapid lactate analyzer in critical care. Intensive Care Med 1999,25(5):508–513. 10.1007/s001340050889
Astles R, Williams CP, Sedor F: Stability of plasma lactate in vitro in the presence of antiglycolytic agents. Clin Chem 1994,40(7 Pt 1):1327–1330.
Andersen O, Haugaard SB, Jorgensen LT, Sorensen S, Nielsen JO, Madsbad S, Iversen J: Preanalytical handling of samples for measurement of plasma lactate in HIV patients. Scand J Clin Lab Invest 2003,63(6):449–454. 10.1080/00365510310005128
Jansen TC, van Bommel J, Bakker J: Blood lactate monitoring in critically ill patients: a systematic health technology assessment. Crit Care Med 2009,37(10):2827–2839. 10.1097/CCM.0b013e3181a98899
Bakker J, Gris P, Coffernils M, Kahn RJ, Vincent JL: Serial blood lactate levels can predict the development of multiple organ failure following septic shock. Am J Surg 1996,171(2):221–226. 10.1016/S0002-9610(97)89552-9
Jansen TC, van Bommel J, Woodward R, Mulder PG, Bakker J: Association between blood lactate levels, sequential organ failure assessment subscores, and 28-day mortality during early and late intensive care unit stay: a retrospective observational study. Crit Care Med 2009,37(8):2369–2374. 10.1097/CCM.0b013e3181a0f919
Jansen TC, van Bommel J, Mulder PG, Rommes JH, Schieveld SJ, Bakker J: The prognostic value of blood lactate levels relative to that of vital signs in the pre-hospital setting: a pilot study. Crit Care 2008,12(6):R160. 10.1186/cc7159
Bakker J, Coffernils M, Leon M, Gris P, Vincent J-L: Blood lactate levels are superior to oxygen-derived variables in predicting outcome in human septic shock. Chest 1991,99(4):956–962. 10.1378/chest.99.4.956
Howell M, Donnino M, Clardy P, Talmor D, Shapiro N: Occult hypoperfusion and mortality in patients with suspected infection. Intensive Care Med 2007,33(11):1892–1899. 10.1007/s00134-007-0680-5
Shapiro NI, Howell MD, Talmor D, Nathanson LA, Lisbon A, Wolfe RE, Weiss JW: Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med 2005,45(5):524–528. 10.1016/j.annemergmed.2004.12.006
Hernandez G, Bruhn A, Castro R, Regueira T: The holistic view on perfusion monitoring in septic shock. Curr Opin Crit Care 2012,18(3):280–286. 10.1097/MCC.0b013e3283532c08
Vincent JL, Ince C, Bakker J: Clinical review: circulatory shock - an update: a tribute to Professor Max Harry Weil. Crit Care 2012,16(6):239.
Polonen P, Ruokonen E, Hippelainen M, Poyhonen M, Takala J: A prospective, randomized study of goal-oriented hemodynamic therapy in cardiac surgical patients. Anesth Analg 2000,90(5):1052–1059. 10.1097/00000539-200005000-00010
Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA: Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010,303(8):739–746. 10.1001/jama.2010.158
Jansen TC, van Bommel J, Schoonderbeek FJ, Sleeswijk Visser SJ, van der Klooster JM, Lima AP, Willemsen SP, Bakker J: Early lactate-guided therapy in intensive care unit patients: a multicenter, open-label, randomized controlled trial. Am J Respir Crit Care Med 2010,182(6):752–761. 10.1164/rccm.200912-1918OC
James JH, Luchette FA, McCarter FD, Fischer JE: Lactate is an unreliable indicator of tissue hypoxia in injury or sepsis. Lancet 1999,354(9177):505–508. 10.1016/S0140-6736(98)91132-1
Vincent JL, Dufaye P, Berre J, Leeman M, Degaute JP, Kahn RJ: Serial lactate determinations during circulatory shock. Crit Care Med 1983,11(6):449–451. 10.1097/00003246-198306000-00012
Lewis RJ: Disassembling goal-directed therapy for sepsis: a first step. JAMA 2010,303(8):777–779. 10.1001/jama.2010.203
Zhang H, Spapen H, Benlabed M, Vincent JL: Systemic oxygen extraction can be improved during repeated episodes of cardiac tamponade. J Crit Care 1993,8(2):93–99. 10.1016/0883-9441(93)90013-B
Spronk PE, Ince C, Gardien MJ, Mathura KR, Oudemans-van Straaten HM, Zandstra DF: Nitroglycerin in septic shock after intravascular volume resuscitation. Lancet 2002,360(9343):1395–1396. 10.1016/S0140-6736(02)11393-6
Lima A, van Genderen M, Van Bommel J, Bakker J: Nitroglycerine dose-dependent improves peripheral perfusion in patients with circulatory shock: results of a prospective cross-over study. Intensive Care Med 2012,38(Suppl 1):S127.
Boerma EC, Koopmans M, Konijn A, Kaiferova K, Bakker AJ, van Roon EN, Buter H, Bruins N, Egbers PH, Gerritsen RT: Effects of nitroglycerin on sublingual microcirculatory blood flow in patients with severe sepsis/septic shock after a strict resuscitation protocol: a double-blind randomized placebo controlled trial. Crit Care Med 2010,38(1):93–100. 10.1097/CCM.0b013e3181b02fc1
Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, Peterson E, Tomlanovich M: Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001,345(19):1368–1377. 10.1056/NEJMoa010307
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JB, MWNN and TCJ all contributed to the paper and wrote equal sections of the manuscript. JB did the final editing of the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bakker, J., Nijsten, M.W. & Jansen, T.C. Clinical use of lactate monitoring in critically ill patients. Ann. Intensive Care 3, 12 (2013). https://doi.org/10.1186/2110-5820-3-12
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2110-5820-3-12