
Abstract
The family of potassium channel tetramerizationdomain (KCTD) proteins consists of 26 members with mostly unknown functions. The name of the protein family is due to the sequence similarity between the conserved N-terminal region of KCTD proteins and the tetramerization domain in some voltage-gated potassium channels. Dozens of publications suggest that KCTD proteins have roles in various biological processes and diseases. In this review, we summarize the character of Bric-a-brack,Tram-track, Broad complex(BTB) of KCTD proteins, their roles in the ubiquitination pathway, and the roles of KCTD mutants in diseases. Furthermore, we review potential downstream signaling pathways and discuss future studies that should be performed.
Similar content being viewed by others


Introduction
The human potassium (K+) channel tetramerization domain (KCTD)family of proteins consists of 26 members that share sequence similarity with the cytoplasmic domain of voltage-gated K+ channels(Kv channels) [1–3]. The KCTD proteins have relatively conserved N-terminal domains and variable C-termini. Comparative analyses of the conserved N-terminal sequence suggest the presence of a common Bric-a-brack,Tram-track, Broad complex (BTB) domain, which is also known as the POZ domain. The BTB domain is a versatile protein-protein interaction motif that facilitates homodimerization or heterodimerization. A variety of functions have been identified for the BTB domain-containing KCTD proteins. These functions include transcriptional repression [4, 5], cytoskeleton regulation [6], tetramerization and gating of ion channels [7, 8], and interaction with the cullin E3 (Cul3) ubiquitin ligase complex [9, 10]. In this review, we will summarize the homology between KCTD family members and some of the key features of KCTD proteins. We will also discuss the roles of mutant KCTDs in disease.
BTB domain and homology between KCTD family members
The human genome includes approximately 400 BTB domain-containing proteins. The BTB domain is a highly conserved motif of about 100 amino acids and can be found at the N-terminusof C2H2-type zinc-finger transcription factors and in some actin-binding proteins [11]. BTB domain-containing proteins include transcription factors, oncogenic proteins, ion channel proteins, and KCTD proteins [2, 12–14]. Many BTB domain-containing proteins contain one or two additional domains, such as kelch repeats, zinc-finger domains, FYVE (Fab1, YOTB, Vac1, and EEA1) fingers which is a novel zinc finger-like domain found in several proteins involved in membrane trafficking, or ankyrin repeats [15]. These special domains provide unique characteristics and functions to the BTB proteins. The BTB domain facilitates protein-protein interactions between KCTD proteins to allow self-assembly or with non-BTB-domain-containing proteins to promote oligomerization [15]. The X-ray crystal structure of KCTD5 also revealed assemblies of five subunits while tetramers were anticipated [16]. A variety of functional roles of KCTD proteins have been identified by different signal pathways, including sonic hedgehog (Shh) [17–19], Wnt/beta-catenin [20], FGF [1], and GABA signaling [21–24]. Alignment of the amino acids in the potassium tetramerization domains of all known KCTD proteins demonstrates that most KCTD proteins can be divided into seven groups by amino acid sequences. The A-group contains KCTD9, KCTD17, KCTD 5, and KCTD 2. The B-group contains KCTD10, KCTD13, and TNFAIP1. The C-group contains KCTD7 and KCTD14. The d-group contains KCTD8, KCTD12, and KCTD16. The E-group contains KCTD11, KCTD21, and KCTD6. Members of the F-group include KCTD1 and KCTD15. And the final group is the G-group, which contains KCTD3 and SHKBP1 and BTB10. KCTD20, KCTD18, KCTD19, and KCTD4 do not belong to these seven groups (Figure 1). The evolutionary tree of the KCTD family proteins is similar to the group that Skoblov M et al. built [25]. We also suggest that homologous KCTD members may share similar functional roles in proliferation, transcription, protein degradation, regulation of G-protein coupled receptors and other molecular or biological processes.

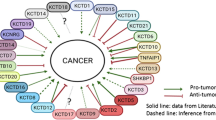
A paralogues tree of the KCTD family proteins as cullin ligase adaptor and their substrate. Left: A paralogues tree built using entire amino acid sequences of the KCTD family proteins; Right: the family of KCTD proteins corresponding to cullin and their substrate.
KCTD proteins as adaptor molecules
BTB-domain-containing KCTD proteins may act as adaptors for interactions between the Cul3 ubiquitin ligase and its substrates. Thus, BTB KCTD proteins may facilitate successful ubiquitination of substrate proteins [26]. Cul3 is one of seven human cullin proteins (Cul1,Cul2,Cul3,Cul4A,Cul4B,Cul5, and Cul7). Most cullins form complexes with substrate proteins by interacting with the BTB domains of adaptor proteins [3]. Thus, the BTB domain is important for the process of ubiquitination and protein degradation. Ubiquitination involves a three-step enzymatic cascade, which is initially activated by ubiquitin-activating enzyme(E1). The substrate is then transferred to ubiquitin-conjugating enzyme(E2) and is finally linked with ubiquitin ligase(E3) [27]. Various cellular functions, including cell proliferation, differentiation, apoptosis, and protein transport, involve protein ubiquitination and de-ubiquitination [28]. Bioinformatics and mutagenesis analyses have demonstrated that the best-characterized member of the KCTD family, KCTD11/REN, is expressed as two alternative variants, sKCTD11 and lKCTD11. Despite the fact that both variants possess a BTB domain in the N-terminus, only the lKCTD11 form has a complete BTB domain. Intriguingly, this has not disturbed the cul3-binding activity of sKCTD11. KCTD11/REN also mediates histone deacetylase (HDAC1) ubiquitination and degradation via cullin binding, resulting in reduced Hh/Gli signaling [18]. The KCTD2 1 and KCTD6 have also been found to have the same features as KCTD11[29]. Thus, KCTD21 and KCTD6 may also facilitate protein degradation and reduced cellular signaling due to associations with ubiquitin ligases. KCTD5 and KCTD7 have also been shown to function as substrate-specific adaptors for cullin3-based E3 ligases [3, 30, 31]. In addition, KCTD7 has been shown to increase potassium conductance due to increased proteasome degradation of an unidentified substrate [30]. Thus, several members of the KCTD family function as critical adaptor molecules for ubiquitin-mediated protein degradation. This function ultimately results in the modulation of important downstream signaling pathways and biological processes. As can be seen from Figure 1, cullin is fairly widely interaction with the family of KCTD proteins. In the future, this novel substrate of KCTD will help to understand the function of the complex of CUL3 –BTB.
KCTDs and disease
KCTD proteins have essential roles in proliferation, differentiation, apoptosis, and metabolism. Improper regulation of KCTD genes has been associated with various diseases, including medulloblastoma [32], breast carcinoma [33], obesity [34, 35], and pulmonary inflammation [36]. Many studies show associations between mutations in individual KCTD genes or allelic loss of KCTDs with specific diseases. For example, a homozygous mutation (R99X) in exon 2 of KCTD7 has been described in progressive myoclonic epilepsy (PME) [37]. A second homozygous missense mutation (R94W) in exon 2 of KCTD7 has also been found in PME [38]. In addition, a heterozygous missense mutation (R84W) and a large heterozygous deletion of exons 3 and 4 of KCTD7 have also been reported in patients with PME [30, 31]. Allelic deletion of human KCTD11 at chromosomal location 17p13.2 has been found in medulloblastoma [19, 39]. In addition, gene copy number variants (CNVs) of KCTD13 mapping to chromosomal location 16p11.2 are considered to be major genetic causes of macrocephaly and microcephaly. Overexpression of KCTD13 induces microcephaly, whereas suppression of the same locus results in a macrocephalic phenotype [40]. Missense mutations in KCTD1 occur in Scalp-ear-nipple (SEN) syndrome [41]. Single nucleotide polymorphisms(SNPs) of KCTD10 (i5642G- > C and V206VT- > C) are associated with altered concentrations of HDL cholesterol, particularly in subjects with high levels of carbohydrate intake [42]. KCTD mutants affect proliferation, differentiation, apoptosis, and metabolism in different tissues. For example, the CNVs of KCTD13 affect the balance of proliferation and apoptosis in neuronal progenitor cells. In addition, deletions in KCTD11 abrogateinhibition of Shh signaling at the outer to inner external granule layer-granule cell progenitor (EGL GCP) transitions by affecting expression of Gli1 and Gli2 [19]. Deletions in KCASH, KCTD21, or KCTD6 block interactions with ubiquitination enzymes, preventing degradation of HDAC1. This leads to increased acetylation of Gli1 and increased Hh/Gli signaling, which drives uncontrolled proliferation and development and progression of medulloblastoma [17, 39]. Not only mutant KCTD could cause diseases, but also the change of KCTD expression involved in different diseases [22, 43–46]. All of the diseases related with KCTD proteins have been list in a Table 1 to make the family more convenient for further study.
Conclusion
There are some features of KCTDs that have not been reviewed in this article. For example, KCTD8, -12, -12b, and-16 form functional oligomers with the GABAB receptor, resulting in the modulation of important signaling pathways [21–24, 47]. In addition, the PDIP1 family members (KCTD10, KCTD13, and TNFAIP1) are tumor necrosis factor-a-inducible proteins that can stimulate the activity of DNA polymerase in DNA replication and repair pathways [48]. Furthermore, interactions between KCTD1, KCTD15, and AP-2 represses the transcriptional activity of AP-2a [13], Finally, KCTD1 has been shown to interact with PrPC [49]. In the review, we summarize the BTB characteristics of the KCTD proteins, their roles in the ubiquitination pathway, and the relevance of KCTD mutations in various diseases. The review highlight the extraordinary possibility of the interaction of cullin-KCTDs to target substrates for ubiquitin-dependent degradation. If BTB-containing KCTD proteins can assemble into Cul3-based complexes, we estimate KCTD proteins can recruit substrates into ubiquitin system. We specifically discuss the role of KCTD1 in the ubiquitination pathway via interaction with cul3. We also hypothesize that KCTD1 mediate prion protein into ubiquitination signal pathway, and deregulation of the KCTD1 mediated prion protein ubiquitination might be both a cause and result of prion disease. Furthermore, we speculate that members of the same sub-groups may have similar roles in biological processes or molecular signaling pathways. We believe that further investigations into the functions of individual KCTD family members are warranted, particularly within the context of specific diseases as described here.
Abbreviations
- KCTD:
-
Potassium channel tetramerization domain
- BTB:
-
Bric-a-brack,Tram-track, Broad complex
- Cul3:
-
Cullin E3 ubiquitin ligase
- Shh:
-
Sonic hedgehog
- E1:
-
Ubiquitin-activating enzyme
- E2:
-
Ubiquitin-conjugating enzyme
- E3:
-
Ubiquitin ligase
- HDAC:
-
Histone deacetylase
- PME:
-
Progressive myoclonic epilepsy
- CNV:
-
Copy number variant
- SEN:
-
Scalp-ear-nipplesyndrome
- SNP:
-
Single nucleotide polymorphism
- EGL GCP:
-
External granule layer-granule cell progenitor
- KCASH:
-
KCTD containing, Cullin3 adaptor, suppressor of Hedgehog
- TNFAIP1:
-
Tumor necrosis factor, alpha-induced protein 1.
References
Takahashi C, Suzuki T, Nishida E, Kusakabe M: Identification and characterization of Xenopus kctd15, an ectodermal gene repressed by the FGF pathway. Int J Dev Biol. 2012, 56 (5): 393-402. 10.1387/ijdb.113333ct
Mei F, Xiang J, Han S, He Y, Lu Y, Xu J, Guo D, Xiao G, Tien P, Sun G: Expression, purification, and secondary structure characterization of recombinant KCTD1. Biochemistry (Mosc). 2012, 77 (8): 941-945. 10.1134/S0006297912080160
Bayon Y, Trinidad AG, De la Puerta ML, Del Carmen RM, Bogetz J, Rojas A, De Pereda JM, Rahmouni S, Williams S, Matsuzawa S, et al: KCTD5, a putative substrate adaptor for cullin3 ubiquitin ligases. FEBS J. 2008, 275 (15): 3900-3910. 10.1111/j.1742-4658.2008.06537.x
Melnick A, Ahmad KF, Arai S, Polinger A, Ball H, Borden KL, Carlile GW, Prive GG, Licht JD: In-depth mutational analysis of the promyelocytic leukemia zinc finger BTB/POZ domain reveals motifs and residues required for biological and transcriptional functions. Mol Cell Biol. 2000, 20 (17): 6550-6567. 10.1128/MCB.20.17.6550-6567.2000
Ahmad KF, Melnick A, Lax S, Bouchard D, Liu J, Kiang CL, Mayer S, Takahashi S, Licht JD, Prive GG: Mechanism of SMRT corepressor recruitment by the BCL6 BTB domain. Mol Cell. 2003, 12 (6): 1551-1564. 10.1016/S1097-2765(03)00454-4
Kang MIKA, Wakabayashi N, Kim SG, Yamamoto M: Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc Natl Acad Sci U S A. 2004, 101: 2046-2051. 10.1073/pnas.0308347100
Kreusch A, Pfaffinger PJ, Stevens CF, Choe S: Crystal structure of the tetramerization domain of the Shaker potassium channel. Nature. 1998, 392 (6679): 945-948. 10.1038/31978
Minor DL, Lin YF, Mobley BC, Avelar A, Jan YN, Jan LY, Berger JM: The polar T1 interface is linked to conformational changes that open the voltage-gated potassium channel. Cell. 2000, 102 (5): 657-670. 10.1016/S0092-8674(00)00088-X
Pintard L, Willems A, Peter M: Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J. 2004, 23 (8): 1681-1687. 10.1038/sj.emboj.7600186
Krek W: BTB proteins as henchmen of Cul3-based ubiquitin ligases. Nat Cell Biol. 2003, 5 (11): 950-951. 10.1038/ncb1103-950
Bardwell VJ, Treisman R: The POZ domain: a conserved protein-protein interaction motif. Genes Dev. 1994, 8 (14): 1664-1677. 10.1101/gad.8.14.1664
Correale S, Pirone L, Di Marcotullio L, De Smaele E, Greco A, Mazza D, Moretti M, Alterio V, Vitagliano L, Di Gaetano S, et al: Molecular organization of the cullin E3 ligase adaptor KCTD11. Biochimie. 2011, 93 (4): 715-724. 10.1016/j.biochi.2010.12.014
Ding X, Luo C, Zhou J, Zhong Y, Hu X, Zhou F, Ren K, Gan L, He A, Zhu J, et al: The interaction of KCTD1 with transcription factor AP-2alpha inhibits its transactivation. J Cell Biochem. 2009, 106 (2): 285-295. 10.1002/jcb.22002
Ding XF, Luo C, Ren KQ, Zhang J, Zhou JL, Hu X, Liu RS, Wang Y, Gao X: Characterization and expression of a human KCTD1 gene containing the BTB domain, which mediates transcriptional repression and homomeric interactions. DNA Cell Biol. 2008, 27 (5): 257-265. 10.1089/dna.2007.0662
Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, Prive GG: Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005, 6 (10): R82. 10.1186/gb-2005-6-10-r82
Dementieva IS, Tereshko V, McCrossan ZA, Solomaha E, Araki D, Xu C, Grigorieff N, Goldstein SA: Pentameric assembly of potassium channel tetramerization domain-containing protein 5. J Mol Biol. 2009, 387 (1): 175-191. 10.1016/j.jmb.2009.01.030
De Smaele E, Di Marcotullio L, Moretti M, Pelloni M, Occhione MA, Infante P, Cucchi D, Greco A, Pietrosanti L, Todorovic J, et al: Identification and characterization of KCASH2 and KCASH3, 2 novel Cullin3 adaptors suppressing histone deacetylase and Hedgehog activity in medulloblastoma. Neoplasia. 2011, 13 (4): 374-385.
Canettieri LDM G, Greco A, Coni S, Antonucci L, Infante P, Pietrosanti EDS L, Ferretti E, Miele E, Pelloni M, De Simone G, Pedone PG EM, Giorgi A, Steinkühler C, Vitagliano L, Pedone C, Schinin IS ME, Gulino A: Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010, 12: 132-142. 10.1038/ncb2013
De Smaele E, Di Marcotullio L, Ferretti E, Screpanti I, Alesse E, Gulino A: Chromosome 17p deletion in human medulloblastoma: a missing checkpoint in the Hedgehog pathway. Cell Cycle. 2004, 3 (10): 1263-1266. 10.4161/cc.3.10.1200
Dutta S, Dawid IB: Kctd15 inhibits neural crest formation by attenuating Wnt/beta-catenin signaling output. Development. 2010, 137 (18): 3013-3018. 10.1242/dev.047548
Seddik R, Jungblut SP, Silander OK, Rajalu M, Fritzius T, Besseyrias V, Jacquier V, Fakler B, Gassmann M, Bettler B: Opposite effects of KCTD subunit domains on GABA(B) receptor-mediated desensitization. J Biol Chem. 2012, 287 (47): 39869-39877. 10.1074/jbc.M112.412767
Sand PG, Langguth B, Itzhacki J, Bauer A, Geis S, Cardenas-Conejo ZE, Pimentel V, Kleinjung T: Resequencing of the auxiliary GABA(B) receptor subunit gene KCTD12 in chronic tinnitus. Front Syst Neurosci. 2012, 6: 41.
Metz M, Gassmann M, Fakler B, Schaeren-Wiemers N, Bettler B: Distribution of the auxiliary GABAB receptor subunits KCTD8, 12, 12b, and 16 in the mouse brain. J Comp Neurol. 2011, 519 (8): 1435-1454. 10.1002/cne.22610
Schwenk J, Metz M, Zolles G, Turecek R, Fritzius T, Bildl W, Tarusawa E, Kulik A, Unger A, Ivankova K, et al: Native GABA(B) receptors are heteromultimers with a family of auxiliary subunits. Nature. 2010, 465 (7295): 231-235. 10.1038/nature08964
Skoblov M, Marakhonov A, Marakasova E, Guskova A, Chandhoke V, Birerdinc A, Baranova A: Protein partners of KCTD proteins provide insights about their functional roles in cell differentiation and vertebrate development. Bioessays. 2013, 35 (7): 586-596. 10.1002/bies.201300002
Furukawa YJH M, Borchers C, Xiong Y: Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat Cell Biol. 2005, 5: 1001-1007.
Hochstrasser M: Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996, 30: 405-439. 10.1146/annurev.genet.30.1.405
Hershko A, Ciechanover A: The ubiquitin system. Annu Rev Biochem. 1998, 67: 425-479. 10.1146/annurev.biochem.67.1.425
De Smaele LDM E, Moretti M, Pelloni M, Occhione MA, Infante P, Cucchi D, Greco A, Pietrosanti L, Todorovic J, Coni S, Canettieri G, Ferretti E, Bei R, Maroder M, Screpanti I, Gulino A: Identification and characterization of KCASH2 and KCASH3, 2 novel Cullin3 adaptors suppressing histone deacetylase and Hedgehog activity in medulloblastoma. Neoplasia. 2010, 13: 374-385.
Azizieh R, Orduz D, Van Bogaert P, Bouschet T, Rodriguez W, Schiffmann SN, Pirson I, Abramowicz MJ: Progressive myoclonic epilepsy-associated gene KCTD7 is a regulator of potassium conductance in neurons. Mol Neurobiol. 2011, 44 (1): 111-121. 10.1007/s12035-011-8194-0
Staropoli JF, Karaa A, Lim ET, Kirby A, Elbalalesy N, Romansky SG, Leydiker KB, Coppel SH, Barone R, Xin W, et al: A homozygous mutation in KCTD7 links neuronal ceroid lipofuscinosis to the ubiquitin-proteasome system. Am J Hum Genet. 2012, 91 (1): 202-208. 10.1016/j.ajhg.2012.05.023
Mancarelli MM, Zazzeroni F, Ciccocioppo L, Capece D, Po A, Murgo S, Di Camillo R, Rinaldi C, Ferretti E, Gulino A, et al: The tumor suppressor gene KCTD11REN is regulated by Sp1 and methylation and its expression is reduced in tumors. Mol Cancer. 2010, 9: 172. 10.1186/1476-4598-9-172
Faryna MF, Konermann C, Aulmann S, Bermejo JL, Brugger M, Diederichs S, Rom J, Weichenhan D, Claus R, Rehli M, Schirmacher P, Sinn HP, Plass C, Gerhauser C: Genome-wide methylation screen in low-grade breast cancer identifies novel epigenetically altered genes as potential biomarkers for tumor diagnosis. FASEB J. 2012, 26: 4937-4950. 10.1096/fj.12-209502
Yoganathan P, Karunakaran S, Ho MM, Clee SM: Nutritional regulation of genome-wide association obesity genes in a tissue-dependent manner. Nutr Metab (Lond). 2012, 9 (1): 65. 10.1186/1743-7075-9-65
Paternoster L, Evans DM, Nohr EA, Holst C, Gaborieau V, Brennan P, Gjesing AP, Grarup N, Witte DR, Jorgensen T, et al: Genome-wide population-based association study of extremely overweight young adults–the GOYA study. PLoS One. 2011, 6 (9): e24303. 10.1371/journal.pone.0024303
Koehler GPD DR, Sweezey NB, Tanswell AK, Hu J: Lung inflammation as a therapeutic target in cystic fibrosis. Cell Mol Biol. 2004, 31: 377-381.
Van Bogaert P, Azizieh R, Desir J, Aeby A, De Meirleir L, Laes JF, Christiaens F, Abramowicz MJ: Mutation of a potassium channel-related gene in progressive myoclonic epilepsy. Ann Neurol. 2007, 61 (6): 579-586. 10.1002/ana.21121
Kousi M, Anttila V, Schulz A, Calafato S, Jakkula E, Riesch E, Myllykangas L, Kalimo H, Topcu M, Gokben S, et al: Novel mutations consolidate KCTD7 as a progressive myoclonus epilepsy gene. J Med Genet. 2012, 49 (6): 391-399. 10.1136/jmedgenet-2012-100859
Di Marcotullio L, Ferretti E, De Smaele E, Argenti B, Mincione C, Zazzeroni F, Gallo R, Masuelli L, Napolitano M, Maroder M, et al: REN(KCTD11) is a suppressor of Hedgehog signaling and is deleted in human medulloblastoma. Proc Natl Acad Sci U S A. 2004, 101 (29): 10833-10838. 10.1073/pnas.0400690101
Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF, et al: KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012, 485 (7398): 363-367. 10.1038/nature11091
Marneros AG, Beck AE, Turner EH, McMillin MJ, Edwards MJ, Field M, De Macena Sobreira NL, Perez AB, Fortes JA, Lampe AK, et al: Mutations in KCTD1 cause scalp-ear-nipple syndrome. Am J Hum Genet. 2013, 92 (4): 621-626. 10.1016/j.ajhg.2013.03.002
Junyent M, Parnell LD, Lai CQ, Lee YC, Smith CE, Arnett DK, Tsai MY, Kabagambe EK, Straka RJ, Province M, et al: Novel variants at KCTD10, MVK, and MMAB genes interact with dietary carbohydrates to modulate HDL-cholesterol concentrations in the genetics of lipid lowering drugs and diet network study. Am J Clin Nutr. 2009, 90 (3): 686-694. 10.3945/ajcn.2009.27738
Hasegawa T, Asanuma H, Ogino J, Hirohashi Y, Shinomura Y, Iwaki H, Kikuchi H, Kondo T: Use of potassium channel tetramerization domain-containing 12 as a biomarker for diagnosis and prognosis of gastrointestinal stromal tumor. Hum Pathol. 2013, 44 (7): 1271-1277. 10.1016/j.humpath.2012.10.013
Lwa TR, Tan CH, Lew QJ, Chu KL, Tan J, Lee YY, Chao SH: Identification of cellular genes critical to recombinant protein production using a Gaussia luciferase-based siRNA screening system. J Biotechnol. 2010, 146 (4): 160-168. 10.1016/j.jbiotec.2010.02.016
Chen T, Zhu L, Zhou Y, Pi B, Liu X, Deng G, Zhang R, Wang Y, Wu Z, Han M, et al: KCTD9 contributes to liver injury through NK cell activation during hepatitis B virus-induced acute-on-chronic liver failure. Clin Immunol. 2013, 146 (3): 207-216. 10.1016/j.clim.2012.12.013
Zhou YY, Zou Y, Chen T, Wang HW, Han MF, Pi B, Yan WM, Xi D, Huang JQ, Luo XP, et al: KCTD9, a novel potassium channel related gene, was highly expressed in hepatic NK cells and T cells of fulminant hepatitis mice induced by MHV-3. Zhonghua Gan Zang Bing Za Zhi. 2011, 19 (11): 833-837.
Hayasaki H, Sohma Y, Kanbara K, Otsuki Y: Heterogenous GABA(B) receptor-mediated pathways are involved in the local GABAergic system of the rat trigeminal ganglion: possible involvement of KCTD proteins. Neuroscience. 2012, 218: 344-358.
Zhou J, Ren K, Liu X, Xiong X, Hu X, Zhang J: A novel PDIP1-related protein, KCTD10, that interacts with proliferating cell nuclear antigen and DNA polymerase delta. Biochim Biophys Acta. 2005, 1729 (3): 200-203. 10.1016/j.bbaexp.2005.05.005
Huang T, Xu J, Xiang J, Lu Y, Chen R, Huang L, Xiao G, Sun G: PrPC interacts with potassium channel tetramerization domain containing 1 (KCTD1) protein through the PrP(51–136) region containing octapeptide repeats. Biochem Biophys Res Commun. 2012, 417 (1): 182-186. 10.1016/j.bbrc.2011.11.081
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 30870113 and 31370187).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ZP, YX, and GS co-wrote this review. All authors read and approved the final manuscript.
Zhepeng Liu, Yaqian Xiang contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Liu, Z., Xiang, Y. & Sun, G. The KCTD family of proteins: structure, function, disease relevance. Cell Biosci 3, 45 (2013). https://doi.org/10.1186/2045-3701-3-45
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2045-3701-3-45