
Abstract
Retinoic acid (RA) is a morphogen derived from retinol (vitamin A) that plays important roles in cell growth, differentiation, and organogenesis. The production of RA from retinol requires two consecutive enzymatic reactions catalyzed by different sets of dehydrogenases. The retinol is first oxidized into retinal, which is then oxidized into RA. The RA interacts with retinoic acid receptor (RAR) and retinoic acid X receptor (RXR) which then regulate the target gene expression. In this review, we have discussed the metabolism of RA and the important components of RA signaling pathway, and highlighted current understanding of the functions of RA during early embryonic development.
Similar content being viewed by others

Introduction
Retinoids refer to those chemicals that are structurally or functionally similar to retinol, or vitamin A [1], which is an essential biomolecule for embryonic development and adult body homeostasis. All retinoids retain the polyene hydrophobic tail attached to a cyclic 6-carbon ring. The polyene tail is characterized by the alternating conjugated carbon-carbon double bonds, which makes retinoids light-sensitive. In contrast with other signaling proteins, retinoids have a much lower molecular weight of approximately 300 Da. Given their molecular structures, retinoids are highly oil-soluble and able to diffuse across the cell membrane. Retinoids are involved in cellular growth, apoptosis, immune response, and epithelial growth [2–7] through the interaction with the nuclear receptors, retinoic acid receptor (RAR) and retinoid X receptor (RXR). During early embryonic development, the major active form of retinoids, all-trans retinoic acid (atRA), regulates germ layer formation, body axis formation, neurogenesis, cardiogenesis, and the development of pancreas, lung, and eye. It is also a critical element for visual function [8]. Because of the wide spectrum of RA functions, the metabolism, regulation, and function of vitamin A have been extensively studied for decades, and here we summarize our current understanding on retinoids metabolic pathways and RA functions during early embryonic development.
2. Metabolism of vitamin A and the production of all-trans retinoic acid
Vitamin A is a necessary dietary vitamin for the normal development and vision. The critical necessity of vitamin A was hinted as early as 1881 by Nikolai Lunin, who discovered that purified protein, fat, and carbohydrate did not sustain the normal growth of mice, unless the diet was supplemented with milk. Elmer Verner McCollum, then determined in 1917 that the critical component concerned in milk was actually a "fat-soluble factor A", named in contrast to the previously discovered "water-soluble factor B", or vitamin B. These discoveries allowed Carl Edvard Bloch, a Denmark paediatrician, to identify vitamin A deficiency as the cause of night blindness, or xerophthalmia [9].
While vitamin A was a necessary dietary vitamin, vitamin A itself is not the main bioactive mediator of its function. The key mediators of vitamin A function were identified as atRA and 11-cis retinal. atRA is a regulator of gene transcription, while 11-cis retinal acts as a chromophore for visual functions [10]. In this section, we will review the metabolic processes of converting vitamin A into various retinoids, with emphasis on the production of atRA (Figure 1).

Schematic diagram showing the metabolic pathways of vitamin A metabolism. This illustration lists the major steps of RA metabolism in cell, please refer to the text for details. Abbreviation: NAD, nicotinamide adenine dinucleotide; NADH, The reduced form of NAD; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, The reduced form of NADP; CRBP: cellular retinol-binding protein; LRAT: lecithin retinol acyltransferase; REH: retinyl ester hydrolase; ADH: alcohol dehydrogenase; RDH: retinol dehydrogenase; SDR: short-chain dehydrogenase/reductases; ALDH: aldehyde dehydrogenase; RALDH: retinaldehyde dehydrogenase. CRAD: cis- retinoid/androgen dehydrogenase. Modified from [18, 25].
2.1 Conversion of Vitamin A (atROL) to all-trans retinal (atRAL)
Vitamin A (hereby referred to as all-trans retinol, atROL) is absorbed in the small intestine and esterified as retinyl esters for the blood stream transport. Retinyl ester is first transported to the liver for storage, mainly in the hepatic stellate cell. Hydrolysis of retinyl esters results in retinol, which then binds to retinol binding protein (RBP). The atROL/RBP complex is the dominant form for systematical and intercellular transport. After target organs take up the atROL/RBP complex, the atROL either is re-esterified into retinyl ester by lecithin retinol acyltransferase (LRAT) or binds to cellular retinol-binding protein (CRBP). The CRBP can prevent intracellular atROL from non-specific oxidation, immobilize intracellular retinol for storage, and acts as a carrier protein to present the atROL to respective retinol dehydrogenase for oxidation, or to LRAT for esterification [11]. Thus, the esterification by LRAT and/or the binding to CRBP represent the most upstream regulation of atROL availability, and therefore atRA metabolism, in the cell [12].
Retinyl ester and CRBP-bound retinol are the main storage forms of atROL. The retinyl ester can be hydrolyzed into 11-cis retinol by isomerohydrolases [13, 14]. The cellular retinaldehyde binding protein (CRALBP) binds to 11-cis retinol with high affinity [15], which leads to the oxidation of 11-cis retinol to 11-cis retinal by 11-cis retinol dehydrogenase [16, 17]. The 11-cis retinal is an essential component for vision. It binds to opsin to form rhodopsin, which can absorb lights within the visible spectrum. When the 11-cis-retinal absorbs a photon, it isomerizes from the 11-cis state to atRAL. The binding of atRAL to opsin is not stable and atRAL is rapidly released from opsin. Such molecule movements cause cell membrane change, and eventually lead to generation of the nerve impulse for vision (Figure 1).
atROL/CRBP complexes are the first substrate in the metabolic pathway which leads to the production of atRA. Using atROL/CRBP as substrate, retinol dehydrogenase (RDH), which belongs to short-chain dehydrogenase/reductase (SDR) family, catalyzes the oxidation of atROL to all-trans retinal (atRAL) [18]. This step is the rate limiting step in the production of atRA [19]. It has also been demonstrated that in vitro free atROL can be converted into atRAL by non-specific enzymes including alcohol dehydrogenase [20]. CRBP therefore provides a selection mechanism for specific RDH-mediated oxidation of atROL. Indeed, RDH has a much higher affinity towards atROL/CRBP complex than free atROL and the reaction depends on protein-protein interaction between RDH and CRBP [21]. It has been demonstrated that the microsomal RDH interacts with atROL/CRBP-I in the presence of co-factor, nicotinamide adenine dinucleotide phosphate ( NADP) [22]. Moreover, atROL/CRBP is the preferred substrate for the retinol dehydrogenase 16 (RDH16. Abbreviated as RoDH-4 in the original article), but not for 3α-hydroxysteroid dehydrogenase (3α-HSD), a similar alcohol dehydrogenase which has an even higher affinity towards free atROL [23]. Thus, CRBP acts as a selection protein for RDH by increasing the substrate affinity for RDH, which in turn prevents non-specific oxidation of atROL by alcohol dehydrogenase.
The conversion of atROL to atRAL is, in fact, reversible. The direction - oxidation or reduction - favored by different members of SDR family depends on the substrate affinity, co-factor affinity, and the rate of reaction. In addition to CRBP that contributes to substrate affinity, another factor that contributes to substrate affinity is the intrinsic substrate binding site on the SDR family of enzymes. Retinal reductase 1 (RalR1), a human SDR, has been shown to be reactive towards both atROL and atRAL in vitro. However, RalR1 has a higher affinity and rate of reduction for atRAL than atROL, indicating that RalR1 is a retinal reductase, rather than retinol dehydrogenase, under physiological conditions [24]. Co-factor affinity of different SDR has also been demonstrated that the co-factor favored by SDR reflects the direction of reaction catalyzed by that particular SDR (reviewed by Pares) [25].
Apart from RalR1, a number of retinal reductases have been identified in the past decades. A human short-chained retinol reductase (retSDR1) identified in a neuroblastoma cell line has been shown to promote the formation of retinyl ester in the presence of exogenous atRAL [26]. A mouse liver peroxisomal SDR termed mouse retinal reductase (RRD) also showed a high atRAL-specific reductase activity in the presence of CRBP in vitro[27]. This enzyme was induced by peroxisome proliferator-activated receptor (PPAR), suggesting a relationship between retinoid metabolism and peroxisome activity [27]. Using in vitro assay, some studies have identified reductases which showed in vitro retinal-reducing activity. For example, human aldose reductase and human small intestine aldose reductase can function as retinal reductase in vitro[28]. Similarly, the mouse short-chained aldehyde reductase (SCALD) could reduce atRAL and 9-cis retinal in vitro[29]. However, these studies are based on in vitro biochemical analysis only and do not take into account the substrate selection by CRBP. Therefore, the enzymatic activities on different retinal reported should not be taken as a direct evidence for physiological retinal reductases.
2.2 Conversion of atRAL to atRA
Similar to atROL, all-trans retinal is also transported by CRBP in the cell, and is then oxidized to atRA. The oxidation from atRAL to atRA has been observed as early as 1960 [30]. The oxidation of atRAL to atRA was mediated by various retinaldehyde dehydrogenases (RALDH). At least 3 RALDHs have been identified in human, mouse, and Xenopus, with different physiological functions [31]. Retinaldehyde dehydrogenase 1 (Raldh1) is highly expressed in the dorsal retina of mouse embryos [32], in epithelial tissues of adult mice and Xenopus[33], and in the stomach and small intestine of adult rats [34]. Xenopus raldh1 has been shown to be an atRA synthesizing enzyme in a retinoic acid responsive cell line [35]. RALDH1-/- mice also suggest that Raldh1 is capable of atRA synthesis [36]. However, knockout of RALDH1 did not severely affect the morphology of the retina although RALDH1 is localized in the dorsal retina [36], indicating that other enzymes might redundantly share the function of RALDH1.
The retinaldehyde dehydrogenase 2 (RALDH2) was identified in human, mouse, chick, zebrafish and Xenopus[37–39]. Interestingly, RALDH2 was identified as a crucial enzyme for atRA synthesis in different organisms. Knockout of RALDH2 was embryonic lethal during the post-implantation period in mice [40], suggesting that atRA is essential for normal embryonic development. The phenotypes of RALDH2 knock-out mice include severely impaired segmentation of rhombomeres, altered homeobox gene expression pattern, and defective neural crest cell migration [41]. In zebrafish, knockdown of raldh2 caused a down-regulation of retinoic acid signaling, malformation in the central nervous system, and disruption of left-right asymmetry [42, 43]. The raldh2 mutant, neckless (nls), displayed a suppressed formation of the midbrain to hindbrain region, as well as segmentation defects in rhombomeres [44]. Such defects were attributed to the reduction in atRA signaling [45], since the spatial and temporal pattern of atRA signaling is maintained mainly by raldh2 and a degradative enzyme cytochrome P450 hydroxylase A1 (cyp26a1) in zebrafish. In Xenopus embryos, ectopic expression of raldh2 caused teratogenic effects such as the expression of posterior neural markers (en2 and krox20) in the anterior region, which is similar to that due to atRA toxicity [38], suggesting that raldh2 is an important enzyme in maintaining atRA homeostasis in embryos. Knockdown of raldh2 in Xenopus embryos caused a shortening of anteroposterior axis and a posterior shift of neural marker en2 and krox20[46]. Collectively, such evidence indicated that raldh2 plays a crucial role in the anteroposterior patterning of the central nervous system and trunk axis through regulation of the RA signaling.
Retinaldehyde dehydrogenase 3 (RALDH3) has been identified in human, chick, mouse, zebrafish, and Xenopus, and is expressed in the ventral retina across various species [47–51]. Studies in mouse have shown that RALDH3 was mainly involved in the frontonasal development and patterning of ocular structures [52]. Mice lacking RALDH3 were neonatal lethal, due to the respiratory tract obstruction in nasal region, and the neonatal lethality could be rescued by atRA supplements [53], suggesting that RALDH3 is an atRA synthesis enzyme. In 2007, Halilagic et al. showed that atRA production by RALDH3 contributed to the correct patterning of the anterior and dorsal boundaries of the developing forebrain [54]. It was further delineated that RALDH3 knockout mice exhibited loss of dopamine receptor D2 in the ventral forebrain. These studies suggest that RALDH3 is essential for the development of the central nervous system and the morphogenesis of anterior head structures [52].
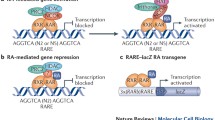
Similar to atROL and atRAL, the metabolism of atRA is also closely related to retinoid binding protein termed cellular retinoic acid binding proteins (CRABPs). CRABPs bind to intracellular RA and prevent it from non-specific degradation [55, 56]. There are two species of CRABP, CRABP-I and CRABP-II. These carrier proteins also ensure the solubility of hydrophobic retinoid in the aqueous intracellular environment. However, a recent study of CRBP-I/CRABP-I/CRABP-II triple knock-out mice has shown that the main regulator of retinoid homeostasis was likely to be CRBPs, with CRABPs playing a minor role in this process. Hoegberg et al. found that the chemical-induced depletion of total retinoids in triple knockout mice was more severe than the wild type and CRABP-I/CRABP-II double knockout mice [57], suggesting that CRBP-I is a more potent regulator of retinoid homeostasis. While CRABPs might not be critical in regulating total retinoids homeostasis, they participate in mediating RA signaling by transporting RA to the nucleus to interact with RARs. CRABP-II was shown to be translocated into nucleus upon the ligand binding [58], which allows atRA to bind to and activate RAR, a transcription factor responsible for the RA signaling (Figure 2). Interestingly, the RA signaling is tightly regulated by negative feedback mechanisms as CRABP-II is negatively regulated by atRA [59]. Elevated RA signaling suppresses the production of CRABPs, which down-regulate the activation of RARs and the RA signaling. CRABP-I, on the other hand, regulates the rate of RA metabolism by presenting RA to the degrading enzyme CYP26A1 [60].

Ilustration of RA and paracrine RA signaling. In serum, retinol is bound to retinol-binding protein 4 (RBP-4) synthesized in the liver. Although retinol is lipid soluble, it enters cells mainly through the interaction with its receptor STRA. In the cell, retinol can either be converted into retinyl esters for storage via lecithin retinol acyltransferase (LRAT) or bind to the cellular retinol binding protein (CRBP). The CRBP-bound retinol is oxidized to retinal by either alcohol dehydrogenase (ADH) or retinol dehydrogenase (RDH), and retinal is oxidized to retinoic acid (RA) by retinaldehyde dehydrogenases (RALDH1/2/3). All-trans retinoic acid (atRA) is the major bioactive component among the retinoids. CYP26 can further oxidize atRA to 4-oxo-RA for degradation. Cellular retinoic acid-binding protein (CRABP) facilitates transportation of atRA into the nucleus where atRA binds its receptors. The ternary complex of ligand-bound RAR and RXR binds to the retinoic acid response element (RARE) and activates the RA target genes. atRA can diffuse to adjacent cells to activate target gene expression in these cells. RAR can also bind to the liver X receptor (LXR), farnesoid X receptor (FXR), and peroxisome proliferator-activated receptor (PPAR) for multiple functions.
2.3 Degradation of atRA
All-trans retinoic acid is degraded by CYP26 enzymes, which belong to cytochrome P450 hydroxylase family. A number of CYP26 family including CYP26A1, B1, C1, and D1 have been characterized and all of them possess the ability to degrade atRA into less bio-active retinoid [61–63]. Rhombomeric alteration defects were only observed by the knockdown of all three cyp26 enzymes in zebrafish [64], suggesting that cyp26a1, b1 and c1 act redundantly in hindbrain patterning. CYP26A1 is induced by atRA while it promotes the hydroxylation of atRA into 4-hydroxy retinoic acid, 4-oxo retinoic acid, and 18-hyroxy retinoic acid [45, 65–67]. Since RALDH2 and CYP26A1 are both regulated by atRA itself, the metabolism of atRA therefore forms an auto-regulatory loop that regulates and balances atRA levels in embryos. Such regulation not only maintains the endogenous atRA level within a normal range, but also allows the organisms to respond to exogenous atRA fluctuation.
3. Retinoic acid receptors
atRA is carried into the nucleus by CRABP-II, and interacts with RARs, which themselves are transcription factors. RARs belong to retinoid receptor family, which also includes another group called retinoid X receptors (RXRs). RARs recognize both atRA and 9-cis retinoic acid, while RXRs only recognize 9-cis retinoic acid. Upon the ligand binding, RAR dimerizes with RXR to form a heterodimer, which then initiate gene transcription by binding to the retinoic acid response element (RARE) in the promoter region of the targets genes (Figure 2). The RAR family consists of RARα/β/γ three members that bind to atRA [68–71]. Single knockout mice that lack each of RARs were not embryonic lethal and did not display the complete spectrum of vitamin A deficiency phenotype. A disruption in RARα did not cause any observable phenotypic change in a mouse model [72]. Knockout of RARβ caused a reduction in the body weight and ocular defect, while limb formation remained normal [73]. Double knockout of two RARγ subtypes caused growth deficiency, cartilage dysmorphogenesis, and vertebrate malformation [74]. These results imply that RARs work redundantly and compensate the function of each other. Indeed, knockdown of RARα caused an increase in the expression level of RARβ and RARγ [75]. Only double knockout mutants showed phenotype close to the symptoms of vitamin A deficiency [76]. The auto-regulatory loop of RAR expression is similar to that regulating the expression of of RALDH2 and CYP26a1. Moreover, RARE has been identified in the promoter regions of RARα and β[77–79], indicating that the expression of these RARs is also under control of atRA.
Similarly, there are also three subtypes of RXRs [80]. RXRα knockout mice were embryonic lethal, potentially due to malformation of the heart in utero [81]. RXRβ knockout mice were 50% embryonic lethal, and the surviving littermates were morphologically normal except spermatogenesis defects which rendered the male sterile [82], while the RXRγ-null mutant mice were morphologically normal when compared with the wild type [83]. Moreover, the mice carrying only one copy of RXRα (RXRα+/-/RXRβ-/-/RXRγ-/-) were viable, suggesting one copy of RXRα is sufficient to carry out most of function of the RXRs [83]. Since atRA-bound RAR can form heterodimer with RXR in the absence of 9-cis retinoic acid and is still active in transcription activities, the importance of RXRs may not be as critical as RARs. This may explain why only one copy of RXRα is sufficient for the mouse embryonic development. Taken together, these results suggest that each of the RAR subtypes function redundantly and most of the RXR subtypes are not critical for the embryonic development.
While RARs mainly mediate the RA signaling, it has been revealed by many studies that ligand-bound RXRs activate other signaling pathways by forming heterodimer with other nuclear receptors such as liver X receptor (LXR), farnesoid X receptor (FXR), and PPAR [84–86] (Figure 2). LXR mainly functions as a sensor of cholesterol levels by recognizing its ligand oxysterols. Overloading cells with cholesterol activates LXR/RXR heterodimers which in turn initiate transcription of target genes, thereby regulate cholesterol transport, uptake, metabolism, and bile acid synthesis in the liver [87, 88]. FXR can recognize free or conjugated bile acid and thus acts as an intracellular sensor of bile acid to regulate the metabolism of bile acid in the liver. Activation of liganded-FXR/RXR promotes bile acid efflux and inhibits bile acid synthesis [89]. PPAR is a lipid sensing nuclear receptor, recognizing a wide range of fatty acids [90]. These interactions between LXR, FXR, PPAR, and RXR reflect the complexity of RXR functions and their potentials on the RA signaling. In addition, not only do RXRs take part promiscuously in multiple signaling pathways, the expression of these nuclear receptors is also under control of complex feedback loops and cross-talks with other signaling pathways [91]. The heterodimerization of RXR with RAR, LXR, FXR, or PPAR is therefore mutually competitive, and atRA signaling not only triggers the transcription of its target genes, but also competitively suppresses the transcription of others. This may explain the board spectrum of atRA-induced teratogenicity observed in embryos.
4. Differential expression and gene regulation of RA metabolic enzymes
The RA metabolic enzymes show distinct differential expression pattern during early embryonic development, and interestingly their expression is regulated by the RA signaling. Detailed descriptions of the expression patterns of these genes are beyond the scope of this review. Schematic drawing of expression of rdh10, dhrs3, raldh2, cyp26 a1, rarα2, and crabp-II at Xenopus gastrula (stage 11) and neurula (stage 14) stages are illustrated in Figure 3, which shows that the RA signaling itself regulates expression of the enzymes for RA biosynthesis and elicits the complexity of RA acting as a morphogen in early embryonic development. Ectopic cyp26 expression can be induced by atRA treatment [92], while the embryos treated with atRA showed down-regulation of raldh2[38] and rdh10[46]. Dhrs3 can also be induced by atRA treatment (RKT Kam, Y Chen, WY Chan and H Zhao. Dhrs3 attenuates the retinoic acid signaling and is required for early embryonic patterning. Submitted). Thus the RA signaling down-regulates the expression of the enzymes for atRA production, but up-regulates enzymes that can reduce atRA level in embryos. Other components in the RA signaling are also responsive to atRA treatment. For example, crabp-II was found to be an atRA-inducible gene [93], and was found to contain a RARE domain in its promoter region [94]. Similarly, rarα2 was found to be inducible by atRA treatment in leukemic cell lines [95], and in rat embryos as well [96]. We summarize the regulation of these genes by RA signaling in Figure 4.

Schematic diagram illustrating expression of the genes that are involved in RA biosynthesis and transportation at gastrula (stage 11) and neurula (stage 14) stages of Xenopus embryos. rdh10 is expressed in the circumblastoporal region of Xenopus gastrula, and the signals at the dorsal side form a zone extending anteriorly. At stage14, the signals are found in trunk paraxial mesoderm region [46]. rdh10 is not expressed in the notochord. raldh2 is expressed in a form of a ring around the vegetal pole with signals more intense in the dorsal blastpore lip. During neurula stages, raldh2 signals are mainly distributed in the trunk paraxial mesoderm, expanding ventrally [38]. rdh10 and raldh2 display overlapping expression pattern in the trunk paraxial mesoderm, with rdh10 expressed more anteriorly than raldh2. cyp26a is expressed in two primary domains at stage11, the posterior domain surrounding the blastopore and the anterior domain covering the anterior part of prospective neural plate. At stage 14, the anterior cyp26a transcripts is developed into three elements corresponding to the cement gland anlage, the mid-/hindbrain boundary, and the auditory placodes, while the posterior expression domain remains in the circumblastoporal area, and the developing neural plate is also covered by a gradient of cyp26a signals with the highest present in posterior region [92]. The expression domains of radlh2 and cyp26a do not overlap at gastrula and neurula stages of Xenopus embryos. rarα2 is expressed in the involuting surface layer surrounding the blastopore, and becomes stronger as the gastrulation proceeds. At stage 14, rarα2 expression is expressed predominantly in the posterior neural plate of the embryos [167]. The expression of the carbp-II is defined into an anterior and a posterior domain at gastrula stages. In the anterior domain, Xenopus crabp-II is limited to the dorsal area which generates prospective head structures. At the neurula stages, crabp-II is expressed in the prospective telencephalon and rhombencephalon, and the most posterior region of the embryos [168]. The dhrs3 signals form a circumblastoporal ring which is similar to rdh10 at stage 11. The signals in the neural plate form two signaling zones and gradually converge towards the midline, forming two signal strips extending posteriorly. In addition, dhrs3 is expressed in the notochord at neurula stages [169]. All these drawings are shown in dorsal view and the blue color represents expression signals.

The RA signaling regulates expression of the components involved in RA synthesis and transportation.
5. Retinoic acid signaling during early embryonic development
The RA signaling pathway has been implicated in various developmental processes. During early embryonic development, retinoids act as an important morphogen across different species from invertebrate to metazoan including human [97, 98]. It participates in regulating various biological processes, such as apoptosis and differentiation, and cell fate specification.
5.1 Axis formation
The RA signaling has been implicated in embryonic axis formation as well. Evidence shows that it interacts with Nodal signaling to regulate dorsoventral axis formation. The mice embryos lacking all three Cyp26 genes displayed secondary body axes due to expansion of the Nodal expression domain. In fact, the mouse Nodal gene contains an RARE in the intron 1 that is highly conserved among mammals [99]. Moreover, pre-gastrulation mouse embryos express Cyp26 s but not Raldh s suggesting that maternal RA is decreased by embryonic Cyp26s for proper Nodal expression during embryonic patterning [99].
In addition to the dorsoventral axis formation, cyp26 is also important for restricting the expression of posterior genes during the anteroposterior patterning [100]. Experiments in zebrafish embryos indicate that reduction of atRA by ectopic cyp26 decreased the expression of posterior genes meis3. Consistent with this, knockdown of cyp26 led to the anterior expansion of meis3. Interestingly, the expression of cyp26 is suppressed by the FGF and Wnt signalings, which are involved in the specification of posterior trunk during the gastrulation. Thus, RA together with FGF and Wnt signalings forms a complex regulatory network to regulate the anteroposterior axis formation in gastrula embryos, the cyp26 being one of the cross-talk genes linking these signaling pathways [100].
The RALDHs and CYP26s enzymes also participate in the anteroposterior patterning of the central nervous system by maintaining a gradient of atRA along the axis [101]. Global application of atRA to mouse embryos during day 7 of gestation caused severe head deformations, such as exencephaly, microcephaly, and anencephaly, whereas exposure of embryos to atRA during day 8 of gestation led to severe caudal truncations reminiscent of the human caudal regression syndrome [102, 103]. Similar phenomena were observed with Xenopus embryos as well [104–106].
Vertebrates display asymmetric placement of various internal organs including the heart, liver, spleen, and gut, and the asymmetric development of paired organs such as brain hemispheres and lungs. Treatment with RA antagonist in mouse led to randomization of heart looping and perturbed sideness of node [107]. Application of RA antagonist revealed that the RA signaling sequentially controlled visceral and heart laterality [108]. On the other hand, the somites obviously avoid the influence of signaling pathways that regulate the left-right asymmetry. The somites, derived from the trunk paraxial mesoderm and sequentially segmented along the anteroposterior axis, are formed as bilaterally paired units. However, the mice embryos that lack Raldh2 exhibited asymmetric somite formation [109], which was due to left-right desynchronization of the segmentation clock oscillations. Such defects were also observed in RA deficient chick [110] and zebrafish [43]. It is therefore postulated that the RA form a protection zone, protecting the somite from the left-right asymmetric signals to maintain the bilateral symmetry of somites columns.
5.2 Neural differentiation
atRA has been known for its ability to induce neural differentiation. During neural differentiation in early embryonic development, the pro-neural induction factor Neurogenin 2 (Neurog2) is required for primary sensory neuron specification [111]. Two RARE identified in the promoter region of mouse Neurog2 provide evidence that atRA directly regulates the expression of Neurog2 and therefore neural differentiation [112]. In addition, it has been shown that atRA treatment can induce pluripotent embryonic carcinoma stem cell line NT2 cells to differentiate into forebrain, hindbrain, and spinal cord neural progenitors [113], and the NT2 cells displayed GABAergic and glutamatergic phenotype. In line with this, mouse embryonic stem cells could be induced into GABAergic neurons by atRA treatment in vitro[114]. These in vitro findings were also supported by an in vivo study that knockdown of RARα abolished the effect of atRA on dendritic growth [115]. Collectively, these studies support the view that atRA is required for neural differentiation in the central nervous system.
5.3 Hindbrain patterning
Vertebrate hindbrain contains seven rhombomeres. Several Hox genes have been shown to be involved in rhombomeres formation and/or provide positional identity to specific rhombomeres [116]. Two RAREs have been identified in Hoxb1, one of which is located 5' to Hoxb1 promoter and is required for the restricted expression in rhombomere 4 [117]. Studies in mouse and fish embryos showed that retinoids could induce ectopic expression of Hoxa1 and Hoxb1, which caused a rhombomere 2 to 4 transformation [118–120]. In addition, the Hnf1b that suppressed Hoxb1 expression in rhombomere 5 was also regulated by the RA signaling [121, 122]. Altogether, these data implicate that the RA signaling is involved in hindbrain patterning. Indeed, knockout mice lacking Raldh2 led to the conversion of caudal hindbrain segments into rhombomere 4 identity [123, 124] and the same is true for RA antagonist treated mice [109]. In chick embryos, treatment by using increasing concentrations of RA antagonist, BMS453, showed that successively more posterior rhombomere boundaries required progressively higher concentration of endogenous retinoic acid for their correct positioning [125]. In zebrafish embryos, rhombomere 5 of the caudal hindbrain is well specified but both rhombomere 4 and 5 were posteriorly expanded in the absence of RA [42, 126]. In Xenopus embryos, injection of cyp26a1 mRNAs into one side of the embryo resulted in alterations of the RA signaling at the injected side as indicated by posteriorization of krox20, a transcription factor marking the rhombomere 3 and 5 [92], whereas increasing the RA signaling via ectopic expression of raldh2 led to anteriorization of both midbrain and hindbrain rhombomere identities in the expense of some forebrain territory [38] (Figure 5). Despite this, studies in mouse and Xenopus showed that decreased RA signaling did not change the mid-/hindbrain boundary (MHB)[41, 92], which requires the orchestration of a complex regulatory network involving the FGF, Wnt, and Shh signalings [127]. On the contrary, the hindbrain/spinal cord boundary seems to be specified depending on the RA signaling. In zebrafish cyp26a1 mutant embryos, the expression domains of hoxb5a and hoxb6a were expanded rostrally, leading to the expansion of rostral spinal cord at the expense of the hindbrain territory [128].

Schematic diagram of krox20 expression in control embryos (A), and in embryos with alterated RA signaling (B, C). Alternation of RA gradient induces shift of the expression domain of krox20. The expression of krox20 labels rhombomeres 3 and 5 (A, yellow). Co-injection of lacZ and cyp26 or rdh10 or rdh2 mRNA into one blastomere of two-cell stage Xenopus embryos is indicated by LacZ staining (B, C, blue). The uninjected side is served as control. Ectopic cyp26 at one side of the embryos causes posterior shift of krox20 (B, yellow). In contrast to cyp26, overexpression of rdh10 and radlh2 leads to anterior shift of krox20 (C, yellow). This diagram is based on the previous reports and our unpublished data [46, 92, 129] (RKT Kam, Y Chen, WY Chan and H Zhao. Dhrs3 attenuates the retinoic acid signaling and is required for early embryonic patterning. Submitted.).
5.4 Pancreas
The RA signaling also plays essential roles in the regionalization of endoderm as early as during gastrulation [129]. Studies in frog, avian, and mice indicate that the RA signaling is essential for dorsal pancreas formation, partially required for ventral pancreas patterning [129–134]. In zebrafish, inhibition of RA signaling before the end of gastrulation inhibits the initial development of both hepatic and pancreatic endoderm [135]. Evidence shows that it is the RA signal emanating from paraxial mesoderm that directly induces endocrine pancreatic precursors in the adjacent endoderm [136]. For example, Mnx1 is identified as an RA downstream gene that controls cell fate choice in the developing endocrine pancreas [137]. Cyp26a1, expressed in endoderm, defines the anterior boundary of dorsal pancreatic anlage by inactivating excess RA [138], while Cdx4, expressed in posterior endoderm, acts to antagonize the RA signaling and thus establishes the posterior boundary of dorsal pancreatic territory in zebrafish embryos (Figure 6; [139]). Moreover, Raldh1 is expressed in developing mouse and human pancreas at stages when mature β-cells are generated [140], suggesting the role of RA in β-cell differentiation. The XPDIp is expressed in the exocrine portion of both dorsal and ventral pancreas. Inhibition of RA signaling by BMS453 treatment completely abolished expression of XPDIp in dorsal pancreas, but had mild effects on the ventral pancreas, suggesting the development of dorsal pancreas is more RA signaling dependent [129].

RA plays an essential role in dorsal pancreas specification. During gastrulation and early neurulation, the field in dorsal endoderm that gives rise to the presumptive dorsal pancreas is just underlying the dorsal mesoderm where the highest concentration of RA is generated. These RA signals can act to underly endoderm either directly, or indirectly via first activating unknown factors (Xs) in mesoderm, which then act on the endoderm for dorsal pancreas formation. As described in the text, Shh and Cyp26a1 expressed in more anterior dorsal endoderm block pancreas formation probably via inhibiting the RA signaling activity or directly degrading RA. Cdx4 expressed in the posterior dorsal endoderm helps to establish the posterior boundary of dorsal pancreas via antagonizing the RA signaling.
In addition, in vitro data have also demonstrated a role of RA in pancreas development. In a defined in vitro explant system, RA treatment of cultured mouse E10.5 dorsal pancreatic buds could induce the generation of Ngn3+ endocrine progenitor cells and stimulate their further differentiation into β-cells by activating a differentiation program that recapitulates the normal temporal program of β-cell differentiation [140]. In zebrafish, exogenous RA led to expansion of both exo- and endocrine cells, while in Xenopus embryos as well as cultured mouse embryonic pancreatic rudiments, exogenous RA led to an expansion of the endocrine cell population at the expense of the exocrine cells in the dorsal pancreas [129, 141, 142]. Similarly, the induction of pancreatic fate by RA in endodermalized naive ectoderm dissected from early Xenopus gastrulae only showed the generation of early insulin-expressing cells [143, 144]. Therefore, cautions should be taken to apply RA at the right time and the right dose (close to physiological concentration in early embryos) for in vitro induction of mature pancreatic β-cells from embryonic stem cells.
5.5 Heart
The effect of RA on cardiogenesis was first demonstrated in quail that vitamin A deficiency could induce defective primitive heart development [145]. Further evidence shows that RA signaling is required for the formation of cardiac progenitors [146], and the correct modeling of the early heart field and the anteroposterior patterning of the heart is controlled by atRA through the action of Isl1 and Fgf8 signaling pathway [147, 148]. Knockout mice embryos lacking Raldh2 showed an abnormal development of the second heart field (SHF) [149], which is a population of undifferentiated cardiac precursor cells originating from the pharyngeal mesoderm and lying medial to the cardiac crescent (the first heart field). The expression of SHF markers including Isl1, Tbx1, and Fgf8, was posteriorly expanded in these embryos [150], indicating that the RA signaling is required for the restriction of SHF. Defects in the formation of a proper heart tube might also partially explain why Raldh2-/- is embryonic lethal [148]. Studies in zebrafish indicated RA functions in the restriction of cardiac specification [149]. During this process, if RA signaling was inhibited in lateral mesoderm, the uncommitted cells could differentiate into myocardial progenitor cells instead of pharyngeal or pancreatic cells [149], leading to the expansion of cardiomyocytes.
In addition to the heart development, the RA signaling has also been implicated in heart regeneration in adult zebrafish. Gene profiling assay indicated that raldh2 was one of the most highly induced genes in the regenerating heart [151]. After the ventricular injury, raldh2 was activated in the endocardium, but its expression was subsequently restricted in endocardial cells at the injury site one day post-trauma as the repairing cardiogenesis began. By 7 days after amputation, the epicardial cells at the injury site also expressed raldh2. Inhibition of RARs or expression of an RA-degrading enzyme blocked regenerative cardiomyocyte proliferation [152]. These findings reveal essential roles of RA in repairing the damaged heart tissue and promoting cardiomyocyte proliferation.
5.6 Kidney
There is increasing evidence indicating that atRA plays important roles in kidney development [153–155]. The kidney field is derived from the intermediate mesoderm. The specification of renal progenitor cells is influenced by RA signals emanating from the paraxial mesoderm [153–155]. Recent studies in Xenopus and zebrafish have demonstrated that ectopic production of RA increased the size of the kidney field, while blocking the pathway inhibited the kidney specification [86, 154]. In Xenopus, pax8 and lhx1, which are the earliest determinants of pronephric fate are both under control of the RA signaling [154, 156], while pteg, an early pronephric marker, is a direct target of the RA signaling and an essential factor for pronephric specification [157]. Another critical factor for the pronephros development, Wilm's tumor suppressor (wt1), is under direct control of the RA signaling as well [158, 159]. Inhibition of RA by BMS453 treatment caused reduction of pronephros field, as revealed by the decreased expression of the pronephros markers including smp-30 and pax2 at the late tailbud stage [154]. Furthermore, in Xenopus embryos, the pluripotent animal cap cells treated with RA and activin could differentiate into pronephros tissue [154, 160]. Likewise, the RA signaling is also essential for directing the proximodistal patterning of pronephric nephron in zebrafish [161]. The exogenous RA could induce proximal segment fates at the expense of distal fates, whereas inhibition of the RA signaling caused a loss of the proximal segments and an expansion of the distal segments.
Interestingly, inactivation of both RARα and RARβ in mice embryos resulted in renal malformations [155]. Further evidence showed that RARα and RARβ were co-expressed with Ret, a receptor tyrosine kinase involved in renal development, in renal stromal mesenchyme, where their deletion led to altered stromal cell patterning, impaired ureteric bud growth, and down-regulation of Ret in the ureteric bud. Moreover, studies in mice indicate that the RA signaling in ureteric bud cells mainly depends on atRA generated through Raldh2 in stromal cells [162].
5.7 Lung
Evidence indicates that disruption of the RA signaling during early development caused abnormalities of lung formation. Either knockout of Raldh2 or treatment with BMS 493, a RA inhibitor, caused defects of lung formation in mice [163]. During primary lung bud morphogenesis, although RA is not required for specification of lung cell fate in the endoderm, it is essential for the induction of the primordial lung buds [163, 164]. During the induction of lung bud, the RA signaling inhibits the expression of Dkk-1, the Wnt signaling inhibitor, in the foregut mesoderm. This inhibition in turn activates Wnt signaling in the lung bud region. At the same time, the RA signaling also inhibits TGF-β signaling. The balanced Wnt and TGF-β signalings coordinate with each other to induce and maintain the optimal expression level of Fgf10 for the induction of the lung primordium [165]. Furthermore, airway branching was accompanied by down-regulation of the RA signaling pathway [166], thus regional mechanisms that control RA availability and utilization are important for the lung morphogenesis.
Concluding remarks
To date, it has been demonstrated that the RA signaling plays sophisticated roles in early embryonic development. In fact, gastrulation is a very critical period for the RA signaling to exert its function in the regionalization of all three germ layers along the anteroposterior axis. RA is mainly generated in the mesoderm and thus can be most efficiently used. It is not clear as to how the RA metabolic enzymes coordinate with each other to generate the RA gradient at right time, right region, and with right strength during early embryonic development. Future studies are needed to identify the immediate and direct RA target genes in distinct RA responsive cells and to define the crosstalks between RA and other signaling pathways, such as hedgehog, FGF, Wnt, and Notch signalings for the formation and regionalization of three germ layers. Such knowledge will be essential for understanding organogenesis and establishing reliable strategies for stem cell differentiation into specific cell types that can be used for treatment of human diseases.
References
Tang XH, Gudas LJ: Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol. 2011, 6: 345-364. 10.1146/annurev-pathol-011110-130303
Duriancik DM, Lackey DE, Hoag KA: Vitamin A as a regulator of antigen presenting cells. J Nutr. 140 (8): 1395-1399.
Gudas LJ, Wagner JA: Retinoids regulate stem cell differentiation. J Cell Physiol. 226 (2): 322-330.
Hogarth CA, Griswold MD: The key role of vitamin A in spermatogenesis. J Clin Invest. 120 (4): 956-962.
Mora JR, Iwata M, von Andrian UH: Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol. 2008, 8 (9): 685-698. 10.1038/nri2378
Mukherjee S, Date A, Patravale V, Korting HC, Roeder A, Weindl G: Retinoids in the treatment of skin aging: an overview of clinical efficacy and safety. Clin Interv Aging. 2006, 1 (4): 327-348. 10.2147/ciia.2006.1.4.327
Pino-Lagos K, Benson MJ, Noelle RJ: Retinoic acid in the immune system. Ann N Y Acad Sci. 2008, 1143: 170-187. 10.1196/annals.1443.017
Davies WL, Hankins MW, Foster RG: Vertebrate ancient opsin and melanopsin: divergent irradiance detectors. Photochem Photobiol Sci. 2010, 9 (11): 1444-1457. 10.1039/c0pp00203h
Wolf G: The discovery of the visual function of vitamin A. J Nutr. 2001, 131 (6): 1647-1650.
D'Ambrosio DN, Clugston RD, Blaner WS: Vitamin A Metabolism: An Update. 2011, 3 (1): 63-103.
Penzes P, Napoli JL: Holo-cellular retinol-binding protein: distinction of ligand-binding affinity from efficiency as substrate in retinal biosynthesis. Biochemistry. 1999, 38 (7): 2088-2093. 10.1021/bi982228t
Liu L, Gudas LJ: Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. J Biol Chem. 2005, 280 (48): 40226-40234. 10.1074/jbc.M509643200
Deigner PS, Law WC, Canada FJ, Rando RR: Membranes as the energy source in the endergonic transformation of vitamin A to 11-cis-retinol. Science. 1989, 244 (4907): 968-971. 10.1126/science.2727688
Mata NL, Moghrabi WN, Lee JS, Bui TV, Radu RA, Horwitz J, Travis GH: Rpe65 is a retinyl ester binding protein that presents insoluble substrate to the isomerase in retinal pigment epithelial cells. J Biol Chem. 2004, 279 (1): 635-643.
Saari JC, Nawrot M, Kennedy BN, Garwin GG, Hurley JB, Huang J, Possin DE, Crabb JW: Visual cycle impairment in cellular retinaldehyde binding protein (CRALBP) knockout mice results in delayed dark adaptation. Neuron. 2001, 29 (3): 739-748. 10.1016/S0896-6273(01)00248-3
Driessen CA, Janssen BP, Winkens HJ, van Vugt AH, de Leeuw TL, Janssen JJ: Cloning and expression of a cDNA encoding bovine retinal pigment epithelial 11-cis retinol dehydrogenase. Invest Ophthalmol Vis Sci. 1995, 36 (10): 1988-1996.
Simon A, Hellman U, Wernstedt C, Eriksson U: The retinal pigment epithelial-specific 11-cis retinol dehydrogenase belongs to the family of short chain alcohol dehydrogenases. J Biol Chem. 1995, 270 (3): 1107-1112. 10.1074/jbc.270.3.1107
Duester G: Families of retinoid dehydrogenases regulating vitamin A function: production of visual pigment and retinoic acid. Eur J Biochem. 2000, 267 (14): 4315-4324. 10.1046/j.1432-1327.2000.01497.x
Agadir A, Nau H, Blaner WS: Retinoids: the biochemical and molecular basis of Vitamin A and retinoid action. 1999, Berlin; New York Springer.
Posch KC, Enright WJ, Napoli JL: Retinoic acid synthesis by cytosol from the alcohol dehydrogenase negative deermouse. Arch Biochem Biophys. 1989, 274 (1): 171-178. 10.1016/0003-9861(89)90428-1
Noy N: Retinoid-binding proteins: mediators of retinoid action. Biochem J. 2000, 348 (Pt 3): 481-495.
Boerman MH, Napoli JL: Characterization of a microsomal retinol dehydrogenase: a short-chain alcohol dehydrogenase with integral and peripheral membrane forms that interacts with holo-CRBP (type I). Biochemistry. 1995, 34 (21): 7027-7037. 10.1021/bi00021a014
Lapshina EA, Belyaeva OV, Chumakova OV, Kedishvili NY: Differential recognition of the free versus bound retinol by human microsomal retinol/sterol dehydrogenases: characterization of the holo-CRBP dehydrogenase activity of RoDH-4. Biochemistry. 2003, 42 (3): 776-784. 10.1021/bi026836r
Belyaeva OV, Stetsenko AV, Nelson P, Kedishvili NY: Properties of short-chain dehydrogenase/reductase RalR1: characterization of purified enzyme, its orientation in the microsomal membrane, and distribution in human tissues and cell lines. Biochemistry. 2003, 42 (50): 14838-14845. 10.1021/bi035288u
Pares X, Farres J, Kedishvili N, Duester G: Medium- and short-chain dehydrogenase/reductase gene and protein families: Medium-chain and short-chain dehydrogenases/reductases in retinoid metabolism. Cell Mol Life Sci. 2008, 65 (24): 3936-3949. 10.1007/s00018-008-8591-3
Cerignoli F, Guo X, Cardinali B, Rinaldi C, Casaletto J, Frati L, Screpanti I, Gudas LJ, Gulino A, Thiele CJ: retSDR1, a short-chain retinol dehydrogenase/reductase, is retinoic acid-inducible and frequently deleted in human neuroblastoma cell lines. Cancer Res. 2002, 62 (4): 1196-1204.
Lei Z, Chen W, Zhang M, Napoli JL: Reduction of all-trans-retinal in the mouse liver peroxisome fraction by the short-chain dehydrogenase/reductase RRD: induction by the PPAR alpha ligand clofibrate. Biochemistry. 2003, 42 (14): 4190-4196. 10.1021/bi026948i
Crosas B, Hyndman DJ, Gallego O, Martras S, Pares X, Flynn TG, Farres J: Human aldose reductase and human small intestine aldose reductase are efficient retinal reductases: consequences for retinoid metabolism. Biochem J. 2003, 373 (Pt 3): 973-979.
Kasus-Jacobi A, Ou J, Bashmakov YK, Shelton JM, Richardson JA, Goldstein JL, Brown MS: Characterization of mouse short-chain aldehyde reductase (SCALD), an enzyme regulated by sterol regulatory element-binding proteins. J Biol Chem. 2003, 278 (34): 32380-32389. 10.1074/jbc.M304969200
Dowling JE, Wald G: The role of vitamin A acid. Vitam Horm. 1960, 18: 515-541.
Duester G, Mic FA, Molotkov A: Cytosolic retinoid dehydrogenases govern ubiquitous metabolism of retinol to retinaldehyde followed by tissue-specific metabolism to retinoic acid. Chem Biol Interact. 2003, 143-144: 201-210.
Haselbeck RJ, Hoffmann I, Duester G: Distinct functions for Aldh1 and Raldh2 in the control of ligand production for embryonic retinoid signaling pathways. Dev Genet. 1999, 25 (4): 353-364. 10.1002/(SICI)1520-6408(1999)25:4<353::AID-DVG9>3.0.CO;2-G
Ang HL, Duester G: Retinoic acid biosynthetic enzyme ALDH1 localizes in a subset of retinoid-dependent tissues during xenopus development. Dev Dyn. 1999, 215 (3): 264-272. 10.1002/(SICI)1097-0177(199907)215:3<264::AID-AJA8>3.0.CO;2-I
Frota-Ruchon A, Marcinkiewicz M, Bhat PV: Localization of retinal dehydrogenase type 1 in the stomach and intestine. Cell Tissue Res. 2000, 302 (3): 397-400. 10.1007/s004410000281
Ang HL, Duester G: Stimulation of premature retinoic acid synthesis in Xenopus embryos following premature expression of aldehyde dehydrogenase ALDH1. Eur J Biochem. 1999, 260 (1): 227-234. 10.1046/j.1432-1327.1999.00139.x
Fan X, Molotkov A, Manabe S, Donmoyer CM, Deltour L, Foglio MH, Cuenca AE, Blaner WS, Lipton SA, Duester G: Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol Cell Biol. 2003, 23 (13): 4637-4648. 10.1128/MCB.23.13.4637-4648.2003
Blentic A, Gale E, Maden M: Retinoic acid signalling centres in the avian embryo identified by sites of expression of synthesising and catabolising enzymes. Dev Dyn. 2003, 227 (1): 114-127. 10.1002/dvdy.10292
Chen Y, Pollet N, Niehrs C, Pieler T: Increased XRALDH2 activity has a posteriorizing effect on the central nervous system of Xenopus embryos. Mech Dev. 2001, 101 (1-2): 91-103. 10.1016/S0925-4773(00)00558-X
Niederreither K, McCaffery P, Drager UC, Chambon P, Dolle P: Restricted expression and retinoic acid-induced downregulation of the retinaldehyde dehydrogenase type 2 (RALDH-2) gene during mouse development. Mech Dev. 1997, 62 (1): 67-78. 10.1016/S0925-4773(96)00653-3
Niederreither K, Subbarayan V, Dolle P, Chambon P: Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet. 1999, 21 (4): 444-448. 10.1038/7788
Niederreither K, Vermot J, Schuhbaur B, Chambon P, Dolle P: Retinoic acid synthesis and hindbrain patterning in the mouse embryo. Development. 2000, 127 (1): 75-85.
Grandel H, Lun K, Rauch GJ, Rhinn M, Piotrowski T, Houart C, Sordino P, Kuchler AM, Schulte-Merker S, Geisler R: Retinoic acid signalling in the zebrafish embryo is necessary during pre-segmentation stages to pattern the anterior-posterior axis of the CNS and to induce a pectoral fin bud. Development. 2002, 129 (12): 2851-2865.
Kawakami Y, Raya A, Raya RM, Rodriguez-Esteban C, Belmonte JC: Retinoic acid signalling links left-right asymmetric patterning and bilaterally symmetric somitogenesis in the zebrafish embryo. Nature. 2005, 435 (7039): 165-171. 10.1038/nature03512
Begemann G, Marx M, Mebus K, Meyer A, Bastmeyer M: Beyond the neckless phenotype: influence of reduced retinoic acid signaling on motor neuron development in the zebrafish hindbrain. Dev Biol. 2004, 271 (1): 119-129. 10.1016/j.ydbio.2004.03.033
Dobbs-McAuliffe B, Zhao Q, Linney E: Feedback mechanisms regulate retinoic acid production and degradation in the zebrafish embryo. Mech Dev. 2004, 121 (4): 339-350. 10.1016/j.mod.2004.02.008
Strate I, Min TH, Iliev D, Pera EM: Retinol dehydrogenase 10 is a feedback regulator of retinoic acid signalling during axis formation and patterning of the central nervous system. Development. 2009, 136 (3): 461-472. 10.1242/dev.024901
Koenig SF, Brentle S, Hamdi K, Fichtner D, Wedlich D, Gradl D: En2, Pax2/5 and Tcf-4 transcription factors cooperate in patterning the Xenopus brain. Dev Biol. 340 (2): 318-328.
Lupo G, Liu Y, Qiu R, Chandraratna RA, Barsacchi G, He RQ, Harris WA: Dorsoventral patterning of the Xenopus eye: a collaboration of Retinoid, Hedgehog and FGF receptor signaling. Development. 2005, 132 (7): 1737-1748. 10.1242/dev.01726
Mic FA, Molotkov A, Fan X, Cuenca AE, Duester G: RALDH3, a retinaldehyde dehydrogenase that generates retinoic acid, is expressed in the ventral retina, otic vesicle and olfactory pit during mouse development. Mech Dev. 2000, 97 (1-2): 227-230. 10.1016/S0925-4773(00)00434-2
Pittlik S, Domingues S, Meyer A, Begemann G: Expression of zebrafish aldh1a3 (raldh3) and absence of aldh1a1 in teleosts. Gene Expr Patterns. 2008, 8 (3): 141-147. 10.1016/j.gep.2007.11.003
Suzuki R, Shintani T, Sakuta H, Kato A, Ohkawara T, Osumi N, Noda M: Identification of RALDH-3, a novel retinaldehyde dehydrogenase, expressed in the ventral region of the retina. Mech Dev. 2000, 98 (1-2): 37-50. 10.1016/S0925-4773(00)00450-0
Molotkova N, Molotkov A, Duester G: Role of retinoic acid during forebrain development begins late when Raldh3 generates retinoic acid in the ventral subventricular zone. Dev Biol. 2007, 303 (2): 601-610. 10.1016/j.ydbio.2006.11.035
Dupe V, Matt N, Garnier JM, Chambon P, Mark M, Ghyselinck NB: A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc Natl Acad Sci USA. 2003, 100 (24): 14036-14041. 10.1073/pnas.2336223100
Halilagic A, Ribes V, Ghyselinck NB, Zile MH, Dolle P, Studer M: Retinoids control anterior and dorsal properties in the developing forebrain. Dev Biol. 2007, 303 (1): 362-375. 10.1016/j.ydbio.2006.11.021
Chytil F, Ong DE: Cellular retinol- and retinoic acid-binding proteins in vitamin A action. Fed Proc. 1979, 38 (11): 2510-2514.
Napoli JL, Boerman MH, Chai X, Zhai Y, Fiorella PD: Enzymes and binding proteins affecting retinoic acid concentrations. J Steroid Biochem Mol Biol. 1995, 53 (1-6): 497-502. 10.1016/0960-0760(95)00096-I
Hoegberg P, Schmidt CK, Fletcher N, Nilsson CB, Trossvik C, Gerlienke Schuur A, Brouwer A, Nau H, Ghyselinck NB, Chambon P: Retinoid status and responsiveness to 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking retinoid binding protein or retinoid receptor forms. Chem Biol Interact. 2005, 156 (1): 25-39. 10.1016/j.cbi.2005.06.006
Sessler RJ, Noy N: A ligand-activated nuclear localization signal in cellular retinoic acid binding protein-II. Mol Cell. 2005, 18 (3): 343-353. 10.1016/j.molcel.2005.03.026
Stachurska E, Loboda A, Niderla-Bielinska J, Szperl M, Juszynski M, Jozkowicz A, Dulak J, Ratajska A: Expression of cellular retinoic acid-binding protein I and II (CRABP I and II) in embryonic mouse hearts treated with retinoic acid. Acta Biochim Pol. 58 (1): 19-29.
Boylan JF, Gudas LJ: The level of CRABP-I expression influences the amounts and types of all-trans-retinoic acid metabolites in F9 teratocarcinoma stem cells. J Biol Chem. 1992, 267 (30): 21486-21491.
Gu X, Xu F, Wang X, Gao X, Zhao Q: Molecular cloning and expression of a novel CYP26 gene (cyp26d1) during zebrafish early development. Gene Expr Patterns. 2005, 5 (6): 733-739. 10.1016/j.modgep.2005.04.005
Sakai Y, Luo T, McCaffery P, Hamada H, Drager UC: CYP26A1 and CYP26C1 cooperate in degrading retinoic acid within the equatorial retina during later eye development. Dev Biol. 2004, 276 (1): 143-157. 10.1016/j.ydbio.2004.08.032
Takeuchi H, Yokota A, Ohoka Y, Iwata M: Cyp26b1 regulates retinoic acid-dependent signals in T cells and its expression is inhibited by transforming growth factor-beta. PLoS One. 6 (1): e16089.
Hernandez RE, Putzke AP, Myers JP, Margaretha L, Moens CB: Cyp26 enzymes generate the retinoic acid response pattern necessary for hindbrain development. Development. 2007, 134 (1): 177-187. 10.1242/dev.02706
White JA, Beckett-Jones B, Guo YD, Dilworth FJ, Bonasoro J, Jones G, Petkovich M: cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450. J Biol Chem. 1997, 272 (30): 18538-18541. 10.1074/jbc.272.30.18538
de Roos K, Sonneveld E, Compaan B, ten Berge D, Durston AJ, van der Saag PT: Expression of retinoic acid 4-hydroxylase (CYP26) during mouse and Xenopus laevis embryogenesis. Mech Dev. 1999, 82 (1-2): 205-211. 10.1016/S0925-4773(99)00016-7
Swindell EC, Thaller C, Sockanathan S, Petkovich M, Jessell TM, Eichele G: Complementary domains of retinoic acid production and degradation in the early chick embryo. Dev Biol. 1999, 216 (1): 282-296. 10.1006/dbio.1999.9487
Giguere V, Ong ES, Segui P, Evans RM: Identification of a receptor for the morphogen retinoic acid. Nature. 1987, 330 (6149): 624-629. 10.1038/330624a0
Petkovich M, Brand NJ, Krust A, Chambon P: A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature. 1987, 330 (6147): 444-450. 10.1038/330444a0
Brand N, Petkovich M, Krust A, Chambon P, de The H, Marchio A, Tiollais P, Dejean A: Identification of a second human retinoic acid receptor. Nature. 1988, 332 (6167): 850-853. 10.1038/332850a0
Ruberte E, Dolle P, Krust A, Zelent A, Morriss-Kay G, Chambon P: Specific spatial and temporal distribution of retinoic acid receptor gamma transcripts during mouse embryogenesis. Development. 1990, 108 (2): 213-222.
Li E, Sucov HM, Lee KF, Evans RM, Jaenisch R: Normal development and growth of mice carrying a targeted disruption of the alpha 1 retinoic acid receptor gene. Proc Natl Acad Sci USA. 1993, 90 (4): 1590-1594. 10.1073/pnas.90.4.1590
Ghyselinck NB, Dupe V, Dierich A, Messaddeq N, Garnier JM, Rochette-Egly C, Chambon P, Mark M: Role of the retinoic acid receptor beta (RARbeta) during mouse development. Int J Dev Biol. 1997, 41 (3): 425-447.
Subbarayan V, Kastner P, Mark M, Dierich A, Gorry P, Chambon P: Limited specificity and large overlap of the functions of the mouse RAR gamma 1 and RAR gamma 2 isoforms. Mech Dev. 1997, 66 (1-2): 131-142. 10.1016/S0925-4773(97)00098-1
Manshouri T, Yang Y, Lin H, Stass SA, Glassman AB, Keating MJ, Albitar M: Downregulation of RAR alpha in mice by antisense transgene leads to a compensatory increase in RAR beta and RAR gamma and development of lymphoma. Blood. 1997, 89 (7): 2507-2515.
Mark M, Ghyselinck NB, Wendling O, Dupe V, Mascrez B, Kastner P, Chambon P: A genetic dissection of the retinoid signalling pathway in the mouse. Proc Nutr Soc. 1999, 58 (3): 609-613. 10.1017/S0029665199000798
Chomienne C, Balitrand N, Ballerini P, Castaigne S, de The H, Degos L: All-trans retinoic acid modulates the retinoic acid receptor-alpha in promyelocytic cells. J Clin Invest. 1991, 88 (6): 2150-2154. 10.1172/JCI115547
Kamei Y, Kawada T, Kazuki R, Sugimoto E: Retinoic acid receptor gamma 2 gene expression is up-regulated by retinoic acid in 3T3-L1 preadipocytes. Biochem J. 1993, 293 (Pt 3): 807-812.
Sucov HM, Murakami KK, Evans RM: Characterization of an autoregulated response element in the mouse retinoic acid receptor type beta gene. Proc Natl Acad Sci USA. 1990, 87 (14): 5392-5396. 10.1073/pnas.87.14.5392
Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ: Nuclear receptors and lipid physiology: opening the X-files. Science. 2001, 294 (5548): 1866-1870. 10.1126/science.294.5548.1866
Kastner P, Messaddeq N, Mark M, Wendling O, Grondona JM, Ward S, Ghyselinck N, Chambon P: Vitamin A deficiency and mutations of RXRalpha, RXRbeta and RARalpha lead to early differentiation of embryonic ventricular cardiomyocytes. Development. 1997, 124 (23): 4749-4758.
Kastner P, Mark M, Leid M, Gansmuller A, Chin W, Grondona JM, Decimo D, Krezel W, Dierich A, Chambon P: Abnormal spermatogenesis in RXR beta mutant mice. Genes Dev. 1996, 10 (1): 80-92. 10.1101/gad.10.1.80
Krezel W, Dupe V, Mark M, Dierich A, Kastner P, Chambon P: RXR gamma null mice are apparently normal and compound RXR alpha +/-/RXR beta -/-/RXR gamma -/- mutant mice are viable. Proc Natl Acad Sci USA. 1996, 93 (17): 9010-9014.
Mello T, Polvani S, Galli A: Peroxisome proliferator-activated receptor and retinoic x receptor in alcoholic liver disease. PPAR Res. 2009, 2009: 748174.
Rizzo G, Renga B, Antonelli E, Passeri D, Pellicciari R, Fiorucci S: The methyl transferase PRMT1 functions as co-activator of farnesoid X receptor (FXR)/9-cis retinoid X receptor and regulates transcription of FXR responsive genes. Mol Pharmacol. 2005, 68 (2): 551-558. 10.1124/mol.105.012104
Wingert RA, Davidson AJ: The zebrafish pronephros: a model to study nephron segmentation. Kidney Int. 2008, 73 (10): 1120-1127. 10.1038/ki.2008.37
Lagu B, Pio B, Lebedev R, Yang M, Pelton PD: RXR-LXR heterodimer modulators for the potential treatment of dyslipidemia. Bioorg Med Chem Lett. 2007, 17 (12): 3497-3503. 10.1016/j.bmcl.2007.01.047
Zhao C, Dahlman-Wright K: Liver X receptor in cholesterol metabolism. J Endocrinol. 204 (3): 233-240.
Desvergne B: RXR: from partnership to leadership in metabolic regulations. Vitam Horm. 2007, 75: 1-32.
Desvergne B, Wahli W: Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999, 20 (5): 649-688. 10.1210/er.20.5.649
Desvergne B, Michalik L, Wahli W: Transcriptional regulation of metabolism. Physiol Rev. 2006, 86 (2): 465-514. 10.1152/physrev.00025.2005
Hollemann T, Chen Y, Grunz H, Pieler T: Regionalized metabolic activity establishes boundaries of retinoic acid signalling. EMBO J. 1998, 17 (24): 7361-7372. 10.1093/emboj/17.24.7361
Astrom A, Pettersson U, Voorhees JJ: Structure of the human cellular retinoic acid-binding protein II gene. Early transcriptional regulation by retinoic acid. J Biol Chem. 1992, 267 (35): 25251-25255.
Durand B, Saunders M, Leroy P, Leid M, Chambon P: All-trans and 9-cis retinoic acid induction of CRABPII transcription is mediated by RAR-RXR heterodimers bound to DR1 and DR2 repeated motifs. Cell. 1992, 71 (1): 73-85. 10.1016/0092-8674(92)90267-G
Zhu J, Heyworth CM, Glasow A, Huang QH, Petrie K, Lanotte M, Benoit G, Gallagher R, Waxman S, Enver T: Lineage restriction of the RARalpha gene expression in myeloid differentiation. Blood. 2001, 98 (8): 2563-2567. 10.1182/blood.V98.8.2563
Takeyama K, Kojima R, Ohashi R, Sato T, Mano H, Masushige S, Kato S: Retinoic acid differentially up-regulates the gene expression of retinoic acid receptor alpha and gamma isoforms in embryo and adult rats. Biochem Biophys Res Commun. 1996, 222 (2): 395-400. 10.1006/bbrc.1996.0755
Halme A, Cheng M, Hariharan IK: Retinoids regulate a developmental checkpoint for tissue regeneration in Drosophila. Curr Biol. 20 (5): 458-463.
Maden M: Retinoids in nonmammalian embryos. Methods Mol Biol. 2008, 461: 541-559. 10.1007/978-1-60327-483-8_37
Uehara M, Yashiro K, Takaoka K, Yamamoto M, Hamada H: Removal of maternal retinoic acid by embryonic CYP26 is required for correct Nodal expression during early embryonic patterning. Genes Dev. 2009, 23 (14): 1689-1698. 10.1101/gad.1776209
Kudoh T, Wilson SW, Dawid IB: Distinct roles for Fgf, Wnt and retinoic acid in posteriorizing the neural ectoderm. Development. 2002, 129 (18): 4335-4346.
White RJ, Schilling TF: How degrading: Cyp26s in hindbrain development. Dev Dyn. 2008, 237 (10): 2775-2790. 10.1002/dvdy.21695
Kessel M, Gruss P: Homeotic transformations of murine vertebrae and concomitant alteration of Hox codes induced by retinoic acid. Cell. 1991, 67 (1): 89-104. 10.1016/0092-8674(91)90574-I
Padmanabhan R: Retinoic acid-induced caudal regression syndrome in the mouse fetus. Reprod Toxicol. 1998, 12 (2): 139-151. 10.1016/S0890-6238(97)00153-6
Durston AJ, Timmermans JP, Hage WJ, Hendriks HF, de Vries NJ, Heideveld M, Nieuwkoop PD: Retinoic acid causes an anteroposterior transformation in the developing central nervous system. Nature. 1989, 340 (6229): 140-144. 10.1038/340140a0
Sive HL, Draper BW, Harland RM, Weintraub H: Identification of a retinoic acid-sensitive period during primary axis formation in Xenopus laevis. Genes Dev. 1990, 4 (6): 932-942. 10.1101/gad.4.6.932
Ruiz i Altaba A, Jessell TM: Retinoic acid modifies the pattern of cell differentiation in the central nervous system of neurula stage Xenopus embryos. Development. 1991, 112 (4): 945-958.
Chazaud C, Chambon P, Dolle P: Retinoic acid is required in the mouse embryo for left-right asymmetry determination and heart morphogenesis. Development. 1999, 126 (12): 2589-2596.
Huang S, Ma J, Liu X, Zhang Y, Luo L: Retinoic acid signaling sequentially controls visceral and heart laterality in zebrafish. J Biol Chem. 286 (32): 28533-28543.
Vermot J, Gallego Llamas J, Fraulob V, Niederreither K, Chambon P, Dolle P: Retinoic acid controls the bilateral symmetry of somite formation in the mouse embryo. Science. 2005, 308 (5721): 563-566. 10.1126/science.1108363
Vermot J, Pourquie O: Retinoic acid coordinates somitogenesis and left-right patterning in vertebrate embryos. Nature. 2005, 435 (7039): 215-220. 10.1038/nature03488
Anderson DJ: Lineages and transcription factors in the specification of vertebrate primary sensory neurons. Curr Opin Neurobiol. 1999, 9 (5): 517-524. 10.1016/S0959-4388(99)00015-X
Ribes V, Stutzmann F, Bianchetti L, Guillemot F, Dolle P, Le Roux I: Combinatorial signalling controls Neurogenin2 expression at the onset of spinal neurogenesis. Dev Biol. 2008, 321 (2): 470-481. 10.1016/j.ydbio.2008.06.003
Coyle DE, Li J, Baccei M: Regional differentiation of retinoic acid-induced human pluripotent embryonic carcinoma stem cell neurons. PLoS One. 2011, 6 (1): e16174. 10.1371/journal.pone.0016174
Shan ZY, Liu F, Lei L, Li QM, Jin LH, Wu YS, Li X, Shen JL: Generation of dorsal spinal cord GABAergic neurons from mouse embryonic stem cells. Cell Reprogram. 2011, 13 (1): 85-91.
Chen N, Napoli JL: All-trans-retinoic acid stimulates translation and induces spine formation in hippocampal neurons through a membrane-associated RARalpha. FASEB J. 2008, 22 (1): 236-245.
Krumlauf R: Hox genes and pattern formation in the branchial region of the vertebrate head. Trends Genet. 1993, 9 (4): 106-112. 10.1016/0168-9525(93)90203-T
Studer M, Lumsden A, Ariza-McNaughton L, Bradley A, Krumlauf R: Altered segmental identity and abnormal migration of motor neurons in mice lacking Hoxb-1. Nature. 1996, 384 (6610): 630-634. 10.1038/384630a0
Marshall H, Nonchev S, Sham MH, Muchamore I, Lumsden A, Krumlauf R: Retinoic acid alters hindbrain Hox code and induces transformation of rhombomeres 2/3 into a 4/5 identity. Nature. 1992, 360 (6406): 737-741. 10.1038/360737a0
Kessel M: Reversal of axonal pathways from rhombomere 3 correlates with extra Hox expression domains. Neuron. 1993, 10 (3): 379-393. 10.1016/0896-6273(93)90328-O
Hill J, Clarke JD, Vargesson N, Jowett T, Holder N: Exogenous retinoic acid causes specific alterations in the development of the midbrain and hindbrain of the zebrafish embryo including positional respecification of the Mauthner neuron. Mech Dev. 1995, 50 (1): 3-16. 10.1016/0925-4773(94)00321-D
Hernandez RE, Rikhof HA, Bachmann R, Moens CB: vhnf1 integrates global RA patterning and local FGF signals to direct posterior hindbrain development in zebrafish. Development. 2004, 131 (18): 4511-4520. 10.1242/dev.01297
Wiellette EL, Sive H: vhnf1 and Fgf signals synergize to specify rhombomere identity in the zebrafish hindbrain. Development. 2003, 130 (16): 3821-3829. 10.1242/dev.00572
Begemann G, Meyer A: Hindbrain patterning revisited: timing and effects of retinoic acid signalling. Bioessays. 2001, 23 (11): 981-986. 10.1002/bies.1142
Gavalas A: ArRAnging the hindbrain. Trends Neurosci. 2002, 25 (2): 61-64. 10.1016/S0166-2236(02)02067-2
Dupe V, Lumsden A: Hindbrain patterning involves graded responses to retinoic acid signalling. Development. 2001, 128 (12): 2199-2208.
Begemann G, Schilling TF, Rauch GJ, Geisler R, Ingham PW: The zebrafish neckless mutation reveals a requirement for raldh2 in mesodermal signals that pattern the hindbrain. Development. 2001, 128 (16): 3081-3094.
Partanen J: FGF signalling pathways in development of the midbrain and anterior hindbrain. J Neurochem. 2007, 101 (5): 1185-1193. 10.1111/j.1471-4159.2007.04463.x
Emoto Y, Wada H, Okamoto H, Kudo A, Imai Y: Retinoic acid-metabolizing enzyme Cyp26a1 is essential for determining territories of hindbrain and spinal cord in zebrafish. Dev Biol. 2005, 278 (2): 415-427. 10.1016/j.ydbio.2004.11.023
Chen Y, Pan FC, Brandes N, Afelik S, Solter M, Pieler T: Retinoic acid signaling is essential for pancreas development and promotes endocrine at the expense of exocrine cell differentiation in Xenopus. Dev Biol. 2004, 271 (1): 144-160. 10.1016/j.ydbio.2004.03.030
Stafford D, Hornbruch A, Mueller PR, Prince VE: A conserved role for retinoid signaling in vertebrate pancreas development. Dev Genes Evol. 2004, 214 (9): 432-441.
Martin M, Gallego-Llamas J, Ribes V, Kedinger M, Niederreither K, Chambon P, Dolle P, Gradwohl G: Dorsal pancreas agenesis in retinoic acid-deficient Raldh2 mutant mice. Dev Biol. 2005, 284 (2): 399-411. 10.1016/j.ydbio.2005.05.035
Molotkov A, Molotkova N, Duester G: Retinoic acid generated by Raldh2 in mesoderm is required for mouse dorsal endodermal pancreas development. Dev Dyn. 2005, 232 (4): 950-957. 10.1002/dvdy.20256
Pan FC, Chen Y, Bayha E, Pieler T: Retinoic acid-mediated patterning of the pre-pancreatic endoderm in Xenopus operates via direct and indirect mechanisms. Mech Dev. 2007, 124 (7-8): 518-531. 10.1016/j.mod.2007.06.003
Bayha E, Jorgensen MC, Serup P, Grapin-Botton A: Retinoic acid signaling organizes endodermal organ specification along the entire antero-posterior axis. PLoS One. 2009, 4 (6): e5845. 10.1371/journal.pone.0005845
Stafford D, Prince VE: Retinoic acid signaling is required for a critical early step in zebrafish pancreatic development. Curr Biol. 2002, 12 (14): 1215-1220. 10.1016/S0960-9822(02)00929-6
Stafford D, White RJ, Kinkel MD, Linville A, Schilling TF, Prince VE: Retinoids signal directly to zebrafish endoderm to specify insulin-expressing beta-cells. Development. 2006, 133 (5): 949-956. 10.1242/dev.02263
Dalgin G, Ward AB, Hao le T, Beattie CE, Nechiporuk A, Prince VE: Zebrafish mnx1 controls cell fate choice in the developing endocrine pancreas. Development. 138 (21): 4597-4608.
Kinkel MD, Prince VE: On the diabetic menu: zebrafish as a model for pancreas development and function. Bioessays. 2009, 31 (2): 139-152. 10.1002/bies.200800123
Kinkel MD, Eames SC, Alonzo MR, Prince VE: Cdx4 is required in the endoderm to localize the pancreas and limit beta-cell number. Development. 2008, 135 (5): 919-929. 10.1242/dev.010660
Ostrom M, Loffler KA, Edfalk S, Selander L, Dahl U, Ricordi C, Jeon J, Correa-Medina M, Diez J, Edlund H: Retinoic acid promotes the generation of pancreatic endocrine progenitor cells and their further differentiation into beta-cells. PLoS One. 2008, 3 (7): e2841. 10.1371/journal.pone.0002841
Tulachan SS, Doi R, Kawaguchi Y, Tsuji S, Nakajima S, Masui T, Koizumi M, Toyoda E, Mori T, Ito D: All-trans retinoic acid induces differentiation of ducts and endocrine cells by mesenchymal/epithelial interactions in embryonic pancreas. Diabetes. 2003, 52 (1): 76-84. 10.2337/diabetes.52.1.76
Shen CN, Marguerie A, Chien CY, Dickson C, Slack JM, Tosh D: All-trans retinoic acid suppresses exocrine differentiation and branching morphogenesis in the embryonic pancreas. Differentiation. 2007, 75 (1): 62-74.
Moriya N, Komazaki S, Takahashi S, Yokota C, Asashima M: In vitro pancreas formation from Xenopus ectoderm treated with activin and retinoic acid. Dev Growth Differ. 2000, 42 (6): 593-602. 10.1046/j.1440-169x.2000.00542.x
Asashima M, Michiue T, Kurisaki A: Elucidation of the role of activin in organogenesis using a multiple organ induction system with amphibian and mouse undifferentiated cells in vitro. Dev Growth Differ. 2008, 50 (Suppl 1): S35-45.
Heine UI, Roberts AB, Munoz EF, Roche NS, Sporn MB: Effects of retinoid deficiency on the development of the heart and vascular system of the quail embryo. Virchows Arch B Cell Pathol Incl Mol Pathol. 1985, 50 (2): 135-152.
Lin SC, Dolle P, Ryckebusch L, Noseda M, Zaffran S, Schneider MD, Niederreither K: Endogenous retinoic acid regulates cardiac progenitor differentiation. Proc Natl Acad Sci USA. 2010, 107 (20): 9234-9239. 10.1073/pnas.0910430107
Sirbu IO, Zhao X, Duester G: Retinoic acid controls heart anteroposterior patterning by down-regulating Isl1 through the Fgf8 pathway. Dev Dyn. 2008, 237 (6): 1627-1635. 10.1002/dvdy.21570
Stachurska E, Loboda A, Niderla-Bielinska J, Szperl M, Juszynski M, Jozkowicz A, Dulak J, Ratajska A: Expression of cellular retinoic acid-binding protein I and II (CRABP I and II) in embryonic mouse hearts treated with retinoic acid. Acta Biochim Pol. 2011, 58 (1): 19-29.
Keegan BR, Feldman JL, Begemann G, Ingham PW, Yelon D: Retinoic acid signaling restricts the cardiac progenitor pool. Science. 2005, 307 (5707): 247-249. 10.1126/science.1101573
Ryckebusch L, Wang Z, Bertrand N, Lin SC, Chi X, Schwartz R, Zaffran S, Niederreither K: Retinoic acid deficiency alters second heart field formation. Proc Natl Acad Sci USA. 2008, 105 (8): 2913-2918. 10.1073/pnas.0712344105
Mathew LK, Sengupta S, Franzosa JA, Perry J, La Du J, Andreasen EA, Tanguay RL: Comparative expression profiling reveals an essential role for raldh2 in epimorphic regeneration. J Biol Chem. 2009, 284 (48): 33642-33653. 10.1074/jbc.M109.011668
Kikuchi K, Holdway JE, Major RJ, Blum N, Dahn RD, Begemann G, Poss KD: Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev Cell. 20 (3): 397-404.
Serluca FC, Fishman MC: Pre-pattern in the pronephric kidney field of zebrafish. Development. 2001, 128 (12): 2233-2241.
Cartry J, Nichane M, Ribes V, Colas A, Riou JF, Pieler T, Dolle P, Bellefroid EJ, Umbhauer M: Retinoic acid signalling is required for specification of pronephric cell fate. Dev Biol. 2006, 299 (1): 35-51. 10.1016/j.ydbio.2006.06.047
Mendelsohn C, Lohnes D, Decimo D, Lufkin T, LeMeur M, Chambon P, Mark M: Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development. 1994, 120 (10): 2749-2771.
Carroll TJ, Vize PD: Synergism between Pax-8 and lim-1 in embryonic kidney development. Dev Biol. 1999, 214 (1): 46-59. 10.1006/dbio.1999.9414
Lee SJ, Kim S, Choi SC, Han JK: XPteg (Xenopus proximal tubules-expressed gene) is essential for pronephric mesoderm specification and tubulogenesis. Mech Dev. 2010, 127 (1-2): 49-61. 10.1016/j.mod.2009.11.001
Bollig F, Perner B, Besenbeck B, Kothe S, Ebert C, Taudien S, Englert C: A highly conserved retinoic acid responsive element controls wt1a expression in the zebrafish pronephros. Development. 2009, 136 (17): 2883-2892. 10.1242/dev.031773
Perner B, Englert C, Bollig F: The Wilms tumor genes wt1a and wt1b control different steps during formation of the zebrafish pronephros. Dev Biol. 2007, 309 (1): 87-96. 10.1016/j.ydbio.2007.06.022
Osafune K, Nishinakamura R, Komazaki S, Asashima M: In vitro induction of the pronephric duct in Xenopus explants. Dev Growth Differ. 2002, 44 (2): 161-167. 10.1046/j.1440-169x.2002.00631.x
Wingert RA, Selleck R, Yu J, Song HD, Chen Z, Song A, Zhou Y, Thisse B, Thisse C, McMahon AP: The cdx genes and retinoic acid control the positioning and segmentation of the zebrafish pronephros. PLoS Genet. 2007, 3 (10): 1922-1938.
Rosselot C, Spraggon L, Chia I, Batourina E, Riccio P, Lu B, Niederreither K, Dolle P, Duester G, Chambon P: Non-cell-autonomous retinoid signaling is crucial for renal development. Development. 137 (2): 283-292.
Desai TJ, Malpel S, Flentke GR, Smith SM, Cardoso WV: Retinoic acid selectively regulates Fgf10 expression and maintains cell identity in the prospective lung field of the developing foregut. Dev Biol. 2004, 273 (2): 402-415. 10.1016/j.ydbio.2004.04.039
Desai TJ, Chen F, Lu J, Qian J, Niederreither K, Dolle P, Chambon P, Cardoso WV: Distinct roles for retinoic acid receptors alpha and beta in early lung morphogenesis. Dev Biol. 2006, 291 (1): 12-24. 10.1016/j.ydbio.2005.10.045
Chen F, Cao Y, Qian J, Shao F, Niederreither K, Cardoso WV: A retinoic acid-dependent network in the foregut controls formation of the mouse lung primordium. J Clin Invest. 120 (6): 2040-2048.
Malpel S, Mendelsohn C, Cardoso WV: Regulation of retinoic acid signaling during lung morphogenesis. Development. 2000, 127 (14): 3057-3067.
Shiotsugu J, Katsuyama Y, Arima K, Baxter A, Koide T, Song J, Chandraratna RA, Blumberg B: Multiple points of interaction between retinoic acid and FGF signaling during embryonic axis formation. Development. 2004, 131 (11): 2653-2667. 10.1242/dev.01129
Ho L, Mercola M, Gudas LJ: Xenopus laevis cellular retinoic acid-binding protein: temporal and spatial expression pattern during early embryogenesis. Mech Dev. 1994, 47 (1): 53-64. 10.1016/0925-4773(94)90095-7
Kam RK, Chen Y, Chan SO, Chan WY, Dawid IB, Zhao H: Developmental expression of Xenopus short-chain dehydrogenase/reductase 3. Int J Dev Biol. 2010, 54 (8-9): 1355-1360. 10.1387/ijdb.092984rk
Acknowledgements
We thank members of our laboratories for valuable discussion. This work is supported by GRF grant from the Research Grants Council of Hong Kong (No. CUHK480709) to HZ, the National Basic Research Program of China (2009CB941202), and the fund from the Key Project of Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2-YW-R-083) to YLC. RKTK was supported by the Graduate Studentships from The Chinese University of Hong Kong.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RKTK and HZ planned the manuscript outline. RKTK, HZ and YLC wrote the draft, YD revised and did proof reading, HZ and YLC finalized the manuscript. All authors read and approve the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kin Ting Kam, R., Deng, Y., Chen, Y. et al. Retinoic acid synthesis and functions in early embryonic development. Cell Biosci 2, 11 (2012). https://doi.org/10.1186/2045-3701-2-11
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2045-3701-2-11