
Abstract
Background
The systemic treatment of malignant endometrial stromal tumors (EST) is not well established. A few reports describe objective responses to imatinib, which suggest a novel therapeutic strategy for these tumors. Due to these facts, we aimed to perform a retrospective analysis of possible molecular targets of tyrosine kinase inhibitors (TKI) in EST: KIT, PDGFRA and EGFR.
Methods
52 endometrial stromal sarcomas and 13 undifferentiated endometrial sarcomas were examined and reviewed. Mutational analysis were performed for exons 9, 11, 13, and 17 of the KIT gene, exons 12 and 18 of the PDGFRA gene and exons 18, 19, 20 and 21 of the EGFR gene. The incidence and distribution of the KIT, PDGFRA, and EGFR expression were examined by immunohistochemistry, and EGFR amplification was assessed by fluorescence in situ hybridization.
Results
No mutations in KIT, PDGFRA and EGFR genes were detected. Overexpression of KIT, PDGFRA, EGFR, was detected in 2 (3%), 23 (35.4%), 7 (10.8%) cases respectively, whereas amplification of EGFR gene was not found.
Conclusions
Absence of significant expression, amplification and activating mutations on these tyrosine kinase receptors suggest that it is unlikely that EST can benefit from therapies such as TKI on the systemic setting.
Similar content being viewed by others


Background
Endometrial stromal sarcoma, low grade (ESS) and undifferentiated endometrial sarcoma (UES) belong to the rare group of endometrial stromal tumors (EST), which represents 15% of uterine sarcomas [1]. ESS presents a cellular background similar to the cells of normal endometrial stroma in proliferative phase. In contrast, UES lacks specific differentiation and bears no histological resemblance to endometrial stroma. Tumor cells are high-grade spindle to polygonal-shaped, with marked nuclear pleomorphism and high mitotic activity. Necrosis and vascular invasion are commonly seen [2]. While ESS is characterized by indolent course and late recurrences, with a 5-year overall survival (OS) up 70%, UES is usually diagnosed at advanced stages, and has a high rate of distant metastasis and a 5-year OS ranging from 25-55% [3–6]. FIGO stage [7] is the strongest prognostic factor for these malignancies [5, 8]. CD10 is the most sensitive marker for ESS [9, 10]. Estrogen and progesterone receptors [11] and aromatase [12] are usually expressed in ESS, and less commonly in UES [13–15]. The rearrangement t(7;17)(p15;q21), which results in JAZF1/JJAZ1 gene fusion, is the cytogenetic hallmark of ESS [16], although other translocations have been reported [17–19]. In contrast, UES is characterized by a complex karyotype [20, 21]. Recently, the t(10;17)(q22;p13) that results in YWHAE-FAM22A/B gene fusion with oncogenic properties was reported in a subset of UES [22], which is associated to the expression of Cyclin D1 [23, 24] and β-catenin [23]; this supports the recent sub-classification of UES [15, 25]. Surgery is the standard treatment, and includes total hysterectomy and bilateral salpingo-oophorectomy. However, due the rarity of these tumors, distinct clinical behavior, and lack of randomized studies including both categories, an appropriate systemic treatment of these malignancies was not been yet established.
Molecular targets of tyrosine kinase inhibitors (TKI) such as imatinib mesylate (Glivec®, STI-571, Novartis, Switzerland), gefitinib (Iressa®, AstraZeneca, Macclesfield, UK) and erlotinib (Tarceva®, OSI-Pharmaceuticals, New York, NY), which includes PDGFRA/B, KIT, C-ABL and EGFR, were reported to be expressed in ESS and UES by immunohistochemistry [26–40], although without presence of any activating mutations [36–39, 41, 42]. Interestingly, a few reports described objective responses with imatinib in patients who express at least one TKI target [36, 37, 43]. Another report described a unique case of UES with EGFR expression and EGFR amplification which temporarily responded to imatinib [42]. Based on these findings, an extensive evaluation of the molecular targets of TKI on EST was carried out to identify a novel therapeutic strategy for these malignancies. In the present study we analyzed the gene status and protein expression of KIT, PDGFRA, and EGFR in a large series of ESS and UES to evaluate their distribution among the distinct subgroups and correlate the immunohistochemical expression with mutational status.
Material and methods
Patient selection and study design
A series of 75 EST was retrieved from Spanish centers associated to Spanish Sarcoma Group (GEIS) and from the Pathology Departments of Complejo Hospitalario Universitario de Badajoz, Badajoz, Spain and Hospital do Espírito Santo E.P.E, Évora, Portugal, and sent to Tumor Bank of the Cancer Research Centre – Salamanca, Spain. The selection of patients was made according to the following inclusion criteria - previous diagnosis of EST (any histological grade) and availability of histological material sufficient to perform the study. After receiving and encoding the samples, cases were reviewed and subclassified by 1 co-author (EA) based on the current WHO classification [2]. The study was approved by the Ethics Committee of Hospital Germans Trias i Pujol (Spain), and was conducted in accordance with the Declaration of Helsinki and Spanish regulative law for Tumor Banks.
Tissue microarray and immunohistochemistry
65 samples were considered valid and included in the study. Ten cases were discarded because of a sample too small to perform the proposed study or non-representative sample or diagnosis different than EST. Before tissue microarray (TMA) construction, representative areas of tumor were selected on hematoxylin and eosin (H&E) section and marked on the paraffin block. For each sample were obtained two cylinders of 1 mm diameter and placed in a recipient block using a tissue microarrayer (Manual Tissue Array; Beecher Instruments Inc. Sun Prairie, Wisconsin, USA). In total two TMAs were constructed according to previously described [44].
The tyrosine kinase receptors (TKR) evaluated were KIT, PDGFRA and EGFR, and to confirm diagnosis the expression of two markers most commonly used on ESS [45, 46], CD10 and Calponin were assessed in each tumor. Immunohistochemistry (IHC) was performed in 3 μm sections. KIT, PDGFRA, CD10 and Calponin immunostaining was performed using a Discovery® Ventana automated immunostainer (Ventana Medical Systems, Tucson, Arizona, USA). Heat-induced antigen retrieval was done with Tris-EDTA buffer (pH 8.0) for KIT, PDGFRA and Calponin and with citrate buffer (pH 6.0) for CD10. Sections were incubated with the primary antibodies Calponin (clone CALP; Dako, Carpinteria, CA, USA), KIT (polyclonal; Dako, Carpinteria, CA, USA), PDGFRA (polyclonal; Thermo Fisher Scientific, Fremont, CA, USA) and CD10 (clone 56C6; Novocastra Laboratoires, Newcastle upon Tyne, UK) at 1:100 dilution. Universal secondary biotinylated antibody (Discovery™ Universal Secondary Antibody, Ventana Medical Systems, Tucson, Arizona, USA) was used and developed for detection using the DAB MAP system (Ventana Medical Systems, Tucson, Arizona, USA). For EGFR analysis expression, a ready-to-use monoclonal antibody (clone 2.1E1; Gennova Scientific, Seville, Spain) was used. Briefly, paraffin-embedded sections were deparaffinized in xylene and rehydrated in downgraded alcohols and distilled water. Antigen retrieval was performed with Proteinase K solution (Dako, Carpinteria, CA, USA). The primary antibody was detected using a secondary antibody- horseradish peroxidase polymer conjugate (Dako REAL™ EnVision™ Detection System; Dako, Carpinteria, CA, USA), and all incubations were done with the Dako Autostainer Plus system (Dako, Carpinteria, CA, USA). All sections were counterstained with hematoxylin, upgraded alcohols, and xylene, mounted, and analyzed by standard light microscopy.
Immunohistochemical evaluation
The expression of CD10 and Calponin was considered positive when over 1% of the tumor cells showed cytoplasmic expression. The TKR expression was detected in the cytoplasm of tumor cells, and each case was interpreted for immunoreactivity using a 0 to 3 semiquantitative scoring system for both the intensity of stain and the percentage of positive cells, as previously reported [47]. The multiplicative index of intensity and labeling was considered for statistic analysis and the expression was defined as weak and/or focal for a multiplicative index 1–3, moderate local or diffuse for a multiplicative index 4–6 and intense and diffuse if the multiplicative index was >6. The IHC analysis was scored by the same co-author who performed the histologic review (EA).
Molecular analysis of tyrosine kinase receptors
DNA extraction
Ten serial 5 μm sections were cut and transferred into 15 ml conical centrifugation tubes and deparaffinized in xylene (two times for 5 minutes) and absolute ethanol (two times for 5 minutes) at 4000 rpm, followed by drying of the samples at room temperature. Subsequently, DNA extractions were performed according to QIAamp DNA Mini Kit protocol (Qiagen, Hilden, Germany), and then quantified by spectrophotometry (SmartSpec Plus Spectrophotometer, Bio-Rad Laboratories Inc., CA, USA).
KIT and PDGFRA
According to previous studies [48–50], exons 9, 11, 13, and 17 of KIT gene and exons 12 and 18 of PDGFRA gene were amplified in order to identify possible mutations. Amplification of GAPDH gene was performed to confirm integrity and quality of extracted DNA. PCR amplification of KIT and PDGFRA was performed using 1–5 μl of genomic DNA, 1X PCR buffer + 2 mM MgCl2 (Roche Applied Science, Mannheim, Germany), 0.15 mM of each primer, 200 μM of each dNTP (GeneAmp® dNTP Blend, 10 mM, ABI, Carlsbad, CA, USA), and 1.5 U of Taq DNA polymerase (Roche Applied Science, Mannheim, Germany) in a total volume of 25 μl. The PCR conditions were 94°C for 10 min, 40 cycles of 1 min at 94°C, 1min30sec to 56°C (KIT) or 65°C (PDGFRA) and 1 min at 72°C, followed by a cycle of 10 min at 72°C. The amplified products were visualized on agarose gel 2%.
Prior to sequence analysis, KIT and PDGFRA PCR products were first purified using a QIAquick® PCR Purification Technology Kit (Qiagen, Hilden, Germany). Direct sequencing of PCR products was performed using ABI PRISM® BigDye Terminator Cycle Sequencing Kit in an ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer instructions. Sequencing primers were the same as those used for PCR, and both strands (forward and reverse) were sequenced.
All sequences obtained were visualized and analyzed in the program Sequence Scanner Software v1.0 (Applied Biosystems, Carlsbad, CA, USA), based on the reference sequences: KIT - ENSG00000157404 and PDGFRA - ENSG00000134853.
EGFR
PCR amplification of exons 18, 19, 20 and 21 of the EGFR gene, which encompass most of the EGFR mutations [51, 52], was carried out using previously described primers [53, 54] (Table 1). Using 4 μl of DNA, PCR was performed in a reaction volume of 15 μl containing 1X PCR buffer (Applied Biosystems, Carlsbad, CA, USA), 2.5 mM MgCl2 (Applied Biosystems, Carlsbad, CA, USA), 0.17 mM of each primer, 200 mM of each dNTP (GeneAmp® dNTP Blend, 10 mM, Applied Biosystems, Carlsbad, CA, USA) and 2 U of Taq DNA polymerase (AmpliTaq Gold® DNA Polymerase, Applied Biosystems, Carlsbad, CA, USA).
The conditions for PCR were 95°C for 12 min, 40 cycles of 30 sec at 95°C, 45 sec at 65°C and 1 min at 72°C, followed by a cycle of 10 min at 72°C. After visualization of amplified products by gel electrophoresis on a 2% agarose, these were purified with USB® ExoSAP-IT® (Affymetrix, Inc., Cleveland, Ohio, USA). Subsequently the purified products were precipitated and labeled with GenomeLab™ DTCS Quick Start Kit (Beckman Coulter Inc, Fullerton, CA, USA) according to the manufacturer’s instructions and were sequenced using GenomeLab™ GeXP Genetic Analysis System (Beckman Coulter Inc, Fullerton, CA, USA). As described above, sequencing was performed in both directions, and the same PCR primers were used.
The sequences were analyzed using Genome Lab Genetic Analysis System v10.0.30 (Beckman Coulter Inc, Fullerton, CA, USA), and compared with the reference sequence ENSG00000146648.
Fluorescence in situ hybridization - EGFR
The ploidy status of EGFR in each tumor was assessed by fluorescence in situ hybridization (FISH) on previously constructed TMAs. Gene amplification was determined by using an EGFR/CEN-7 FISH Probe Mix (Y5500, Dako, Carpinteria, CA, USA) containing Texas Red-labeled DNA probe covering the full EGFR region and a mixture of fluorescein-labeled PNA probes targeted at the centromeric region of chromosome 7 (Chr7).
The FISH was performed using the Histology FISH Accessory Kit (Dako, Carpinteria, CA, USA), according to the manufacturer’s instructions.
Hybridization signals were visualized using fluorescence microscope equipped with a IAI monochrome progressive scan (IAI Company, Taiwan) and run by image analysis software Cytovision® (Leica Microsystems, Wetzlar, Germany). 20 tumour nuclei/case were scored, and the tumor cells in which the signals of EGFR and CEP7 were increased equally were classified as polysomy 7 and those for which there was a double signal for EGFR or CEP7 were considered diploid. To assess gene amplification, we calculated the ratio of EGFR to CEP7 and evaluated in accordance with the criteria of FISH scoring system of Colorado Group [55]. Evaluation of the ploidy status of EGFR was performed by a single pathologist (EA).
Statistical analysis
The association between the expression of TKR and ploidy of EGFR with different histological types of EST, as well the mutational status of TKR were assessed. All statistical analyses were performed based on contingency tables, with SPSS software v18.0 statistics (Chicago, Illinois).
Results
Classification of endometrial stromal tumors
From 65 EST cases, 80% (52/65) were diagnosed as ESS, and 20% (13/65) were UES. As expected, the majority of ESS presented expression of CD10, 51.9% (27/52), which was lower in UES cases (23.1%; 3/13).
Analysis of expression of tyrosine kinase receptors
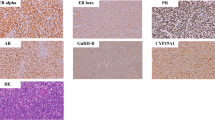
The expression of KIT, PDGFRA and EGFR was evaluated in all cases of EST, and an example of the pattern of expression of these markers is shown in Figure 1.

Immunohistochemical expression of tyrosine kinase receptors in endometrial stromal tumors. Endometrial stromal sarcoma, low grade (A) and undifferentiated endometrial sarcoma (B). Negative expression of KIT in ESS (C) and very focal expression in UES (D). Weak and/or focal PDGFRA and EGFR expression in ESS (E, G) and UES (F,H).
KIT stromal expression was found in only 2 out of 65 cases, and it was weak and/or focal. In contrast, PDGFRA showed positive stromal expression in 35.4% of cases (23/65), mostly weak and/or focal; its distribution was similar in both histological subtypes. Interestingly, expression was not intense and/or diffuse in any case. For EGFR, a minority of cases were classified as positive, representing 10.8% (7/65).
Mutational status of tyrosine kinase receptors
The molecular study was performed in 62 cases. Sequencing of exons 9, 11, 13 and 17 of KIT only revealed the presence of a silent base substitution (ATC>ATT) in exon 17 at codon 798 (I798I) in two cases (ESS). The PDGFRA analysis showed the presence of several silent mutations, a base substitution (CCA>CCG) in exon 12 at codon 567 (P567P) in all cases, 32.3% (21/65) showed a silent base substitution (GTC>GTT) in exon 18 at codon 824 (V824V) and 1 case had a double silent base substitution at codons 824 and 838 (GTC>GTT and GGC>GGT; V824V and G838G respectively) both in exon 18. There were no somatic mutations in the genes KIT and PDGFRA.
EGFR mutational analysis was performed only in cases with expression of EGFR, and in which DNA was available for molecular studies. Two cases without EGFR expression were included as control. There were no mutations in EGFR exons 18, 19, 20 and 21. In 6 of 7 cases silent base substitution were identified at codon 787 (CAG>CAA; Q787Q), and in one of these cases were identified another silent base substitution at codon 790 (ACG>ACA; T790T) both in exon 20.
EGFR amplification by FISH
EGFR gene amplification analysis was performed in all cases included in the study (65 cases), except in one which was considered not evaluable. No amplification of EGFR gene was observed. Numerous cases (58/64, 90.6%) were diploid (1–2 copies), and few cases (6/64, 9.4%) were polyploid (3–4 copies), and the distribution was similar in both histological types.
Discussion
Systemic therapy in EST has a marginal efficacy. Low grade ESS can achieve control with hormonal treatments (progestins, aromatase inhibitors) and high grade undifferentiated uterine sarcomas are included in clinical trials together with leiomyosarcomas. Tanner et al. reported efficacy of docetaxel and gemcitabine or adriamycin for advanced cases of high grade endometrial tumors [56]. Indeed, the indication of systemic treatment of EST is controversial. Hormonal treatment can depend on hormonal receptor status and is associated to light side-effects and occasionally indicated. Responses were reported in a few ESS cases treated with progestin or aromatase inhibitors [57–64], but prospective larger trials are needed. Chemotherapy showed no apparent benefit in ESS [34, 65–67]. In the case of UES some objective and partial responses were observed [68–70]. In a retrospective study including only 21 patients was described a response rate of 62% with gemcitabine/docetaxel and doxorubicin-based regimens in first-line, whereas in second- or additional chemotherapy for progressive disease, the response rate was around 19% [56]. On the other hand, radiotherapy seems to have a significant effect on local-regional control rate with a low impact on survival improvement [3, 4, 71–74]. Due to these facts several reports address the need to explore the role of TKR in these malignancies. The possible clinical efficacy of imatinib in the treatment of EST and the contribution of its TKR-target in disease progression are the key questions to solve [34].
Previous reports showed the presence at least one TKR in EST, but available data just refer a few case reports and small series, which makes it difficult to draw conclusions. Based on this, we performed an extensive evaluation of molecular and immunohistochemical expression of TKR KIT, PDGFRA and EGFR in a large series of ESS and UES.
KIT and PDGFRA cytoplasmic expression was observed in 3% and 35.4% respectively. In the literature, KIT expression has been reported to be quite variable (0-100%) (Table 2) and this might be due to differences in the choice of antibody clones, tissue pretreatment, sensitivity of immunohistochemical procedures, sample selection and use of whole tissue sections or TMA sections, which was also suggested in previous studies [75, 76]. On the other hand, cytoplasmic PDGFRA expression in ESS, although mostly weak and/or focal, lies within the range reported in previous studies [34, 37–39, 77, 78].
In our series, mutational analysis of hot spots of KIT and PDGFRA genes did not reveal any somatic mutation, which is consistent with previous studies [36–39, 41, 42, 77]. Constitutive activation via autocrine/paracrine stimulation of the receptor by its ligand was observed in several human cancers and is related with their tumorigenesis [82, 83]. Probably, expression of related ligands of KIT and PDGFRA, which were not evaluated in this series, might be the reason for expression of both receptors in these tumors, although more studies are needed. Second, the work of Kang et al. demonstrated that relative KIT expression ratio was fivefold higher in cases with KIT mutation, than in GISTs lacking KIT mutation and the mutation status of KIT and PDGFRA was directly related to the different expression levels of activated KIT and PDGFRA [84]. Furthermore, with exception of GISTs, the presence of overexpression of KIT and PDGFRA in human cancers was not correlated with presence of activating mutations [85]. These facts support our results, once that KIT and PDGFRA expression observed was mostly weak and/or focal and lacks activating mutations on its related genes. Furthermore, the consistent expression of KIT and PDGFR receptors and their ligands was seen in a phase II trial in women with ovarian cancer, together with the absence of mutations in these genes and with lack of response to imatinib [86]. In other two phase II clinical trials in lung cancer [87] and uterine carcinosarcomas [88], the high expression of KIT did not correlate with response to imatinib. These clinical findings support the hypothesis that overexpression of these TKR does not confer sensitivity to imatinib, which was previously suggested for other uterine sarcomas [89].
However, the expression of at least one of imatinib-target (KIT, PDGFRA and PDGFRB) without evidence of activating mutations was reported in EST patients who responded to imatinib treatment [36, 37, 43]. Tumor shrinkage was the main indicator of objective response accompanied by stable disease in two cases [36, 37]. One explanation for these results may be due to the autocrine/paracrine signaling of PDGFRB. In dermatofibrosarcoma protuberans (DFSP), t(17;22)(q22;q13) results in a COL1A1-PDGFB gene fusion. This cytogenetic abnormality is essential for pathogenesis of the disease and responsible by constitutive activation of PDGFRB. Imatinib inhibits the activity of the dysregulated PDGFRB [90], decreases enzymatic activity in DFSP cells and inhibits their ability to divide and grow [91] and induces apoptosis in tumor cells [92], which may have effects on decreasing tumor size [93]. In fact, an effect of imatinib on tumor shrinkage was observed in patients with DFSP [94, 95], and therefore imatinib was approved for unresectable, recurrent, and/or metastatic DFSP in adults. However, in ESS and UES cases reported as responsive to imatinib, PDGFRB was only determined in one case reported by Trojan et al.[37], which makes difficult to draw any conclusions.
Concerning EGFR expression, our study reveals lower cytoplasmic expression of EGFR in EST cases (around 10.8%), in contrast with the results obtained by other authors [40, 77] (Table 2). This contradiction may be a consequence of the same variables indicated for the immunoexpression of KIT. Since i) a correlation between an increased EGFR copy number and gefitinib was proposed in non-small-cell lung cancer (NSCLC) [96] and ii) immunohistochemistry is not a reliable approach for this determination, we decided to evaluate EGFR amplification status in our series by FISH. The EGFR/Chr7 ratio was below 2, which is indicative of non-amplification, and only a small percentage of the cases presented polysomy. Previous evaluation of EGFR amplification was only performed in a UES case which presented a temporary response to imatinib. This case presented a low-level amplification (mean ratio 2.9) according to the evaluation criteria of the authors [42]. Although no consensus has been reached on how to assess the presence and extent of EGFR status dysregulation in solid tumors by FISH analysis [97], it seems likely that gene amplification found in NSCLC and glioblastoma is an uncommon event in EST. EGFR mutational status also did not show any somatic mutation in exons 18–21. In fact, all these data confirm that it is unlikely that EGFR activation could play a role in tumorigenesis of EST.
Conclusions
In summary, we performed an extensive molecular and expression analysis of KIT, PDGFRA and EGFR in the largest series of EST studied so far (Table 2). Our findings reveal a lack of significant expression, amplification and activating mutations on these receptors and related genes, which suggests that it is unlikely that EST can benefit from therapies such as imatinib or EGFR inhibitors in advanced disease. However, the role of imatinib in the management of unresectable tumors needs to be clarified through evaluation of expression of constitutive activation of PDGFRB by its ligands, or other genetic alterations in ESS and UES. The recent discovery of a chromosomal rearrangement t(10;17)(q22;p13) that results in a 14-3-3 fusion protein with oncogenic properties in a subgroup of UES [22], and the nuclear expression of β-catenin, a member of Wnt and E-cadherin signaling pathways in EST need to be better explored, since they can represent an entry point for targeted therapeutic strategies.
References
Koivisto-Korander R, Butzow R, Koivisto AM, Leminen A: Clinical outcome and prognostic factors in 100 cases of uterine sarcoma: experience in Helsinki University Central Hospital 1990–2001. Gynecol Oncol. 2008, 111: 74-81. 10.1016/j.ygyno.2008.06.002
WHO: Pathology and genetics of tumours of the breast and female genital organs. 2003, Lyon: IARC Press,
Li N, Wu LY, Zhang HT, An JS, Li XG, Ma SK: Treatment options in stage I endometrial stromal sarcoma: a retrospective analysis of 53 cases. Gynecol Oncol. 2008, 108: 306-11. 10.1016/j.ygyno.2007.10.023
Schick U, Bolukbasi Y, Thariat J, Abdah-Bortnyak R, Kuten A, Igdem S, Caglar H, Ozsaran Z, Lossl K, Schleicher U: Outcome and prognostic factors in endometrial stromal tumors: a rare cancer network study. Int J Radiat Oncol Biol Phys. 2012, 82: e757-63. 10.1016/j.ijrobp.2011.11.005
Abeler VM, Royne O, Thoresen S, Danielsen HE, Nesland JM, Kristensen GB: Uterine sarcomas in Norway. A histopathological and prognostic survey of a total population from 1970 to 2000 including 419 patients. Histopathology. 2009, 54: 355-64. 10.1111/j.1365-2559.2009.03231.x
Nordal RR, Kristensen GB, Kaern J, Stenwig AE, Pettersen EO, Trope CG: The prognostic significance of surgery, tumor size, malignancy grade, menopausal status, and DNA ploidy in endometrial stromal sarcoma. Gynecol Oncol. 1996, 62: 254-9. 10.1006/gyno.1996.0224
Prat J: FIGO staging for uterine sarcomas. Int J Gynaecol Obstet. 2009, 104: 177-8. 10.1016/j.ijgo.2008.12.008
Chan JK, Kawar NM, Shin JY, Osann K, Chen LM, Powell CB, Kapp DS: Endometrial stromal sarcoma: a population-based analysis. Br J Cancer. 2008, 99: 1210-5. 10.1038/sj.bjc.6604527
Toki T, Shimizu M, Takagi Y, Ashida T, Konishi I: CD10 is a marker for normal and neoplastic endometrial stromal cells. Int J Gynecol Pathol. 2002, 21: 41-7. 10.1097/00004347-200201000-00008
Chu PG, Arber DA, Weiss LM, Chang KL: Utility of CD10 in distinguishing between endometrial stromal sarcoma and uterine smooth muscle tumors: an immunohistochemical comparison of 34 cases. Mod Pathol. 2001, 14: 465-71. 10.1038/modpathol.3880335
Reich O, Regauer S, Urdl W, Lahousen M, Winter R: Expression of oestrogen and progesterone receptors in low-grade endometrial stromal sarcomas. Br J Cancer. 2000, 82: 1030-4.
Reich O, Regauer S: Aromatase expression in low-grade endometrial stromal sarcomas: an immunohistochemical study. Mod Pathol. 2004, 17: 104-8. 10.1038/modpathol.3800031
Ioffe YJ, Li AJ, Walsh CS, Karlan BY, Leuchter R, Forscher C, Cass I: Hormone receptor expression in uterine sarcomas: prognostic and therapeutic roles. Gynecol Oncol. 2009, 115: 466-71. 10.1016/j.ygyno.2009.08.014
D’Angelo E, Spagnoli LG, Prat J: Comparative clinicopathologic and immunohistochemical analysis of uterine sarcomas diagnosed using the World Health Organization classification system. Hum Pathol. 2009, 40: 1571-85. 10.1016/j.humpath.2009.03.018
Kurihara S, Oda Y, Ohishi Y, Iwasa A, Takahira T, Kaneki E, Kobayashi H, Wake N, Tsuneyoshi M: Endometrial stromal sarcomas and related high-grade sarcomas: immunohistochemical and molecular genetic study of 31 cases. Am J Surg Pathol. 2008, 32: 1228-38. 10.1097/PAS.0b013e31816a3b42
Koontz JI, Soreng AL, Nucci M, Kuo FC, Pauwels P, van Den Berghe H, Dal Cin P, Fletcher JA, Sklar J: Frequent fusion of the JAZF1 and JJAZ1 genes in endometrial stromal tumors. Proc Natl Acad Sci USA. 2001, 98: 6348-53. 10.1073/pnas.101132598
Micci F, Panagopoulos I, Bjerkehagen B, Heim S: Consistent rearrangement of chromosomal band 6p21 with generation of fusion genes JAZF1/PHF1 and EPC1/PHF1 in endometrial stromal sarcoma. Cancer Res. 2006, 66: 107-12. 10.1158/0008-5472.CAN-05-2485
Panagopoulos I, Mertens F, Griffin CA: An endometrial stromal sarcoma cell line with the JAZF1/PHF1 chimera. Cancer Genet Cytogenet. 2008, 185: 74-7. 10.1016/j.cancergencyto.2008.04.020
Panagopoulos I, Micci F, Thorsen J, Gorunova L, Eibak AM, Bjerkehagen B, Davidson B, Heim S: Novel fusion of MYST/Esa1-associated factor 6 and PHF1 in endometrial stromal sarcoma. PLoS One. 2012, 7: e39354- 10.1371/journal.pone.0039354
Gil-Benso R, Lopez-Gines C, Navarro S, Carda C, Llombart-Bosch A: Endometrial stromal sarcomas: immunohistochemical, electron microscopical and cytogenetic findings in two cases. Virchows Arch. 1999, 434: 307-14. 10.1007/s004280050346
Halbwedl I, Ullmann R, Kremser ML, Man YG, Isadi-Moud N, Lax S, Denk H, Popper HH, Tavassoli FA, Moinfar F: Chromosomal alterations in low-grade endometrial stromal sarcoma and undifferentiated endometrial sarcoma as detected by comparative genomic hybridization. Gynecol Oncol. 2005, 97: 582-7. 10.1016/j.ygyno.2005.01.002
Lee CH, Ou WB, Marino-Enriquez A, Zhu M, Mayeda M, Wang Y, Guo X, Brunner AL, Amant F, French CA: 14-3-3 fusion oncogenes in high-grade endometrial stromal sarcoma. Proc Natl Acad Sci USA. 2012, 109: 929-34. 10.1073/pnas.1115528109
Kurihara S, Oda Y, Ohishi Y, Kaneki E, Kobayashi H, Wake N, Tsuneyoshi M: Coincident expression of beta-catenin and cyclin D1 in endometrial stromal tumors and related high-grade sarcomas. Mod Pathol. 2010, 23: 225-34. 10.1038/modpathol.2009.162
Lee CH, Ali RH, Rouzbahman M, Marino-Enriquez A, Zhu M, Guo X, Brunner AL, Chiang S, Leung S, Nelnyk N: Cyclin D1 as a Diagnostic Immunomarker for Endometrial Stromal Sarcoma with YWHAE-FAM22 Rearrangement. Am J Surg Pathol. 2012, 36: 1562-70. 10.1097/PAS.0b013e31825fa931
Lee CH, Marino-Enriquez A, Ou W, Zhu M, Ali RH, Chiang S, Amant F, Gilks CB, van de Rijn M, Oliva E: The clinicopathologic features of YWHAE-FAM22 endometrial stromal sarcomas: a histologically high-grade and clinically aggressive tumor. Am J Surg Pathol. 2012, 36: 641-53. 10.1097/PAS.0b013e31824a7b1a
Wang L, Felix JC, Lee JL, Tan PY, Tourgeman DE, O’Meara AT, Amezcua CA: The proto-oncogene c-kit is expressed in leiomyosarcomas of the uterus. Gynecol Oncol. 2003, 90: 402-6. 10.1016/S0090-8258(03)00274-9
Rushing RS, Shajahan S, Chendil D, Wilder JL, Pulliam J, Lee EY, Ueland FR, van Nagell JR, Ahmed MM, Lele SM: Uterine sarcomas express KIT protein but lack mutation(s) in exon 11 or 17 of c-KIT. Gynecol Oncol. 2003, 91: 9-14. 10.1016/S0090-8258(03)00442-6
Klein WM, Kurman RJ: Lack of expression of c-kit protein (CD117) in mesenchymal tumors of the uterus and ovary. Int J Gynecol Pathol. 2003, 22: 181-4. 10.1097/00004347-200304000-00011
Leath CA, Straughn JM, Conner MG, Barnes MN, Alvarez RD, Partridge EE, Huh WK: Immunohistochemical evaluation of the c-kit proto-oncogene in sarcomas of the uterus: a case series. J Reprod Med. 2004, 49: 71-5.
Geller MA, Argenta P, Bradley W, Dusenbery KE, Brooker D, Downs LS, Judson PL, Carson LF, Boente MP: Treatment and recurrence patterns in endometrial stromal sarcomas and the relation to c-kit expression. Gynecol Oncol. 2004, 95: 632-6. 10.1016/j.ygyno.2004.07.049
Zafrakas M, Theodoridis TD, Zepiridis L, Venizelos ID, Agorastos T, Bontis J: KIT protein expression in uterine sarcomas: an immunohistochemical study and review of the literature. Eur J Gynaecol Oncol. 2008, 29: 264-6.
Martin J, Ramesh A, Kuruvilla S, Lalitha D: Uterine C-Kit positive low grade stromal sarcoma. Indian J Med Paediatr Oncol. 2009, 30: 113-5. 10.4103/0971-5851.64258
Koivisto-Korander R, Butzow R, Koivisto AM, Leminen A: Immunohistochemical studies on uterine carcinosarcoma, leiomyosarcoma, and endometrial stromal sarcoma: expression and prognostic importance of ten different markers. Tumour Biol. 2011, 32: 451-9. 10.1007/s13277-010-0138-1
Cheng X, Yang G, Schmeler KM, Coleman RL, Tu X, Liu J, Kavanagh JJ: Recurrence patterns and prognosis of endometrial stromal sarcoma and the potential of tyrosine kinase-inhibiting therapy. Gynecol Oncol. 2011, 121: 323-7. 10.1016/j.ygyno.2010.12.360
Caudell JJ, Deavers MT, Slomovitz BM, Lu KH, Broaddus RR, Gershenson DM, Ramondetta LM: Imatinib mesylate (gleevec)–targeted kinases are expressed in uterine sarcomas. Appl Immunohistochem Mol Morphol. 2005, 13: 167-70. 10.1097/01.pai.0000129057.38941.a1
Salvatierra A, Tarrats A, Gomez C, Sastre JM, Balana C: A case of c-kit positive high-grade stromal endometrial sarcoma responding to Imatinib Mesylate. Gynecol Oncol. 2006, 101: 545-7. 10.1016/j.ygyno.2006.01.024
Trojan A, Montemurro M, Kamel M, Kristiansen G: Successful PDGFR-{alpha}/{beta} targeting with imatinib in uterine sarcoma. Ann Oncol. 2009, 20: 1898-9. 10.1093/annonc/mdp431
Liegl B, Gully C, Reich O, Nogales FF, Beham A, Regauer S: Expression of platelet-derived growth factor receptor in low-grade endometrial stromal sarcomas in the absence of activating mutations. Histopathology. 2007, 50: 448-52. 10.1111/j.1365-2559.2007.02634.x
Adams SF, Hickson JA, Hutto JY, Montag AG, Lengyel E, Yamada SD: PDGFR-alpha as a potential therapeutic target in uterine sarcomas. Gynecol Oncol. 2007, 104: 524-8. 10.1016/j.ygyno.2006.09.013
Moinfar F, Gogg-Kamerer M, Sommersacher A, Regitnig P, Man YG, Zatloukal K, Denk H, Tavassoli FA: Endometrial stromal sarcomas frequently express epidermal growth factor receptor (EGFR, HER-1): potential basis for a new therapeutic approach. Am J Surg Pathol. 2005, 29: 485-9. 10.1097/01.pas.0000155149.83541.24
Murray S, Linardou H, Mountzios G, Manoloukos M, Markaki S, Eleutherakis-Papaiakovou E, Dimopoulos MA, Papadimitriou CA: Low frequency of somatic mutations in uterine sarcomas: a molecular analysis and review of the literature. Mutat Res. 2010, 686: 68-73. 10.1016/j.mrfmmm.2010.01.019
Mitsuhashi T, Nakayama M, Sakurai S, Fujimura M, Shimizu Y, Ban S, Ogawa F, Hirose T, Ishihara O, Shimizu M: KIT-negative undifferentiated endometrial sarcoma with the amplified epidermal growth factor receptor gene showing a temporary response to imatinib mesylate. Ann Diagn Pathol. 2007, 11: 49-54. 10.1016/j.anndiagpath.2006.03.012
Kalender ME, Sevinc A, Yilmaz M, Ozsarac C, Camci C: Detection of complete response to imatinib mesylate (Glivec/Gleevec) with 18 F-FDG PET/CT for low-grade endometrial stromal sarcoma. Cancer Chemother Pharmacol. 2009, 63: 555-9. 10.1007/s00280-008-0786-7
Parsons M, Grabsch H: How to make tissue microarrays. Diagn Histopathol. 2009, 15: 142-50. 10.1016/j.mpdhp.2009.01.010.
Agoff SN, Grieco VS, Garcia R, Gown AM: Immunohistochemical distinction of endometrial stromal sarcoma and cellular leiomyoma. Appl Immunohistochem Mol Morphol. 2001, 9: 164-9. 10.1097/00022744-200106000-00009
Zhu XQ, Shi YF, Cheng XD, Zhao CL, Wu YZ: Immunohistochemical markers in differential diagnosis of endometrial stromal sarcoma and cellular leiomyoma. Gynecol Oncol. 2004, 92: 71-9. 10.1016/j.ygyno.2003.08.038
de Alava E, Ocana A, Abad M, Montero JC, Esparis-Ogando A, Rodriguez CA, Otero AP, Hernandez T, Cruz JJ, Pandiella A: Neuregulin expression modulates clinical response to trastuzumab in patients with metastatic breast cancer. J Clin Oncol. 2007, 25: 2656-63. 10.1200/JCO.2006.08.6850
Corless CL, McGreevey L, Haley A, Town A, Heinrich MC: KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol. 2002, 160: 1567-72. 10.1016/S0002-9440(10)61103-0
Lasota J, Wozniak A, Sarlomo-Rikala M, Rys J, Kordek R, Nassar A, Sobin LH, Miettinen M: Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. A study of 200 cases. Am J Pathol. 2000, 157: 1091-5. 10.1016/S0002-9440(10)64623-8
Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A: PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003, 299: 708-10. 10.1126/science.1079666
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ: EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004, 304: 1497-500. 10.1126/science.1099314
Takano T, Ohe Y, Sakamoto H, Tsuta K, Matsuno Y, Tateishi U, Yamamoto S, Nokihara H, Yamamoto N, Sekine I: Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005, 23: 6829-37. 10.1200/JCO.2005.01.0793
van Zandwijk N, Mathy A, Boerrigter L, Ruijter H, Tielen I, de Jong D, Baas P, Burgers S, Nederlof P: EGFR and KRAS mutations as criteria for treatment with tyrosine kinase inhibitors: retro- and prospective observations in non-small-cell lung cancer. Ann Oncol. 2007, 18: 99-103.
Jang TW, Oak CH, Chang HK, Suo SJ, Jung MH: EGFR and KRAS mutations in patients with adenocarcinoma of the lung. Korean J Intern Med. 2009, 24: 48-54. 10.3904/kjim.2009.24.1.48
Varella-Garcia M: Stratification of non-small cell lung cancer patients for therapy with epidermal growth factor receptor inhibitors: the EGFR fluorescence in situ hybridization assay. Diagn Pathol. 2006, 1: 19- 10.1186/1746-1596-1-19
Tanner EJ, Garg K, Leitao MM, Soslow R, Hensley ML: High grade undifferentiated uterine sarcoma: Surgery, treatment, and survival outcomes. Gynecol Oncol. 2012, 127: 27-31. 10.1016/j.ygyno.2012.06.030
Pink D, Lindner T, Mrozek A, Kretzschmar A, Thuss-Patience PC, Dorken B, Reichardt P: Harm or benefit of hormonal treatment in metastatic low-grade endometrial stromal sarcoma: single center experience with 10 cases and review of the literature. Gynecol Oncol. 2006, 101: 464-9. 10.1016/j.ygyno.2005.11.010
Maluf FC, Sabbatini P, Schwartz L, Xia J, Aghajanian C: Endometrial stromal sarcoma: objective response to letrozole. Gynecol Oncol. 2001, 82: 384-8. 10.1006/gyno.2001.6238
Sylvestre VT, Dunton CJ: Treatment of recurrent endometrial stromal sarcoma with letrozole: a case report and literature review. Horm Cancer. 2010, 1: 112-5. 10.1007/s12672-010-0007-9
Spano JP, Soria JC, Kambouchner M, Piperno-Neuman S, Morin F, Morere JF, Martin A, Breau JL: Long-term survival of patients given hormonal therapy for metastatic endometrial stromal sarcoma. Med Oncol. 2003, 20: 87-93. 10.1385/MO:20:1:87
Leunen M, Breugelmans M, De Sutter P, Bourgain C, Amy JJ: Low-grade endometrial stromal sarcoma treated with the aromatase inhibitor letrozole. Gynecol Oncol. 2004, 95: 769-71. 10.1016/j.ygyno.2004.07.063
Altman AD, Nelson GS, Chu P, Nation J, Ghatage P: Uterine sarcoma and aromatase inhibitors: tom baker cancer centre experience and review of the literature. Int J Gynecol Cancer. 2012, 22: 1006-12. 10.1097/IGC.0b013e31825b7de8
Leiser AL, Hamid AM, Blanchard R: Recurrence of prolactin-producing endometrial stromal sarcoma with sex-cord stromal component treated with progestin and aromatase inhibitor. Gynecol Oncol. 2004, 94: 567-71. 10.1016/j.ygyno.2004.03.025
Krauss K, Bachmann C, Hartmann JT, Siegmann K, Sotlar K, Wallwiener D, Huober J: Management of late recurrence of a low-grade endometrial stromal sarcoma (LGESS): treatment with letrozole. Anticancer Res. 2007, 27: 3477-80.
Klaritsch P, Reich O, Regauer S, Bauernhofer T: Recurrent endometrial stromal sarcoma after treatment with high-dose chemotherapy and autologous stem-cell support: A case report. Eur J Gynaecol Oncol. 2006, 27: 297-8.
Leyvraz S, Zweifel M, Jundt G, Lissoni A, Cerny T, Sessa C, Fey M, Dietrich D, Honegger HP: Long-term results of a multicenter SAKK trial on high-dose ifosfamide and doxorubicin in advanced or metastatic gynecologic sarcomas. Ann Oncol. 2006, 17: 646-51. 10.1093/annonc/mdl020
Sutton G, Blessing JA, Park R, DiSaia PJ, Rosenshein N: Ifosfamide treatment of recurrent or metastatic endometrial stromal sarcomas previously unexposed to chemotherapy: a study of the gynecologic oncology group. Obstet Gynecol. 1996, 87: 747-50.
Yamawaki T, Shimizu Y, Hasumi K: Treatment of stage IV “high-grade” endometrial stromal sarcoma with ifosfamide, adriamycin, and cisplatin. Gynecol Oncol. 1997, 64: 265-9. 10.1006/gyno.1996.4537
Numa F, Umayahara K, Ogata H, Nawata S, Sakaguchi Y, Emoto T, Kawasaki K, Hirakawa H, Sase M, Oga A, Kato H: De novo uterine sarcoma with good response to neo-adjuvant chemotherapy. Int J Gynecol Cancer. 2003, 13: 364-7.
Szlosarek PW, Lofts FJ, Pettengell R, Carter P, Young M, Harmer C: Effective treatment of a patient with a high-grade endometrial stromal sarcoma with an accelerated regimen of carboplatin and paclitaxel. Anticancer Drugs. 2000, 11: 275-8. 10.1097/00001813-200004000-00008
Valduvieco I, Rovirosa A, Colomo L, De San Juan A, Pahisa J, Biete A: Endometrial stromal sarcoma. Is there a place for radiotherapy?. Clin Transl Oncol. 2010, 12: 226-30. 10.1007/s12094-010-0493-9
Weitmann HD, Knocke TH, Kucera H, Potter R: Radiation therapy in the treatment of endometrial stromal sarcoma. Int J Radiat Oncol Biol Phys. 2001, 49: 739-48. 10.1016/S0360-3016(00)01369-9
Gadducci A, Sartori E, Landoni F, Zola P, Maggino T, Urgesi A, Lissoni A, Losa G, Fanucchi A: Endometrial stromal sarcoma: analysis of treatment failures and survival. Gynecol Oncol. 1996, 63: 247-53. 10.1006/gyno.1996.0314
Ferrer F, Sabater S, Farrus B, Guedea F, Rovirosa A, Anglada L, Delannes M, Marin S, DuBois JB, Daly-Schveitzer N: Impact of radiotherapy on local control and survival in uterine sarcomas: a retrospective study from the grup oncologic catala-occita. Int J Radiat Oncol Biol Phys. 1999, 44: 47-52. 10.1016/S0360-3016(98)00515-X
Went PT, Dirnhofer S, Bundi M, Mirlacher M, Schraml P, Mangialaio S, Dimitrijevic S, Kononen J, Lugli A, Simon R, Sauter G: Prevalence of KIT expression in human tumors. J Clin Oncol. 2004, 22: 4514-22. 10.1200/JCO.2004.10.125
Nakayama M, Mitsuhashi T, Shimizu Y, Ban S, Ogawa F, Ishihara O, Shimizu M: Immunohistochemical evaluation of KIT expression in sarcomas of the gynecologic region. Int J Gynecol Pathol. 2006, 25: 70-6. 10.1097/01.pgp.0000183047.45459.36
Cossu-Rocca P, Contini M, Uras MG, Muroni MR, Pili F, Carru C, Bosincu L, Massarelli G, Nogales FF, De Miglio MR: Tyrosine kinase receptor status in endometrial stromal sarcoma: an immunohistochemical and genetic-molecular analysis. Int J Gynecol Pathol. 2012, 31: 570-579. 10.1097/PGP.0b013e31824fe289
Park JY, Kim KR, Nam JH: Immunohistochemical analysis for therapeutic targets and prognostic markers in low-grade endometrial stromal sarcoma. Int J Gynecol Cancer. 2013, 23: 81-9. 10.1097/IGC.0b013e3182738361
Oliva E, Young RH, Amin MB, Clement PB: An immunohistochemical analysis of endometrial stromal and smooth muscle tumors of the uterus: a study of 54 cases emphasizing the importance of using a panel because of overlap in immunoreactivity for individual antibodies. Am J Surg Pathol. 2002, 26: 403-12. 10.1097/00000478-200204000-00001
Hornick JL, Fletcher CD: Immunohistochemical staining for KIT (CD117) in soft tissue sarcomas is very limited in distribution. Am J Clin Pathol. 2002, 117: 188-93. 10.1309/LX9U-F7P0-UWDH-8Y6R
Winter WE, Seidman JD, Krivak TC, Chauhan S, Carlson JW, Rose GS, Birrer MJ: Clinicopathological analysis of c-kit expression in carcinosarcomas and leiomyosarcomas of the uterine corpus. Gynecol Oncol. 2003, 91: 3-8. 10.1016/j.ygyno.2003.06.001
Inoue M, Kyo S, Fujita M, Enomoto T, Kondoh G: Coexpression of the c-kit receptor and the stem cell factor in gynecological tumors. Cancer Res. 1994, 54: 3049-53.
Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin CH, Westermark B, Nister M: Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992, 52: 3213-9.
Kang HJ, Nam SW, Kim H, Rhee H, Kim NG, Hyung WJ, Noh SH, Kim JH, Yun CO, Liu ET: Correlation of KIT and platelet-derived growth factor receptor alpha mutations with gene activation and expression profiles in gastrointestinal stromal tumors. Oncogene. 2005, 24: 1066-74. 10.1038/sj.onc.1208358
Burger H, den Bakker MA, Kros JM, van Tol H, de Bruin AM, Oosterhuis W, van den Ingh HF, van der Harst E, de Schipper HP, Wiemer EA, Nooter K: Activating mutations in c-KIT and PDGFRalpha are exclusively found in gastrointestinal stromal tumors and not in other tumors overexpressing these imatinib mesylate target genes. Cancer Biol Ther. 2005, 4: 1270-4. 10.4161/cbt.4.11.2253
Schilder RJ, Sill MW, Lee RB, Shaw TJ, Senterman MK, Klein-Szanto AJ, Miner Z, Vanderhyden BC: Phase II evaluation of imatinib mesylate in the treatment of recurrent or persistent epithelial ovarian or primary peritoneal carcinoma: a gynecologic oncology group study. J Clin Oncol. 2008, 26: 3418-25. 10.1200/JCO.2007.14.3420
Krug LM, Crapanzano JP, Azzoli CG, Miller VA, Rizvi N, Gomez J, Kris MG, Pizzo B, Tyson L, Dunne M, Heelan RT: Imatinib mesylate lacks activity in small cell lung carcinoma expressing c-kit protein: a phase II clinical trial. Cancer. 2005, 103: 2128-31. 10.1002/cncr.21000
Huh WK, Sill MW, Darcy KM, Elias KM, Hoffman JS, Boggess JF, Alvarez RD, Long HJ, O’Malley DM, Birrer MJ: Efficacy and safety of imatinib mesylate (gleevec) and immunohistochemical expression of c-Kit and PDGFR-beta in a gynecologic oncology group phase Il trial in women with recurrent or persistent carcinosarcomas of the uterus. Gynecol Oncol. 2010, 117: 248-54. 10.1016/j.ygyno.2010.01.002
Serrano C, Mackintosh C, Herrero D, Martins AS, de Alava E, Hernandez T, Perez-Fontan J, Abad M, Perez A, Serrano E: Imatinib is not a potential alternative treatment for uterine leiomyosarcoma. Clin Cancer Res. 2005, 11: 4977-9. author reply 79–80, 10.1158/1078-0432.CCR-04-2461
Kim GK: Status report on the management of dermatofibrosarcoma protuberans: is there a viable role for the use of imatinib mesylate? In which cases may it be therapeutically helpful and in which cases not?. J Clin Aesthet Dermatol. 2011, 4: 17-26.
Greco A, Roccato E, Miranda C, Cleris L, Formelli F, Pierotti MA: Growth-inhibitory effect of STI571 on cells transformed by the COL1A1/PDGFB rearrangement. Int J Cancer. 2001, 92: 354-60. 10.1002/ijc.1190
Sjoblom T, Shimizu A, O’Brien KP, Pietras K, Dal Cin P, Buchdunger E, Dumanski JP, Ostman A, Heldin CH: Growth inhibition of dermatofibrosarcoma protuberans tumors by the platelet-derived growth factor receptor antagonist STI571 through induction of apoptosis. Cancer Res. 2001, 61: 5778-83.
Llombart B, Sanmartin O, Lopez-Guerrero JA, Monteagudo C, Serra C, Requena C, Poveda A, Vistos JL, Almenar S, Llombart-Bosch A, Guillen C: Dermatofibrosarcoma protuberans: clinical, pathological, and genetic (COL1A1-PDGFB ) study with therapeutic implications. Histopathology. 2009, 54: 860-72. 10.1111/j.1365-2559.2009.03310.x
Maki RG, Awan RA, Dixon RH, Jhanwar S, Antonescu CR: Differential sensitivity to imatinib of 2 patients with metastatic sarcoma arising from dermatofibrosarcoma protuberans. Int J Cancer. 2002, 100: 623-6. 10.1002/ijc.10535
Rubin BP, Schuetze SM, Eary JF, Norwood TH, Mirza S, Conrad EU, Bruckner JD: Molecular targeting of platelet-derived growth factor B by imatinib mesylate in a patient with metastatic dermatofibrosarcoma protuberans. J Clin Oncol. 2002, 20: 3586-91. 10.1200/JCO.2002.01.027
Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, Haney J, Witta S, Danenberg K, Domenichini I: Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005, 97: 643-55. 10.1093/jnci/dji112
Martin V, Mazzucchelli L, Frattini M: An overview of the epidermal growth factor receptor fluorescence in situ hybridisation challenge in tumour pathology. J Clin Pathol. 2009, 62: 314-24. 10.1136/jcp.2008.059592
Acknowledgements
We would like to thank Professor Russell Alpizar-Jara (Department of Mathematics, University of Évora, Portugal) for his statistical advice.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Support for this study was obtained from Novartis.
Authors’ contributions
RS carried out the molecular genetic studies, performed the sequence alignment and data analyses and drafted the manuscript. TH and SF carried out the TMA construction and immunohistochemistry assays. TH carried out fluorescent hybridization assay and participated in the sequence alignment. FT, AV, MCG, AA, NH, JS, JO, LG and RR provided the samples for the study. CB and EA conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Carmen Balañá and Enrique de Álava contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sardinha, R., Hernández, T., Fraile, S. et al. Endometrial stromal tumors: immunohistochemical and molecular analysis of potential targets of tyrosine kinase inhibitors. Clin Sarcoma Res 3, 3 (2013). https://doi.org/10.1186/2045-3329-3-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2045-3329-3-3