
Abstract
Background
microRNA regulation plays an important role in the remodeling that occurs in response to pathologic and physiologic stimuli in skeletal muscle. In response to stress, microRNAs are dynamically regulated, resulting in a widespread “fine-tuning” of gene expression. An understanding of this dynamic regulation is critical to targeting future therapeutic strategies. Experiments elucidating this dynamic regulation have typically relied on in vitro reporter assays, ex vivo sample analysis, and transgenic mouse studies. Surprisingly, no experimental method to date allows rapid in vivo analysis of microRNA activity in mammals.
Methods
To improve microRNA studies we have developed a novel reporter assay for the measurement of skeletal muscle microRNA activity in vivo. To minimize muscle damage, hydrodynamic limb vein injection was used for the introduction of plasmid DNA encoding bioluminescent and fluorescent reporters, including click-beetle luciferase and the far-red fluorescent protein mKATE. We then applied this technique to the measurement of miR-206 activity in dystrophic mdx4cv animals.
Results
We found that hydrodynamic limb vein injection is minimally damaging to myofibers, and as a result no induction of muscle-specific miR-206 (indicative of an injury response) was detected. Unlike intramuscular injection or electroporation, we found that hydrodynamic limb vein injection results in dispersed reporter expression across multiple hindlimb muscle groups. Additionally, by utilizing click-beetle luciferase from Pyrophorus plagiophthalamus as a reporter and the far-red fluorescent protein mKATE for normalization, we show as a proof of principle that we can detect elevated miR-206 activity in mdx4cv animals when compared to C57Bl/6 controls.
Conclusion
Hydrodynamic limb vein injection of plasmid DNA followed by in vivo bioluminescent imaging is a novel assay for the detection of reporter activity in skeletal muscle in vivo. We believe that this method will allow for the rapid and precise detection of both transcriptional and post-transcriptional regulation of gene expression in response to skeletal muscle stress. Additionally, given the post-mitotic status of myofibers and stable expression of plasmid DNA, we believe this method will reduce biological variability in animal studies by allowing longitudinal studies of the same animal cohort.
Similar content being viewed by others


Background
Skeletal muscle displays a remarkable ability to remodel in response to pathologic and physiologic stimuli ([1–3] and reviewed in [4]). How changes in gene expression mediate these processes by post-transcriptional regulation has been heavily studied in the context of diseased, hypertrophic, and regenerating skeletal muscle [5–7]. While recent studies have greatly enhanced our understanding of these dynamic processes, measuring post-transcriptional changes that occur in vivo is challenging and this has limited the experimental avenues available. Additionally, the discovery of post-transcriptional regulation by microRNAs (miRNAs) in skeletal muscle has increased the complexity of these processes.
miRNAs are a type of short, non-coding RNA that post-transcriptionally regulate gene expression. miRNAs function by guiding the RNA-induced silencing complex (RISC) to target mRNAs, dictated by base pairing to target sites primarily in their 3′-untranslated regions (UTRs). miRNAs can downregulate the expression of several to hundreds of target genes by repressing translation and/or destabilizing target mRNAs [8, 9]. miRNAs are required for normal skeletal muscle development in mice [10], and several miRNAs have been shown to be dynamically regulated during hypertrophy [11], acute exercise [12], regeneration after injury [13], and in the remodeling that occurs in response to genetic muscle disease [14–17]. In particular, miR-206 is expressed specifically in skeletal muscle, and is highly expressed in regenerating fibers [13]. miR-206 has been shown to promote terminal differentiation of myoblasts by regulating the expression of genes including connexin43 [18], utrophin [19], pax3 [20], pax7 [21] and DNA polymerase α [22]. Many of these studies rely on data obtained from transgenic mouse studies, and ex vivo sample analysis - both currently invaluable to the study of miRNAs. The study of miRNA regulation in skeletal muscle, however, would benefit from a system that enabled rapid, reproducible, longitudinal in vivo reporter assays, at a fraction of the cost of transgenic mouse production and analysis, and with fewer animals needed than for ex vivo studies.
Several studies have shown that stable gene expression can be achieved in post-mitotic myofibers in vivo by the introduction of plasmid DNA (pDNA) [23–26]. Unlike viral DNA vectors, pDNA is non-immunogenic [23], easily manipulated, and relatively inexpensive to produce. Methods for introducing naked pDNA into myofibers include intramuscular injection [24], electroporation [26, 27], and more recently, hydrodynamic limb vein (HLV) injection [28, 29]. While these methods all result in efficient myofiber transduction, only HLV injection is minimally damaging to the muscle [30], enabling studies of muscle remodeling to take place in the absence of widespread myofiber regeneration. Moreover, HLV injection results in more widespread pDNA distribution than intramuscular injection or electroporation, which are limited to the site of injection, or single muscle groups, respectively [24, 26].
In vivo bioluminescent and fluorescent imaging (BLI) of genetically encoded reporters has been a useful tool in murine xenograft studies including cancer [31], chondrogenic differentiation [32], viral infection [33], and even in studies of miRNA biogenesis and post-transcriptional regulation [34, 35]. All of these studies, however, have relied on the expression of reporter genes in cells cultured in vitro, and none have demonstrated the ability to quantify skeletal muscle reporter gene expression in situ. We therefore developed a novel system using HLV injection of reporter pDNA into skeletal muscle in combination with in vivo BLI in order to quantify miRNA activity. Before testing, we anticipated that animal-to-animal variation in injection efficiency would confound true changes in reporter expression. While several groups have reported the utility of Renilla luciferases as a normalizer in in vivo reporter studies [32, 36], we found that the fast kinetics and requirement for intravenous substrate delivery made this approach technically challenging. To solve this problem, we employed a dual-reporter approach consisting of a high-efficiency click-beetle luciferase from Pyrophorus plagiophthalamus (CBG99) [37], and the far-red fluorescent protein mKATE [38] for signal normalization. mKATE has been shown to have a maximum emission wavelength (635 nm) that is optimal for tissue penetration, and to have high pH and photostability, making this an ideal protein for in vivo BLI.
As a proof of principle, we measured miR-206 activity and show that the activity measured using this technique reproduces the levels obtained from quantitative reverse transcription PCR (qRT-PCR) measurements. We believe this technique will prove useful not only in the quantification of real-time miRNA activity, but also in studies of transcriptional and post-transcriptional regulation.
Methods
Plasmid construction
Two perfectly complementary miR-206 [NCBI:NR_029593] binding sites were inserted between the Xba1 and Fse1 restriction sites downstream of the CBG99 stop codon in pCBG99-Control (Promega, Madison, WI, USA) using the following oligonucleotide sequences: 5′-CTAGACCACACACTTCCTTACATTCCAAAACCACACACTTCCTTACATTCCAGGCCGG, and 5′-CCTGGAATGTAAGGAAGTGTGTGGTTTTGGAATGTAAGGAAGT GTGTGGT. pcDNA-mKATE was a kind gift from Amy Palmer (University of Colorado at Boulder, Boulder, CO, USA), and pCMV-eGFP from Stephen Langer (University of Colorado at Boulder, Boulder, CO, USA).
Hydrodynamic limb vein injection
All animal experiments were performed using protocols approved by University of Colorado and Colorado State University Institutional Animal Care and Use Committees (IACUC). Mice were injected according to a protocol modified from Hagstrom, et al. [29]. Briefly, after sedation with 1 to 4% inhaled isoflurane, a tourniquet was secured around the upper hindlimb to restrict blood flow for 1 to 2 minutes prior to injection, and 2 minutes after injection. The hindlimb was first cleaned with 70% ethanol, and an incision was made with surgical scissors on the medial surface of the leg to expose the great saphenous vein. A ½ inch 30-gauge needle connected by catheter to a syringe was then inserted in an anterograde direction into the vein. Endotoxin-free pDNA isolated using Endofree® Plasmid Maxi Kit (Qiagen, Hilden, Germany) and diluted in sterile saline solution (volume determined according to the formula: 1 + (((body weight in grams − 25)/25) × 1/2)mL), was delivered at a rate of 7 mL/minute by a programmable syringe pump (KD Scientific, Holliston, MA, USA). Two minutes after the injection was completed, the tourniquet was released and the incision was closed with nonabsorbable 6–0 silk suture (Davis-Geck, Brooklyn, NY, USA). Mice recovered on a 37°C heat block and were monitored for adverse effects.
Barium chloride injury
To induce muscle degeneration, mice were first anesthetized with 1 to 4% inhaled isoflurane, and the right hindlimb was shaved. After cleaning the area with 70% ethanol, the right gastrocnemius was injected with 50 μL of a 1.2% barium chloride solution in normal saline using a 27-gauge insulin syringe. Mice recovered on a 37°C heat block and were monitored for adverse effects.
In vivo bioluminescent and fluorescent imaging
Animals were anesthetized using 1 to 4% isoflurane prior to imaging and placed in the chamber of an IVIS 100 in vivo imaging system (Caliper Biosciences, Hopkinton, MA, USA) housed at Colorado State University’s Animal Cancer Center (Fort Collins, CO, USA). The mKATE fluorescent signal was then collected using sequential mode with a 1-s exposure time and the Cy5.5 excitation/emission filter set. Animals were then removed from the imaging chamber and injected intraperitoneally with 200 μL of 30 mg/mL D-luciferin. Ten minutes after substrate injection, bioluminescent signal was collected for 1 minute using the open filter mode. Images were analyzed using LivingImage software (Caliper Biosciences) and photons/sec/cm2/steradian were quantified using region-of-interest (ROI) analysis. Mice recovered from anesthesia on a 37°C heat block.
Immunofluorescence
Muscles for immunofluorescence were frozen in liquid nitrogen-cooled isopentane and mounted in optimal cutting temperature (OCT) medium (Sakura Finetek, Torrance, CA, USA). Cryosections (12 μm thick) were fixed in 4% paraformaldehyde for 10 minutes at room temperature, then blocked for 1 hour with 5% goat serum in PBS and 0.1% Triton X-100. Following blocking, sections were stained with anti-laminin at 1:500 (L-9393, Sigma, St. Louis, MO, USA) or anti-desmin at 1:20 (D-8281, Sigma) overnight at 4°C. After washing several times in PBS with 0.1% Triton X-100, anti-rabbit Texas red secondary antibody was applied at 1:100 dilution for 1 hour at 37°C (711-075-152, Jackson, West Grove, PA, USA). After washing again in PBS with 0.1% Triton X-100, sections were counterstained with 300 nM 4′,6-diamidino-2-phenylindole (DAPI) for 5 minutes at room temperature (Sigma D-9542), and mounted using Fluoromount G (Southern Biotech, Birmingham, AL, USA).
Quantification of injection efficiency
To determine HLV injection efficiency, percentages of GFP-positive fibers were quantified using immunofluorescence on lower hindlimb sections from pCMV-eGFP-injected animals. Following imaging on an inverted epifluorescent microscope (Eclipse TE2000, Nikon, Melville, NY, USA), GFP, laminin, and DAPI images were merged to produce a composite (ImageJ software, National Institutes of Health, Bethesda, MD, USA). GFP-positive fibers were counted when GFP signal across a myofiber was greater than background levels. Total fiber number was counted using laminin staining to demarcate fiber boundaries. Percentages are reported as an average of GFP-positive fibers for ten fields of view for the gastrocnemius, and five fields each for the soleus and tibialis anterior (TA).
qRT-PCR
To measure miRNA expression, total RNA was first isolated from snap-frozen skeletal muscles using TRI reagent (Molecular Research Center, Cincinnati, OH, USA). Then, 7 ng of total RNA per reaction was reverse-transcribed and PCR-amplified on a CFX96 thermocycler (Bio-Rad, Hercules, CA, USA) using Taqman miRNA assays (Invitrogen, Grand Island, NY, USA). Relative miRNA expression was determined using the 2-ΔΔCt method [39], using sno202 as a reference gene.
Statistical analysis
For qRT-PCR experiments, relative miR-206 expression values were compared to controls (for assessment of muscle damage, controls were uninjured/uninjected contralateral limbs for each time point, and for miR-206 reporter measurements, controls were C57Bl/6 animals) using the unpaired Student’s t- test. For BLI measurements, statistics were performed by comparing mean normalized photons/s/cm2/steradian (CBG99/mKATE) of pCBG99-2x-miR-206-injected hindlimbs, to mean normalized photons/s/cm2/steradian of pCBG99-Control-injected hindlimbs for mdx4cv and C57Bl/6 animals using the unpaired Student’s t-test.
Results
HLV injection does not induce myofiber regeneration
Prior to employing HLV to deliver miRNA reporters for miR-206 activity, it was important to determine whether the injection technique itself would induce muscle regeneration and consequently increase the levels of miR-206. To test this, we performed HLV injection (Figure 1A) on wild-type C57Bl/6 mice using saline only and compared miR-206 expression to that in animals injured with BaCl2. miR-206 levels decreased slightly (P =0.02) on the third day after saline injection, likely due to residual edema from the high-volume injection, but remained unchanged (day 7 animals trended towards a 1.7-fold increase, P = 0.051) on subsequent days (Figure 1B). Conversely, barium chloride injury strongly induced miR-206 approximately 11-fold (P = 0.0003) as previously reported [13], suggesting that HLV injection does not activate a widespread program of muscle regeneration.

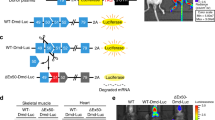
miR-206 levels after hydrodynamic limb vein injection. (A) Schematic depiction of hydrodynamic limb vein injection (HLV) of plasmid DNA followed by in vivo bioluminescent and fluorescent imaging (BLI). (B) miR-206 expression in 3 to 4 month-old C57Bl/6 mice receiving either HLV injection of saline solution or BaCl2 injury, sacrificed at the indicated time points, normalized to sno202. Relative levels measured in the right (treated) gastrocnemius are displayed as mean values normalized to contralateral controls; n= 3 or 4 animals/group, Error bars = standard error of the mean. *P ≤ 0.05, **P ≤ 0.001.
Reporter expression is well distributed and is quantifiable using in vivo imaging
To test the muscle distribution of reporter pDNA, we next injected C57Bl/6 mice with 100 μg/animal of pCMV-eGFP. After 7 days, we performed immunofluorescence on fixed cryosections and found that GFP expression was visible in the sarcoplasm of myofibers in the gastrocnemius, soleus, and TA muscles (Figure 2). To determine injection efficiency, we quantified GFP-positive myofibers and found that 7.7%, 15.9% and 6.1% of fibers in the soleus, gastrocnemius, and TA, respectively, expressed the reporter. We also observed an absence of centrally located nuclei in GFP-positive myofibers, further supporting the finding that HLV injection itself causes minimal muscle injury. Next, we tested whether reporter gene expression can be quantified from both bioluminescent and fluorescent reporters using in vivo imaging. To this end, we injected either 100 μg each of pCBG99-Luc-Control and pcDNA-mKATE, or pcDNA-mKATE alone into the right hindlimbs of C57Bl/6 mice and collected images 7 days later using BLI. The signal was easily visible and localized to the hindlimb skeletal muscle for both reporters, although mKATE background signal was frequently observed from the ventilation nosepiece and/or the distal parts of the hindlimb (Figure 3). We also found that mKATE fluorescence did not bleed into the luciferase channel, making it ideal for in vivo use in combination with CBG99-luciferase.

Reporter distribution. Reporter expression after hydrodynamic limb vein injection. Left column: green fluorescent protein (GFP) epifluorescent signal displayed as inverted grayscale image, percentages are an average of GFP-positive fibers for indicated muscles. Right column: immunostaining for laminin (red) and epifluorescence for GFP (green) and 4′,6-diamidino-2-phenylindole (DAPI) (blue) to show distribution of GFP-positive myofibers in indicated muscles; n=1 animal. Scale bars = 100 μm. TA, tibialis anterior.

Reporter detection using in vivo bioluminescent imaging. Mice were injected with pcDNA-mKATE (top) or pcDNA-mKATE and pCBG99-Luc-Control (bottom). Data collected using Cy5.5 excitation/emission filters is shown in the left column, and after D-luciferin injection in the right column.
miRNA activity measurements are consistent with miRNA qRT-PCR measurements
In order to measure miRNA activity in vivo, we inserted two perfectly complementary miRNA binding sites downstream of the CBG99 luciferase coding sequence, creating pCBG99-2x-miR-206. We next co-injected 100 μg of pcDNA-mKATE along with either pCBG99-2x-miR-206 (left hindlimbs) or pCBG99-Luc-Control (right hindlimbs) into C57Bl/6 or dystrophic mdx4cv mice. Seven days later, we measured the signal using in vivo BLI followed by ROI analysis (Figure 4B). To ensure that we minimized the effect of differential plasmid distribution and injection efficiency, the same region size was used for quantification in all animals, and the luciferase signal was normalized to mKATE. After normalizing, the CBG99:mKATE ratio from left hindlimbs was compared to control right hindlimbs. In agreement with a 5-fold increase in miR-206 expression in mdx4cv animals by qRT-PCR (P = 0.0001) (Figure 4A), the normalized bioluminescent signal in the left hindlimbs of mdx4cv mice was reduced 3.3-fold (P = 0.02) (Figure 2C). While the decreased average signal in C57Bl/6 did not reach significance (P = 0.15), the downward trend is likely due to the high abundance of miR-206 in skeletal muscle, and the decreased miR-206 reporter signal in mdx4cv animals is due to increased miR-206 expression. Given that miR-206 is highly expressed in the regenerating fibers of dystrophic mice [13], this result suggests that pDNA injected using HLV is also expressed in these fibers. To test this, we injected mdx4cv animals with 100 μg of pCMV-eGFP and collected tissues for immunofluorescence 7 days later. As expected, we found GFP expression in small, desmin-positive, regenerating myofibers with centrally located nuclei (Figure 5, arrowheads), indicating that the decrease in luciferase signal measured using BLI likely corresponds to a loss of luciferase activity in these fibers.

Quantification of miR-206 reporter activity in mdx4cv mice. (A) miR-206 expression measured in the gastrocnemius muscles of 3 month-old mdx4cv animals in comparison to C57Bl/6. miR-206 expression is normalized to sno202 ( n = 4 animals/group), mean values are displayed; error bars = standard error of the mean (SEM), *P ≤0.05. (B) Bioluminescent imaging of mdx4cv animals showing CBG99 luciferase and mKATE expression. Left hindlimbs were co-injected with pCBG99-2x-miR-206 and pcDNA-mKATE, and right hindlimbs were co-injected with pCBG99-Control and pcDNA-mKATE. (C) Region of interest (ROI) analysis of (B). The same region size was used for all animals (n= 4 animals/group; mean values are displayed; error bars = SEM, *P≤ 0.05.

Reporter expression in regenerating mdx4cv fibers. pCMV-eGFP-injected mdx4cv (left column) and C57Bl/6 (right column) gastrocnemius sections showing desmin immunostaining (red), green fluorescent protein (GFP) epifluorescence (green), and 4′,6-diamidino-2-phenylindole (DAPI) (blue) demonstrate GFP expression in regenerating fibers. Upper row images were taken using 10× objective lens, lower row with 20× objective. Scale bars = 100 μm.
Discussion
Several groups have reported the safety and efficacy of pDNA injections using HLV for gene therapy approaches [23, 28, 29]. We found this technique also to be useful in studies requiring minimally damaging introduction of genetically encoded reporters into skeletal muscle and have used it in combination with BLI and ROI analysis to analyze Myh7b promoter activity in vivo[40]. We also found that this technique induces minimal muscle regeneration as assessed by miR-206 induction, and to our knowledge this is the first report of using an in vivo transfection method that does not induce myofiber regeneration for luciferase reporter studies.
The distribution of GFP-positive myofibers after HLV injection that we have seen is similar to that reported by Wooddell, et al. [41] although the percentage of transfected fibers we report is lower. This may be due to decreased sensitivity of detecting a fluorescent reporter (GFP) versus a colorimetric stain (β-galactosidase). Similar to this report, we find the highest percentage (15.9%) of transfected myofibers in the posterior lower leg (gastrocnemius) muscles and the lowest percentage (6.1%) in the anterior lower leg (TA) muscles (Figure 3). Additionally, others have observed reporter gene expression up to 49 weeks after HLV injection [41], suggesting that this method could be used for longer-term regulatory studies, provided that the luciferase and mKATE signals follow similar expression profiles.
Conclusion
In summary, HLV injection of pDNA reporters into skeletal muscle followed by BLI is a useful technique for studies of gene regulation. Here, we show that it is possible to measure miRNA activity in myofibers in situ, without activation of a regeneration response. This technique has the added benefit of reducing the cost associated with producing transgenic animals for skeletal muscle studies, and will likely allow reduced animal numbers and decreased variability in future experiments by enabling longitudinal studies of the same animal cohort.
Abbreviations
- BLI:
-
Bioluminescent and fluorescent imaging
- DAPI:
-
4′,6-diamidino-2-phenylindole
- GFP:
-
Green fluorescent protein
- HLV:
-
hydrodynamic limb vein
- miRNA:
-
microRNA
- OCT:
-
Optimum cutting temperature
- PBS:
-
Phosphate-buffered saline
- pDNA:
-
Plasmid DNA
- qRT-PCR:
-
Quantitative reverse transcription polymerase chain reaction
- RISC:
-
RNA-induced silencing complex
- ROI:
-
Region of interest
- UTR:
-
Untranslated region
- TA:
-
Tibialis anterior.
References
Porter JD, Khanna S, Kaminski HJ, Rao JS, Merriam AP, Richmonds CR, Leahy P, Li J, Guo W, Andrade FH: A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum Mol Genet. 2002, 11: 263-272. 10.1093/hmg/11.3.263.
Haddad F, Herrick RE, Adams GR, Baldwin KM: Myosin heavy chain expression in rodent skeletal muscle: effects of exposure to zero gravity. J Appl Physiol. 1993, 75: 2471-2477.
Daugaard JR, Nielsen JN, Kristiansen S, Andersen JL, Hargreaves M, Richter EA: Fiber type-specific expression of GLUT4 in human skeletal muscle: influence of exercise training. Diabetes. 2000, 49: 1092-1095. 10.2337/diabetes.49.7.1092.
Bassel-Duby R, Olson EN: Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006, 75: 19-37. 10.1146/annurev.biochem.75.103004.142622.
Farina NH, Hausburg M, Dalla Betta N, Pulliam C, Srivastava D, Cornelison DD, Olwin BB: A role for RNA post-transcriptional regulation in satellite cell activation. Skeletal Muscle. 2012, 2: 1-10.1186/2044-5040-2-1.
Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barceló J, O’Hoy K: Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992, 255: 1253-1255. 10.1126/science.1546325.
Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I: A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011, 147: 358-369. 10.1016/j.cell.2011.09.028.
Djuranovic S, Nahvi A, Green R: MiRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science. 2012, 336: 237-240. 10.1126/science.1215691.
Bazzini AA, Lee MT, Giraldez AJ: Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science. 2012, 336: 233-237. 10.1126/science.1215704.
O’Rourke JR, Georges SA, Seay HR, Tapscott SJ, McManus MT, Goldhamer DJ, Swanson MS, Harfe BD: Essential role for Dicer during skeletal muscle development. Dev Biol. 2007, 311: 359-368. 10.1016/j.ydbio.2007.08.032.
McCarthy JJ, Esser KA: MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J Appl Physiol. 2007, 102: 306-313.
Safdar A, Abadi A, Akhtar M, Hettinga BP, Tarnopolsky MA, Lucia A: miRNA in the regulation of skeletal muscle adaptation to acute endurance exercise in C57Bl/6J male mice. PLoS One. 2009, 4: e5610-e5610. 10.1371/journal.pone.0005610.
Yuasa K, Hagiwara Y, Ando M, Nakamura A, Takeda S, Hijikata T: MicroRNA-206 is highly expressed in newly formed muscle fibers: implications regarding potential for muscle regeneration and maturation in muscular dystrophy. Cell Struct Funct. 2008, 33: 163-169. 10.1247/csf.08022.
McCarthy JJ, Esser KA, Andrade FH: MicroRNA-206 is overexpressed in the diaphragm but not the hindlimb muscle of mdx mouse. Am J Physiol Cell Physiol. 2007, 293: C451-C457. 10.1152/ajpcell.00077.2007.
Liu N, Williams AH, Maxeiner JM, Bezprozvannaya S, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN: microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J Clin Invest. 2012, 122: 2054-2065. 10.1172/JCI62656.
Eisenberg I, Eran A, Nishino I, Moggio M, Lamperti C, Amato AA, Lidov HG, Kang PB, North KN, Mitrani-Rosenbaum S, Flanigan KM, Neely LA, Whitney D, Beggs AH, Kohane IS, Kunkel LM: Distinctive patterns of microRNA expression in primary muscular disorders. Proc Natl Acad Sci USA. 2007, 104: 17016-17021. 10.1073/pnas.0708115104.
Gambardella S, Rinaldi F, Lepore SM, Viola A, Loro E, Angelini C, Vergani L, Novelli G, Botta A: Overexpression of microRNA-206 in the skeletal muscle from myotonic dystrophy type 1 patients. J Transl Med. 2010, 8: 48-10.1186/1479-5876-8-48.
Anderson C, Catoe H, Werner R: MIR-206 regulates connexin43 expression during skeletal muscle development. Nucleic Acids Res. 2006, 34: 5863-5871. 10.1093/nar/gkl743.
Rosenberg MI, Georges SA, Asawachaicharn A, Analau E, Tapscott SJ: MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J Cell Biol. 2006, 175: 77-85. 10.1083/jcb.200603039.
Boutet SC, Cheung TH, Quach NL, Liu L, Prescott SL, Edalati A, Iori K, Rando TA: Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem Cell. 2012, 10: 327-336. 10.1016/j.stem.2012.01.017.
Dey BK, Gagan J, Dutta A: MiR-206 and −486 induce myoblast differentiation by downregulating Pax7. Mol Cell Biol. 2010, 31: 1-46.
Kim HK, Lee YS, Sivaprasad U, Malhotra A, Dutta A: Muscle-specific microRNA miR-206 promotes muscle differentiation. J Cell Biol. 2006, 174: 677-687. 10.1083/jcb.200603008.
Sebestyen MG, Hegge JO, Noble M, Lewis D, Herweijer H, Wolff JA: Progress toward a non-viral gene therapy protocol for the treatment of anemia. Hum Gene Ther. 2007, 18: 269-285. 10.1089/hum.2006.186.
Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, Felgner PL: Direct gene transfer into mouse muscle in vivo. Science. 1990, 247: 1465-1468. 10.1126/science.1690918.
Fabre EE, Bigey P, Orsini C, Scherman D: Comparison of promoter region constructs for in vivo intramuscular expression. J Gene Med. 2006, 8: 636-645. 10.1002/jgm.878.
Aihara H, Miyazaki J-I: Gene transfer into muscle by electroporation in vivo. Nat Biotechnol. 1998, 16: 867-870. 10.1038/nbt0998-867.
Deleuze V, Scherman D, Bureau MF: Interleukin-10 expression after intramuscular DNA electrotransfer: kinetic studies. Biochem Biophys Res Commun. 2002, 299: 29-34. 10.1016/S0006-291X(02)02580-9.
Suda T, Liu D: Hydrodynamic gene delivery: its principles and applications. Mol Ther. 2007, 15: 2063-2069. 10.1038/sj.mt.6300314.
Hagstrom JE, Hegge J, Zhang G, Noble M, Budker V, Lewis DL, Herweijer H, Wolff JA: A facile nonviral method for delivering genes and siRNAs to skeletal muscle of mammalian limbs. Mol Ther. 2004, 10: 386-398. 10.1016/j.ymthe.2004.05.004.
Toumi H, Hegge J, Subbotin V, Noble M, Herweijer H, Best TM, Hagstrom JE: Rapid intravascular injection into limb skeletal muscle: a damage assessment study. Mol Ther. 2006, 13: 229-236. 10.1016/j.ymthe.2005.07.699.
Kim HJ, Chung J-K, Hwang DW, Lee DS, Kim S: In vivo imaging of miR-221 biogenesis in papillary thyroid carcinoma. Mol Imaging Biol. 2009, 11: 71-78. 10.1007/s11307-008-0188-6.
Vilalta M, Jorgensen C, Degano IR, Chernajovsky Y, Gould D, Noel D, Andrades JA, Becerra J, Rubio N, Blanco J: Dual luciferase labelling for non-invasive bioluminescence imaging of mesenchymal stromal cell chondrogenic differentiation in demineralized bone matrix scaffolds. Biomaterials. 2009, 30: 4986-4995. 10.1016/j.biomaterials.2009.05.056.
Luker KEK, Luker GDG: Applications of bioluminescence imaging to antiviral research and therapy: multiple luciferase enzymes and quantitation. Antiviral Res. 2008, 78: 9-9. 10.1016/j.antiviral.2008.01.004.
Lee JY, Kim S, Hwang DW, Jeong JM, Chung J-K, Lee MC, Lee DS: Development of a dual-luciferase reporter system for in vivo visualization of MicroRNA biogenesis and posttranscriptional regulation. J Nucl Med. 2008, 49: 285-294. 10.2967/jnumed.107.042507.
Kim S, Hwang DW, Lee DS: A study of microRNAsin silicoandin vivo: bioimaging of microRNA biogenesis and regulation. FEBS J. 2009, 276: 2165-2174. 10.1111/j.1742-4658.2009.06935.x.
Bhaumik S, Gambhir SS: Optical imaging of Renilla luciferase reporter gene expression in living mice. Proc Natl Acad Sci USA. 2002, 99: 377-382. 10.1073/pnas.012611099.
Miloud T, Henrich C, Hämmerling GJ: Quantitative comparison of click beetle and firefly luciferases for in vivo bioluminescence imaging. J Biomed Opt. 2007, 12: 054018-10.1117/1.2800386.
Shcherbo D, Merzlyak EM, Chepurnykh TV, Fradkov AF, Ermakova GV, Solovieva EA, Lukyanov KA, Bogdanova EA, Zaraisky AG, Lukyanov S, Chudakov DM: Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 2007, 4: 741-746. 10.1038/nmeth1083.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001, 25: 402-408. 10.1006/meth.2001.1262.
Yeung F, Chung E, Guess MG, Bell ML, Leinwand LA: Myh7b/miR-499 gene expression is transcriptionally regulated by MRFs and Eos. Nucleic Acids Res. 2012, 40: 7303-7318. 10.1093/nar/gks466.
Wooddell CI, Hegge JO, Zhang G, Sebestyén MG, Noble M, Griffin JB, Pfannes LV, Herweijer H, Hagstrom JE, Braun S, Huss T, Wolff JA: Dose response in rodents and nonhuman primates after hydrodynamic limb vein delivery of naked plasmid DNA. Hum Gene Ther. 2011, 22: 889-903. 10.1089/hum.2010.160.
Acknowledgements
We would like to thank Mark Noble for assistance with HLV injection, Adam Chicco for assistance with animal protocols, and Margaret Isenhart for animal care. This work was funded by NIH R01GM029090 to LL and NIH 5T32GM007135-36 to MG.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors have no competing interests to declare.
Authors’ contributions
MG designed the experiments, wrote the paper, and carried out the qRT-PCR, injections, imaging, and molecular cloning. KB played a key role in the project’s conception and provided useful discussion and critical reading of the manuscript. EP provided assistance with in vivo BLI and assembling figures. LL provided mentoring and discussion about all experiments and data. All authors have read and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Guess, M.G., Barthel, K.K., Pugach, E.K. et al. Measuring microRNA reporter activity in skeletal muscle using hydrodynamic limb vein injection of plasmid DNA combined with in vivo imaging. Skeletal Muscle 3, 19 (2013). https://doi.org/10.1186/2044-5040-3-19
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2044-5040-3-19