
Abstract
Background
Hutchinson-Gilford progeria syndrome is a rare dominant human disease of genetic origin. The average life expectancy is about 20 years, patients’ life quality is still very poor and no efficient therapy has yet been developed. It is caused by mutation of the LMNA gene, which results in accumulation in the nuclear membrane of a particular splicing form of Lamin-A called progerin. The mechanism by which progerin perturbs cellular homeostasis and leads to the symptoms is still under debate.
Micro-RNAs are able to negatively regulate transcription by coupling with the 3’ UnTranslated Region of messenger RNAs. Several Micro-RNAs recognize the same 3’ UnTranslated Region and each Micro-RNA can recognize multiple 3’ UnTranslated Regions of different messenger RNAs. When different messenger RNAs are co-regulated via a similar panel of micro-RNAs, these messengers are called Competing Endogenous RNAs, or ceRNAs.
The 3’ UnTranslated Region of the longest LMNA transcript was analysed looking for its ceRNAs. The aim of this study was to search for candidate genes and gene ontology functions possibly influenced by LMNA mutations that may exert a role in progeria development.
Results
11 miRNAs were isolated as potential LMNA regulators. By computational analysis, the miRNAs pointed to 17 putative LMNA ceRNAs. Gene ontology analysis of isolated ceRNAs showed an enrichment in RNA interference and control of cell cycle functions.
Conclusion
This study isolated novel genes and functions potentially involved in LMNA network of regulation that could be involved in laminopathies such as the Hutchinson-Gilford progeria syndrome.
Similar content being viewed by others


Background
Lamins are intermediate filament proteins associated with the inner nuclear membrane and are structural components of the nuclear lamina. Interestingly, they can also be found in the nucleoplasm, where they might have regulatory functions that are still poorly investigated [1–3]. Lamins are structural components of the nuclear membrane, but they are also essential for many nuclear functions [1, 3]. Lamins can bind to specific DNA sequences, chromatin modifications, and chromatin associated proteins or complexes either directly or through lamin-interacting proteins [1–4]. It has been reported that lamin functions are involved in transcriptional regulation, DNA replication and repair, epigenetic modifications, chromatin remodelling, and transition between euchromatin and heterochromatin conformation [1, 3, 4]. Lamins are present in almost all pluricellular organisms, with the exception of plants, and are usually absent in unicellular organisms [5, 6]. Generally, lamins are divided into types A and B. In humans, A-type lamins are divided into A and C lamins, both derived by alternative splicing from the LMNA gene [5, 6]. Interestingly, in humans, stem cells and undifferentiated cells seem to lack Lamin-A and Lamin-C. In this perspective, LMNA expressed lamins behave as markers of differentiation [7].
The Hutchinson-Gilford progeria Syndrome (HGPS) is a very rare human disease of genetic origin that leads to very severe premature ageing. HGPS is caused by several mutations in the LMNA gene, the most common of which is the point mutation C1824T, which leads to the accumulation in the nuclear membrane of a rare splicing form of the Lamin-A called “progerin”, and alterations in nuclear shape and structure like the typical nuclear bubbling, the cytological hallmark of HGPS [8]. The accumulation of progerin is due to the impossibility of physiological cleavage of the mature wild type Lamin-A protein. Usually, Lamin-A is farnesylated and incorporated into the nuclear membrane, and later is cleaved and released from the nuclear lamina. The classical mutation in HGPS enhances the activity of a cryptic splicing site that increases the production of progerin and lessens the production of Lamin-A [8, 9], acting as a dominant mutation. Progerin lacks the cleavage site, so the protein is farnesylated and loaded into the membrane but cannot be removed efficiently any more, so it accumulates [8, 10]. It is to be noted that progerin physiologically accumulates in the cells of ageing individuals, with a positive correlation with chronological age [9–11]. HGPS affected individuals have a life expectancy of about twenty years and a very poor quality of life [12]. No efficient healing therapy has yet been developed and the main focus of current pharmacological strategies is on farnesylation inhibitors that lessen the progerin load in the nuclear membrane [10]. Though an amelioration of the symptoms has been reported, farnesylation inhibitors have not led to a definitive solution [10]. The mechanism by which progerin accumulation perturbs cellular homeostasis and leads to the symptoms is still under debate [10]. The aim of this study was to look for candidate genes and gene ontology functions influenced by LMNA mutations that in turn may have a role in progeria development.
The ceRNA (competing endogenous RNAs) hypothesis is based on the rationale that RNA molecules can regulate one another via microRNAs (miRNAs or miRs) and that messengers RNAs (mRNAs) can be positively co-regulated if they share miRNA target sequences amongst their 3’UnTranslated regions (3’UTR), because there is a limited amount of miRNAs within each cell, and each mRNA can act as a quencher for shared miRNAs [13]. Following this rationale, genes whose mRNAs share miRNAs targets in their 3’UTRs might be post-transcriptionally co-regulated. For a more exhaustive description of ceRNA rationale see [2, 13]. The study reported on here follows another study [2] on LMNA interactome. This study focuses on an analysis of the Lamin-A ceRNAs network of interactions.
Methods
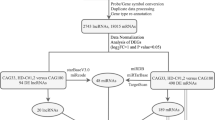
Using the miRWalk [14] database for predicted gene targets, miRNAs of a minimum of 7 matching nucleotides on the longest human LMNA transcript 3’UTR with a maximum p value of 0.05 were isolated. The settings chosen were the standard settings for the software used [14]. The 3’UTR analysed is the same in Lamin-A and progerin transcripts; the 3’ UTR of Lamin-C is shorter and different, and not included in this study. The work was performed on predicted gene targets because there are no validated targets reported for LMNA transcripts in the miRWalk database. The miRNAs considered as putatively recognizing the 3’UTR of the LMNA mRNA were 11 and reported in Table 1. Table 1 also shows a mimiRNA analysis [15] of the compared expression profiles of LMNA and each miRNA in human tissues and cell lines collected in the database. The set of miRNAs in Table 1 was inserted into the miRWalk [14] MicroRNA validated targets analysing tool to discover any human gene mRNA 3’UTR that has been reported to have been recognized by any of them. The genes isolated and the related bait miRNAs are shown in Table 2. The genes collected were organized in a hierarchical order for the number of validated microRNA hits (Table 3). The more microRNAs are shared between the 3’UTR of the longest LMNA transcript and the 3’UTRs of the candidate genes, the higher the possibility that the gene transcripts can act as LMNA ceRNAs. The maximum number of hits is 5. Genes that share in their transcripts 3 to 5 validated microRNAs with the predicted microRNAs with the 3’UTR of the longest LMNA transcript were arbitrarily considered as potential ceRNAs for further analyses. 17 genes have these characteristics out of a total of 335. These 17 genes were analysed using the GeneMANIA [16] tool that helps to predict the functions of a set of genes and to predict in which Gene Ontology (GO) functions the set of genes might be involved. The results are reported in Table 4. Table 4 also shows the GO functions from the ones with the lowest False Discovery Rate (FDR) till a FDR < 0.1. All analyses were updated to September 13th 2012.
Results and discussion
A ceRNA analysis might reveal genes that are co-regulated post-transcriptionally and that can be functionally correlated [13]. The 3’UTR of the longest transcript of the LMNA human gene was selected for the analysis because the vast majority of miRNAs recognize and regulate the efficiency of transcription by binding to this portion of messenger RNAs [13]. Moreover, the 3’UTR of the longest transcript is shared between Lamin-A and progerin splicing form, and it is different form the shorter 3’UTR of the Lamin-C transcript, not used in this study. The miRWalk database and tools [14] were used to perform the analysis. The database reported no validated miRNAs that bind to the 3’UTR of the main transcript of the LMNA gene. By contrast, in this study, predicted miRNAs able to bind to the 3’UTR of the main transcript of the LMNA gene were used. The predicted miRNAs are shown in Table 1.
Although the database does not report any validated miRNAs, it is to be noted that miR-9, one of the predicted miRNAs isolated, has recently been validated [17].
Amongst the isolated miRNAs worth noting is miR-214, clearly involved in cancer and stemness [18, 19]; miR-9, involved in neurogenesis [20] and, very interestingly, protective in neural cells of HGPS patients against the effects of progeria [21]; miR-298, involved in the regulation of beta-amyloid precursor protein converting enzyme messenger RNA translation, and thus in Alzheimer’s disease [22]; the tumour suppressor miR-34a [23]; and miR-449a involved in differentiation [24] and pRB mediated cell cycle arrest [25].
The correlation of the expression levels of LMNA and each miRNA was analysed using the mimiRNA software [15], which allows one to compare the expression profiles reported in different human tissues and cell lines. The data are reported in Table 1. One should expect a negative correlation between a miRNA and its regulated genes. Only 9 of the 11 miRNAs isolated could be analysed using the mimiRNA software; and amongst them 6 out of 9 showed a negative correlation, as expected, with the exception of miR-214, miR-24a and miR-532-3p.
The miRNAs in Table 1 were used as baits to find any human gene whose 3’ UTRs are recognized by them as literature validated targets, again using the miRWalk database and tools [14]. 670 couples of bait miRNA-gene were recovered. They are reported in Table 2.
Following the ceRNA hypothesis, the more miRNA were shared between the transcripts 3’UTRs, the stronger their interaction could be. The data reported in Table were organized on the basis of the quantity of miRNA reported in Table 1 able to hit the transcripts from each gene. Multiple hits from the same miRNA on the same gene were weighted as unique. Genes were then organized from the most hit to the less hit.
The results shown in Table 3 show only one gene transcript hit 5 times (DICER1); 4 gene transcripts hit 4 times (CDKN1A, NFKB1, TP53, VEGFA); 12 gene transcripts hit 3 times (APC, BCL2, CD44, CDC25A, CDK6, EIF2C1, EIF2C2, HDAC9, IL1B, KRAS, MYC, RNASEN); 51 gene transcripts hit 2 times; and 267 gene transcripts hit only once.
Arbitrarily, further analyses were performed focusing on genes whose transcripts were hit from 3 to 5 times, because they more likely constitute ceRNAs for the LMNA transcripts. This choice was made because the higher the number of shared miRNAs, the higher the possibility that the discovered genes could act as ceRNAs. Using genes that are hit by few miRNAs could raise the risk of picking up false positives. Unfortunately, no standardization can be used on the basis of previously published data. The cut-off point was chosen taking into account ceRNAs recognized by more than 50% of miRNAs of the most hit genes: specifically genes hit by more than 2.5 miRNAs, that is 3 to 5 miRNAs, as stated. Of course this arbitrary decision could have affected the subsequent analyses, increasing the risk of false negatives, but a highly stringent strategy was preferred, taking the number of total putative ceRNAs from 422 to 17. In the literature the analysis of these 17 genes strikingly highlights a possible relationship between LMNA gene functions and RNA interference machinery: DICER1, EIF2C1, EIF2C2 and RNASEN code respectively for Dicer, Argonaute1, Argonaute2 and Drosha, the key effectors of the RNA interference machinery [26]. Interestingly, there are no data reporting a relationship between Lamin-A and RNA interference machinery. Another network involved in LMNA functions is p53 and the control of cell cycle: TP53 coding for p53 itself, while CDKN1A, CDC25A and CDK6 code respectively for p21, the phosphatase CDC25A and the cyclin-dependent protein kinase 6, differently expressed during the cell cycle and involved in bypassing the G1/S checkpoint. p53 directly regulates p21 [27] and CDC25A [28] which indirectly regulates the cyclin-dependent protein kinase 6 [29]. Notably, the expression of TP53, APC, BCL2, KRAS and MYC is very often altered in several types of human cancer [30–34]. Interestingly, clear involvement of LMNA mutations in the control of cell cycle and in cancer has been reported [35]. NFKB1, IL1B and VEGFA products are involved in inflammation and cancer-related inflammation and angiogenesis [36–38]. It is worth noting that coronary artery atherosclerosis typically leads to the death of HGPS patients [39] and that LMNA mutations have been linked to inflammatory myopathy [40]. Notably, it has been suggested that the splicing factor SRSF1 may have a role in driving alternative splicing forms of both LMNA and VEGFA during the senescence of endothelial cells [41].
HDAC9 codes for a histone deacetylase, a class of the epigenetic covalent modifiers, involved in the risk of ischemic stroke [42]. It has been reported that Lamin-A/C deficiency causes chromosome condensation that can be reversed by histone acetylase inhibitors [43].
CD44 codes for a surface marker of hematopoietic stem and progenitor cells and is also involved in cancer [44].
With the aim of analysing the 17 putative ceRNAs isolated, the GeneMANIA [16] tool was used. It helps to predict the functions of a set of genes and to predict in which Gene Ontology (GO) functions they might be involved. The results are reported in Table 4 from the most statistically significant to the less, up to a False Discovery Rate (FDR) < 0.1. Interestingly, posttranscriptional gene silencing by RNA, ageing, cellular senescence, G1/S transition of mitotic cell cycle and angiogenesis are all statistically significant, amongst many other GO functions.
It is to be noted that in a previous study [2] the authors used the predicted miRNAs that hit the 3’UTR of the longest transcript of the LMNA gene to find any human gene whose 3’ UTRs are recognized by them as predicted targets, not literature validated ones as in the present study, again using the miRWalk database and tools [14]. Moreover, in that study the GeneMANIA [16] tool was also used together with the Osprey visualization tool [45] of the BioGRID database [46]. The result of that study highlighted a central role of RB1 and HTATIP genes, possible involvement in prostate cancer, and a possible role of LMNA in epigenetic modification, especially acetylation events, and ATP dependent chromatin remodelling via the chromatin remodelling complexes PBAF and SWI/SNF. Taken together, these studies suggest a possible role of LMNA gene products in the control of cell cycle and tumorigenesis, chromatin remodelling, epigenetic modifications, especially acetylation via HTATIP/HDAC9, and a close link with the RNA interference machinery.
Conclusion
In HGPS there is accumulation in the nuclear membrane of the Lamin-A rare splicing form called progerin. How the accumulation of progerin leads to progeria is still under debate. The aim of this article was to investigate the network of interactions of LMNA gene to find candidate genes and gene ontology functions involved in Lamin-A functions and in turn possibly perturbed in progeria. To search for possible partners of the LMNA gene involved in HGPS and normal ageing, bioinformatics analyses of the network of interactions of the Lamin-A/progerin were performed looking for LMNA ceRNAs using Lamin-A/progerin 3’UTR as a bait. The analysis might be quite useful because it allows one to isolate possible interactors for the gene being examined and to discover novel functions in which it might be involved.
LMNA is mutated in several human diseases of genetic origin with very variable phenotypes, suggesting a multiple role of LMNA gene products in cell homeostasis and functions [47, 48]. There have been reported laminopathies due to LMNA mutations such as Emery-Dreifuss muscular dystrophy, familial partial lipodystrophy, limb girdle muscular dystrophy, dilated cardiomyopathy, Charcot-Marie-Tooth disease, and Hutchinson-Gilford progeria syndrome, which is the main focus of this study. This study might help to understand why different mutations of a single gene can induce many different phenotypes in affected patients. The wide range of functions possibly involved in the LMNA network of interactions can give a plausible explanation of the many phenotypes due to LMNA mutations in humans.
The possible involvement of LMNA in the network of regulation of key cellular functions such as control of the cell cycle, epigenetics and chromatin remodelling, and RNA interference highlights a central role of Lamins in the nuclear functions. Further studies are required to better understand the relationship between LMNA products and the cellular components isolated, but a better understanding of the LMNA network might improve the quality of life of many patients affected by laminopathies. It might also enhance our comprehension of how the structural components and the functional components of the eukaryote nuclei interact.
Authors’ information
WA is a PostDoc at the Section of Endocrinology, Diabetology & Metabolism, Dipartimento Biomedico di Medicina Interna e Specialistica (Di.Bi.M.I.S.), University of Palermo, Italy. GP is a researcher at the Section of Endocrinology, Diabetology & Metabolism, Dipartimento Biomedico di Medicina Interna e Specialistica (Di.Bi.M.I.S.), University of Palermo, Italy. CG is a professor of endocrinology at the Section of Endocrinology, Diabetology & Metabolism, Dipartimento Biomedico di Medicina Interna e Specialistica (Di.Bi.M.I.S.), University of Palermo, Italy.
Abbreviations
- ceRNA:
-
Competing endogenous RNA
- FDR:
-
False Discovery Rate
- GO:
-
Gene Ontology
- HGPS:
-
Hutchinson-Gilford Progeria Syndrome
- miRNA:
-
Micro RNA
- miR:
-
Micro RNA
- mRNA:
-
Messenger RNA
- 3’UTR:
-
3’ UnTranslated Region.
References
Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD: Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22 (7): 832-853. 10.1101/gad.1652708.
Arancio W: A bioinformatics analysis of Lamin-A regulatory network: a perspective on epigenetic involvement in Hutchinson-Gilford progeria syndrome. Rejuvenation Res. 2012, 15 (2): 123-127. 10.1089/rej.2011.1250.
Shimi T, Pfleghaar K, Kojima S, Pack CG, Solovei I, Goldman AE, Adam SA, Shumaker DK, Kinjo M, Cremer T, Goldman RD: The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev. 2008, 22 (24): 3409-3421. 10.1101/gad.1735208.
Ottaviani A, Schluth-Bolard C, Rival-Gervier S, Boussouar A, Rondier D, Foerster AM, Morere J, Bauwens S, Gazzo S, Callet-Bauchu E, Gilson E, Magdinier F: Identification of a perinuclear positioning element in human subtelomeres that requires A-type lamins and CTCF. EMBO J. 2009, 28 (16): 2428-2436. 10.1038/emboj.2009.201.
Batsios P, Peter T, Baumann O, Stick R, Meyer I, Gräf R: A lamin in lower eukaryotes. Nucleus. 2012, 3 (3): 237-243.
Peter A, Reimer S: Evolution of the lamin protein family: what introns can tell. Nucleus. 2012, 3 (1): 44-59. 10.4161/nucl.18927.
Constantinescu D, Gray HL, Sammak PJ, Schatten GP, Csoka AB: Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells. 2006, 24 (1): 177-185. 10.1634/stemcells.2004-0159.
Gonzalez JM, Pla D, Perez-Sala D, Andres V: A-type lamins and Hutchinson-Gilford progeria syndrome: pathogenesis and therapy. Front Biosci (Schol Ed). 2011, 3: 1133-1146. Review
Lopez-Mejia IC, Vautrot V, De Toledo M, Behm-Ansmant I, Bourgeois CF, Navarro CL, Osorio FG, Freije JM, Stévenin J, De Sandre-Giovannoli A, Lopez-Otin C, Lévy N, Branlant C, Tazi J: A conserved splicing mechanism of the LMNA gene controls premature aging. Hum Mol Genet. 2011, 20 (23): 4540-4555. 10.1093/hmg/ddr385.
Reddy S, Comai L: Lamin A, farnesylation and aging. Exp Cell Res. 2012, 318 (1): 1-7. 10.1016/j.yexcr.2011.08.009. Review
McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS, Djabali K: The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS One. 2007, 2 (12): e1269-10.1371/journal.pone.0001269.
Ackerman J, Gilbert-Barness E: Hutchinson-Gilford progeria syndrome: a pathologic study. Pediatr Pathol Mol Med. 2002, 21 (1): 1-13.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP: A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language?. Cell. 2011, 146 (3): 353-358. 10.1016/j.cell.2011.07.014.
Dweep H, Sticht C, Pandey P, Gretz N: miRWalk - database: prediction of possible miRNA binding sites by "walking" the genes of 3 genomes. J Biomed Inform. 2011, 10.1016/ j.jbi.2011.05.002 (JBI PMID:21605702). http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/,
Ritchie W, Flamant S, Rasko JE: mimiRNA: a microRNA expression profiler and classification resource designed to identify functional correlations between microRNAs and their targets. Bioinformatics. 2010, 26 (2): 223-227. 10.1093/bioinformatics/btp649. Epub 2009 Nov 17. http://mimirna.centenary.org.au/mep/formulaire.html,
Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, Maitland A, Mostafavi S, Montojo J, Shao Q, Wright G, Bader GD, Morris Q: The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38 (Suppl): W214-220.
Jung HJ, Coffinier C, Choe Y, Beigneux AP, Davies BS, Yang SH, Barnes RH, Hong J, Sun T, Pleasure SJ, Young SG, Fong LG: Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc Natl Acad Sci USA. 2012, 109 (7): E423-431. 10.1073/pnas.1111780109.
Xia H, Ooi LL, Hui KM: MiR-214 Targets β-Catenin Pathway to Suppress Invasion, Stem-Like Traits and Recurrence of Human Hepatocellular Carcinoma. PLoS One. 2012, 7 (9): e44206-10.1371/journal.pone.0044206.
Xu CX, Xu M, Tan L, Yang H, Permuth-Wey J, Kruk PA, Wenham RM, Nicosia SV, Lancaster JM, Sellers TA, Cheng JQ: MiR-214 regulates ovarian cancer cell stemness by targeting p53/nanog. J Biol Chem. 2012, 287 (42): 34970-8. 10.1074/jbc.M112.374611.
Coolen M, Thieffry D, Drivenes Q, Becker TS, Bally Cuif L: miR-9 controls the timing of neurogenesis through the direct inhibition of antagonistic factors. Dev Cell. 2012, 22 (5): 1052-1064. 10.1016/j.devcel.2012.03.003.
Nissan X, Blondel S, Navarro C, Maury Y, Denis C, Girard M, Martinat C, De Sandre-Giovannoli A, Levy N, Peschanski M: Unique Preservation of Neural Cells in Hutchinson- Gilford Progeria Syndrome Is Due to the Expression of the Neural-Specific miR-9 MicroRNA. Cell Rep. 2012, 2 (1): 1-9. 10.1016/j.celrep.2012.05.015.
Provost P: Interpretation and applicability of microRNA data to the context of Alzheimer's and age-related diseases. Aging (Albany NY). 2010, 2 (3): 166-169.
Hermeking H: The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010, 17 (2): 193-199. 10.1038/cdd.2009.56. Epub 2009 May 22. Review
Paik S, Jung HS, Lee S, Yoon DS, Park MS, Lee JW: miR-449a Regulates the Chondrogenesis of Human Mesenchymal Stem Cells Through Direct Targeting of Lymphoid Enhancer-Binding Factor-1. Stem Cells Dev. 2012, 21 (18): 3298-308. 10.1089/scd.2011.0732.
Noonan EJ, Place RF, Basak S, Pookot D, Li LC: miR-449a causes Rb-dependent cell cycle arrest and senescence in prostate cancer cells. Oncotarget. 2010, 1 (5): 349-358.
Sashital DG, Doudna JA: Structural insights into RNA interference. Curr Opin Struct Biol. 2010, 20 (1): 90-97. 10.1016/j.sbi.2009.12.001.
Abbas T, Dutta A: p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009, 9 (6): 400-14. 10.1038/nrc2657.
Rother K, Kirschner R, Sänger K, Böhlig L, Mössner J, Engeland K: p53 downregulates expression of the G1/S cell cycle phosphatase Cdc25A. Oncogene. 2007, 26 (13): 1949-1953. 10.1038/sj.onc.1209989. Epub 2006 Sep 25
Grossel MJ, Hinds PW: From cell cycle to differentiation: an expanding role for cdk6. Cell Cycle. 2006, 5 (3): 266-270. 10.4161/cc.5.3.2385.
Pietsch EC, Humbey O, Murphy ME: Polymorphisms in the p53 pathway. Oncogene. 2006, 25 (11): 1602-1611. 10.1038/sj.onc.1209367. Review
Kwong LN, Dove WF: APC and its modifiers in colon cancer. Adv Exp Med Biol. 2009, 656: 85-106. 10.1007/978-1-4419-1145-2_8.
Frenzel A, Grespi F, Chmelewskij W, Villunger A: Bcl2 family proteins in carcinogenesis and the treatment of cancer. Apoptosis. 2009, 14 (4): 584-96. 10.1007/s10495-008-0300-z.
Jancík S, Drábek J, Radzioch D, Hajdúch M: Clinical relevance of KRAS in human cancers. J Biomed Biotechnol. 2010, 2010: 150960-
Dang CV: MYC on the path to cancer. Cell. 2012, 149 (1): 22-35. 10.1016/j.cell.2012.03.003.
Redwood AB, Gonzalez-Suarez I, Gonzalo S: Regulating the levels of key factors in cell cycle and DNA repair: new pathways revealed by lamins. Cell Cycle. 2011, 10 (21): 3652-3657. 10.4161/cc.10.21.18201.
Del Prete A, Allavena P, Santoro G, Fumarulo R, Corsi MM, Mantovani A: Molecular pathways in cancer-related inflammation. Biochem Med (Zagreb). 2011, 21 (3): 264-75.
Lopez-Castejon G, Brough D: Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011, 22 (4): 189-195. 10.1016/j.cytogfr.2011.10.001.
Stewart MW: Aflibercept (VEGF-TRAP): the next anti-VEGF drug. Inflamm Allergy Drug Targets. 2011, 10 (6): 497-508. 10.2174/187152811798104872.
Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, Nabel EG, Gordon LB: Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. 2010, 30 (11): 2301-2309. 10.1161/ATVBAHA.110.209460.
Komaki H, Hayashi YK, Tsuburaya R, Sugie K, Kato M, Nagai T, Imataka G, Suzuki S, Saitoh S, Asahina N, Honke K, Higuchi Y, Sakuma H, Saito Y, Nakagawa E, Sugai K, Sasaki M, Nonaka I, Nishino I: Inflammatory changes in infantile-onset LMNA-associated myopathy. Neuromuscul Disord. 2011, 21 (8): 563-568. 10.1016/j.nmd.2011.04.010.
Blanco FJ, Bernabéu C: The Splicing Factor SRSF1 as a Marker for Endothelial Senescence. Front Physiol. 2012, 3: 54-
Hacke W, Grond-Ginsbach C: Commentary on a GWAS: HDAC9 and the risk for ischaemic stroke. BMC Med. 2012, 10: 70-10.1186/1741-7015-10-70.
Galiová G, Bártová E, Raska I, Krejcí J, Kozubek S: Chromatin changes induced by lamin A/C deficiency and the histone deacetylase inhibitor trichostatin A. Eur J Cell Biol. 2008, 87 (5): 291-303. 10.1016/j.ejcb.2008.01.013. Epub 2008 Apr 8
Sackstein R: The biology of CD44 and HCELL in hematopoiesis: the 'step 2-bypass pathway' and other emerging perspectives. Curr Opin Hematol. 2011, 18 (4): 239-248. 10.1097/MOH.0b013e3283476140.
Breitkreutz BJ, Stark C, Tyers M: Osprey: A Network Visualization System. Genome Biol. 2003, 4 (3): R22-10.1186/gb-2003-4-3-r22. http://biodata.mshri.on.ca/osprey/servlet/Index,
Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M: Biogrid: A General Repository for Interaction Datasets. Nucleic Acids Res. 34: D535-539. http://thebiogrid.org/,
Bertrand AT, Chikhaoui K, Yaou RB, Bonne G: Clinical and genetic heterogeneity in laminopathies. Biochem Soc Trans. 2011, 39 (6): 1687-1692. 10.1042/BST20110670.
Genschel J, Schmidt HH: Mutations in the LMNA gene encoding lamin A/C. Hum Mutat. 2000, 16 (6): 451-9. 10.1002/1098-1004(200012)16:6<451::AID-HUMU1>3.0.CO;2-9.
Ackowledgements
Our thanks go to Dr. S.I. Genovese and D. Gailor for the help and advice given and to Giordano Bruno, Nikola Tesla and Barbara McClintock for their inspiring vision.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
WA conceived of the study, carried out the bioinformatics studies and drafted the manuscript. GP participated in the design of the study and helped to draft the manuscript. CG participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Arancio, W., Giordano, C. & Pizzolanti, G. A ceRNA analysis on LMNA gene focusing on the Hutchinson-Gilford progeria syndrome. J Clin Bioinform 3, 2 (2013). https://doi.org/10.1186/2043-9113-3-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2043-9113-3-2