
Abstract
Depression is a common mental disorder that co-occurs in other neurological and somatic diseases. Further, sex differences exist in the prevalence rates of many of these diseases, as well as within non-disease associated depression. In this review, the case is made for needing a better recognition of the source of the symptoms of depression with respect to the sex of the individual; in that, some disease states, which includes the neuroendocrine and immune reactions to the underlying pathophysiology of the disease, may initiate depressive symptoms more often in one sex over the other. The diseases specifically addressed to make this argument are: epilepsy, Alzheimer’s disease, cancer, and cardiovascular disease. For each of these conditions, a review of the following are presented: prevalence rates of the conditions within each sex, prevalence rates of depressive symptoms within the conditions, identified relationships to gonadal hormones, and possible interactions between gonadal hormones, adrenal hormones, and immune signaling. Conclusions are drawn suggesting that an evaluation of the root causes for depressive symptoms in patients with these conditions is necessary, as the underlying mechanisms for eliciting the depressive symptoms may be qualitatively different across the four diseases discussed. This review attempts to identify and understand the mechanisms of depression associated with these diseases, in the context of the known sex differences in the disease prevalence and its age of onset. Hence, more extensive, sex-specific model systems are warranted that utilize these disease states to elicit depressive symptoms in order to create more focused, efficient, and sex-specific treatments for patients suffering from these diseases and concurrent depressive symptoms.
Similar content being viewed by others

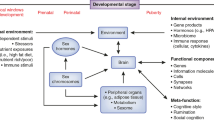
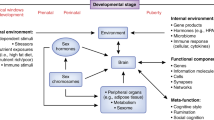
Review
Depression is the leading cause of disability in the United States for individuals 15–44 years of age [1]. In addition, depression is predictive of suicide [2], and suicide accounts for more deaths each year than auto accidents [3]. Despite the high incidence of depression and the increased risk of death in depressed individuals, our understanding of the causes of depression is incomplete, limiting the efficacy of available treatments. A more complete appreciation for the range of possible neural substrates that underlie depression may improve our understanding and thereby our treatment of the disorder. Although the phenotype of depression is somewhat consistent, the mechanisms that account for the manifestation of that phenotype are varied. By analogy, one could travel from North America to Australia by many different routes and by many different methods, but once the journey was complete, the view upon arrival would be the same. In the case of depression, the mechanisms that mediate depression in a young woman with a history of chronic abuse and a recent psychological trauma, are likely to differ from the mechanisms mediating depression in an elderly man who recently had a stroke but who has no history of trauma or abuse. While the mechanisms mediating the depression may differ, the core mental and behavioral symptoms for these two individuals could be nearly identical. Two of the most obvious trends that suggest that depression is not a singular entity are the pervasive sex difference in the incidence of depression and the interactions among depression and diseases.
Women are at a two-fold greater risk of developing depression compared to men [4, 5] and though it is well established that the prevalence, incidence, and morbidity risk for females is greater throughout life, the underlying mechanisms of this sex difference are relatively undefined. Women experience a steep increase in rates of depression beginning at mid-puberty [5, 6]; similarly, women experience fluctuations of sex steroids and corresponding vulnerabilities to mood disorders during and after pregnancy and again at menopause [4]. The pattern of hormonal change parallel to the biological susceptibility to the development of mood disorders in females paints a dramatic role for sex steroids in depression. Despite this relationship, other variables implicated in the pathogenesis of depression, including inflammation [7], socio-demographic demands [5], and a multi-factorial genetic predisposition [4], likely interact with sex steroids to influence specific disease manifestation across individuals. Further highlighting the role of sex steroids in disease, several common medical disorders are exacerbated by hormonal changes associated with the menstrual cycle [8], vulnerabilities that are thought to be related to estrogen’s interactions with both the immune and neuroendocrine systems.
Both neural (e.g. epilepsy [9] and Alzheimer’s disease [10]) and somatic disease (e.g. cancer [11] and cardiovascular disease [12]) are accompanied by a greater prevalence of comorbid mood disorders (see Table 1). The lifetime prevalence of depression among those diagnosed with epilepsy may be as high as 55% depending on the type and severity of epilepsy [9, 13]. Pre-existing depression among patients with new-onset epilepsy is up to seven times more common than in a control population (reviewed in Kanner [14]). Depressive episodes in life confer an increased risk of developing dementia or Alzheimer’s disease later in life, while depression is prevalent in 35-50% of patients already diagnosed with Alzheimer’s disease [10]. Among cancer patients, the prevalence of major depression can be greater than 40% depending on the method of diagnosis and type of cancer [11]. Similarly, the prevalence of major depression among cardiac patients is three-fold higher than that observed in the general population [12], and, indicating the bidirectionality of the comorbidity, patients with depression are at a greater risk for a cardiac event in a dose–response manner.
From a distance, the interaction between disease and depressive disorders confers no pattern of protection or susceptibility in terms of sex steroids; rather, the unique properties of the disease states in conjunction with sex steroids interact to modulate disease symptomology, progression, and mortality. In this review, we will discuss how sex differences observed within depressive disorders vary in the context of concomitant disease and explore how sex steroids may differentially modulate depression within disease.
Depression and neural disease
Epilepsy, sex, and depression
Epilepsy, or chronically recurring unprovoked cortical seizures, affects approximately two million Americans, and about 140,000 new cases of epilepsy are diagnosed in the United States each year [15]. Over two-thirds of epileptic seizures begin in childhood, and incidence levels in adulthood increase again beginning at age 60 [16]. According to a recent meta-analysis [17], females may have a slightly lower susceptibility to epilepsy than males (46.2 vs. 50.7 per 100,000), though the greater incidence in men may be, in part, attributed to the higher number of risk factors in men, including head injury and stroke. When only idiopathic seizures were considered, however, the sex difference remained [17].
The relationship between depression and epilepsy is not new; Hippocrates himself has been attributed with noting this bidirectionality: “Melancholics ordinarily become epileptics, and epileptics melancholics: what determines the preference is the direction the malady takes; if it bears upon the body, epilepsy, if upon the intelligence, melancholy” (translated and cited in Kanner, 2003 and Mula, 2012 [9, 18]). The lifetime prevalence of depression among those diagnosed with epilepsy is 6-55% depending on the type and severity of epilepsy [9, 13], and the rate of suicide among epileptic patients may be five to 25 times higher than that of the general population [19] (see Table 1). Conversely, a pre-existing diagnosis of depression among patients with new-onset epilepsy is up to seven times more common than in a control population [14]. Left temporal lobe epilepsy is commonly associated with the greatest propensity for depression [20–22]; however, some have argued that bilateral epilepsy may be somewhat protective against depressive symptoms as reported on the Beck Depressive Inventory [20] and others have reported no difference in laterality and depression [23]. Most depression characterized in epilepsy is interictal, occurring between seizure bouts, though postictal depression, defined explicitly as a depressive mood in the 72 hours following a seizure, has also been identified in a large portion of epileptic patients [9].
Animal models recapitulate the increased susceptibility to depressive-like behaviors in both status epilepticus [24] and absence models [25, 26] and may offer insight into the mechanism underlying the association between epilepsy and depression. Mazarati and colleagues [24] showed increased immobility in the forced swim test and decreased saccharin consumption after lithium and pilocarpine-induced status epilepticus, coexisting with decreased hippocampal serotonin and decreased serotonergic release from the dorsal raphe, all indicative of a depressive-like phenotype. Intriguingly, treatment with the selective serotonin reuptake inhibitor, fluoxetine, decreased brain excitability associated with status epilepticus but did not alter performance in the forced swim test [24]. Conversely, the genetically epilepsy prone rat (GEPR) has been shown to be susceptible to the proconvulsant activity of antidepressants, as have cats and rabbits at supratherapeutic doses of these drugs (reviewed in Jobe and Browning [27]). Moreover, the WAG/Rij rat strain is used as a model of absence epilepsy and shows increased immobility in the forced swim test and decreased sucrose consumption, an effect that can be blocked by treatment with the antiepileptic drug, ethosuximide [28]. Russo and colleagues [29] sought to determine if this simultaneously antidepressant and anticonvulsant effect of ethosuximide, a purported calcium channel inhibitor, was specific to that particular drug, which is only effective in absence seizures. Their study revealed that while levetiracetam and zonisamide, other compounds that target calcium channels (but not the sodium-channel targeting carbamezapine), lowered the incidence of spike-wave discharges (SWDs), only ethosuximide lowered SWDs and reduced depressive behavior in the forced swim test [29]. Noradrenergic and serotonergic deficits are believed to cause the seizure predisposition in GEPR and other mammals, which may be linked to depressive vulnerability [27]. Antidepressant anticonvulsant activity may be mediated by the restoration of these deficits; yet, simultaneously, antidepressants can enhance glutamatergic activity and inhibit GABAergic activity, which may lower seizure threshold [27]. These data hint that while depression and epilepsy may share common mechanisms, when combined, the neuropathology is more complex than in a typical depressive pattern alone.
In spite of the two-fold greater prevalence of depression in females in general, several studies have found an absence of a consistent sex difference in epilepsy-associated depression [21, 30]. In one large population-based study examining nearly 37,000 people of which 0.6% were diagnosed with epilepsy of heterogenous types, the odds ratio for anxiety disorders or suicidal thoughts among epileptic patients was 2.4 (95% Confidence interval = 1.5-3.8) and 2.2 (1.4-3.3), respectively [30]. Major depressive disorder had a significant age by sex interaction, such that male epileptic patients became more susceptible to depression with age while women became less susceptible [30]. However, this study did not factor in the type of epilepsy, and the greatest sex differences have been seen specifically with left temporal lobe epilepsy (TLE; [20, 22]). Left TLE has been associated with a male-specific vulnerability to depression [20, 22] as well as more severe depressive symptoms on the Beck Depressive Inventory [20]. This data is particularly interesting, and relevant for clinicians, given the dramatic suicide data among epileptics [19] and the sex disparity in suicide [31]. Collectively, the clinical data points toward a diminished if not altered sex difference in epilepsy-associated depression.
What then causes this alteration of the typical sex difference in depression when associated with epilepsy? It is possible that females simply experience less severe seizures on average. Both in humans and in animal models, progesterone can be anticonvulsant and estrogen proconvulsant, though rats typically experience reductions in seizures during proestrus, when both hormones are at their peak (reviewed in Frye [32]). Estrogen potentiates non-NMDA glutamatergic transmission, but reduces seizures associated with GABAA or NMDA activity [32]. Progesterone’s effects are at least in part due to actions by its metabolite, allopregnanolone, 3α-hydroxy-5α-pregnan-20-one (3α,5α-THP), which is not affected by a menopausal or menstrual state [32, 33]. Allopregnanolone is a powerful ligand for the GABAA/benzodiazepine receptor complex, which may explain its importance in seizure reduction [32]. Testosterone can provoke proconvulsant activity; castration on the day of birth reduces susceptibility to seizures in the substantia nigra pars reticulata [34], but this effect can be reversed by administration of testosterone [35]. It is possible that while females show a baseline vulnerability to depression, they are somewhat protected from the exacerbation of altered noradrenergic and serotonergic signaling caused by seizure activity due to the effects of ovarian hormones (or their metabolites). Males, on the other hand, may show increased susceptibility, ending up with a final erasure of the sex disparity in depression when associated with epilepsy. This highlights the important fact that depression is mediated by numerous mechanisms – including neurochemical, hormonal, and inflammatory pathways – that may be differentially affected by sex and disease states.
Alzheimer’s disease, sex, and depression
Advanced age is the number one risk factor for developing the spectrum of Alzheimer’s disease (AD) related pathologies and risk increases with age [36–38]. Women are at a greater risk of Alzheimer’s pathology [39], although men carry a greater risk of death from the disease [36]. Clinical and pre-clinical studies have shown that females carry an increased risk of developing AD pathology compared to males, even after controlling for increased life span [40, 41]. The changing status of sex hormones in aging contributes to the differential diagnoses and prognoses of men and women, as the sudden loss of ovarian hormones following menopause in women is attributed with the increased prevalence of the disease and an observed greater deposition of beta-amyloid (Aβ) plaques in the brains of AD positive women [37]. Depression is diagnosed in 35-50% of Alzheimer’s patients [10], though depressive symptoms are reported in up to 86% of patients [39] (see Table 1). The co-expression of depression with AD is associated with decreased quality of life, more severe cognitive deficits, and decreased longevity [38, 39, 42] though the diagnosis of depression with AD is a difficult one to make [42]. Depression and AD share similar genetic and environmental susceptibilities [10], which may speak to the bidirectionality of disease expression, yet the underlying mechanisms of the relationship are unclear. Mood disturbances, including depression, are reported to be three to four times as prevalent among the AD population [42], while similarly, a history of mood disorders increases the risk of the development of AD [43]. One study evaluating more than 1,300 patients over 14 years found depression to be a significant risk factor for the development of AD in men, but not women [10], which is an interesting finding in light of the increased rates and pathological load of AD in females. The exact interaction of AD pathology and sex steroids is complex and not well understood, though a role for sex steroids in the epidemiology and pathology of AD is undeniable. Generally speaking, estrogens and androgens are well recognized for their neuroprotective properties, including involvement in promoting cell survival, neuronal differentiation, and reducing inflammation [37, 44]. Compared to age-matched controls, reduced circulating levels of 17 beta-estradiol and testosterone are observed in female and male patients with AD, respectively [37, 44]. On a cellular level, decreased levels of sex steroids correlate with the increased load of Aβ in both sexes and the increased reactivity of estrogen receptors in women [37]. Again indicative of the dense relationship between the hypothalamic-pituitary-adrenal (HPA) and hypothalamic-pituitary-gonadal (HPG) axes, the HPA axis also alters the relationship between sex and AD. AD patients are reported to exhibit a delayed cortisol recovery following a stressor, and AD patients with higher levels of cortisol perform worse on cognitive tasks [45, 46]. Alternatively, age-related mitochondrial dysfunction and consequent increases in reactive oxygen species have been proposed to contribute to the neurodegeneration observed in AD [47].
Depression and somatic disease
As discussed above, diseases with direct effects on the brain, such as Alzheimer’s disease, are well documented to cause changes in mental status, including the onset of depressive symptoms, as the disease progresses [48, 49]. However, the link between neural diseases and depression is more intuitive than those of non-neuronal diseases. Regardless, connections between peripheral non-neuronal diseases and brain-associated changes paralleling what is observed in depressed subjects are still evident. These studies have identified patient populations with conditions such as peripheral cancers or cardiovascular disease, as well as an incidence of depressive symptoms, and they have then assessed the patient’s brains for neuronal markers of depression. Thus, the changes in neurochemistry and neuroanatomy identified in this way suggest a biological link between these somatic diseases and the abnormal brain activity thought to underlie symptoms of depression.
Cancer, sex, and depression
Cancer is the second leading cause of death in the United States after heart disease, and kills more than half a million Americans every year [50]. The top five cancer sites differ in incidence and mortality between men and women but show some overlap: the cancers with the highest incidence in men are, in order, prostate, lung, colorectal, bladder, and melanoma; and those with the highest incidence in women are breast, lung, colorectal, uterine, and thyroid [50]. Lung cancer kills the most men and women each year, and colorectal cancer and pancreatic cancer are also among the top five deadliest cancers for both men and women [50].
Both the cancer disease and the treatments of that disease are documented to cause significant changes in neuroanatomy and neurochemistry. For instance, Cousins and Harper describe a case study where chemotherapy treatment for breast cancer caused a significant reduction in neurotransmitter-related chemistry: myo-inositol, choline and glutamate [51]. Another breast-cancer case study found depressed activation of the anterior cingulate cortex (subgenual cingulate cortex), which was subsequently elevated following behavioral therapy [52]. This provides evidence for both disease and disease-treatment associated effects upon the brain that parallel those observed in depression, and strengthens the proposed association between the occurrence of peripheral cancers and mood disorders.
Depression and cancer often co-exist, with major depression prevalence reportedly greater than 40% depending on the method of diagnosis and type of cancer [11] (see Table 1). Major depression is particularly highly associated with oropharyngeal, pancreatic, breast, and lung cancers, and the prevalence of depression increases with disease severity and progression [11, 53]. While the underlying mechanism and the direction of causality in this association remain to be elucidated, several groups have pointed toward HPA axis disruption and immune activation in cancer-related depression. As often seen in non-cancer-related major depression, depressed patients with gynecologic and breast cancer do not suppress cortisol levels after dexamethasone administration [54, 55]. Depressed patients with metastatic breast cancer have shown decreased HPA axis response to awakening, and both depressed and non-depressed patients in this study also had a blunted cortisol response to acute stress [56]. In addition, depressed cancer patients display elevated levels of IL-6 relative to both non-depressed cancer patients [55, 57] and depressed non-cancer patients [57]. In contrast to this inflammatory state, cancer and stress can also induce immune suppression. Psychological stress, in patients treated surgically for breast cancer, can lower natural killer (NK) cell lysis and response to IFN-γ [58]. As NK cells attack transformed and dying cells without antigen presentation, they may provide an important role in cancer surveillance and thus impaired NK activity is relevant to cancer prognosis [53].
The potential iatrogenic role of cancer treatment to promote immune dysfunction and subsequent depression cannot be ignored. IL-2 and IFN-α are commonly used as cancer treatments and can induce cognitive disturbance, psychological symptoms, and neurovegetative symptoms of depression including loss of appetite, fatigue, and altered sleep [59–61]. Mechanistically, these elevated cytokines may generate glucocorticoid resistance, perhaps by interfering with glucocorticoid receptor translocation [62, 63], and thus leading to chronically elevated plasma cortisol associated with the depressive state. Additionally, elevated cytokines can downregulate serotonin [64] and promote indoleamine 2,3 dioxygenase activity, which would further reduce serotonin synthesis by promoting a switch in tryptophan metabolism towards kynurenine and quinolinic acid [65]. This hints at directionality in the link between cancer and depression, suggesting that cancer-associated inflammation promotes neurochemical alterations associated with depression.
While cancer has on the one hand been proposed to promote depressive symptoms by altering HPA and immune function, depression and associated HPA and immune alterations have, on the other hand, themselves been put forth as potential modulators of cancer risk and/or morbidity [53]. Depressed patients are less likely to adhere to medical therapies [66], which may account for some reports of worse outcomes in depressed cancer patients [67]. Sapolsky and Donnelly [68, 69] showed that stress-induced growth of a virally-derived tumor is accelerated in aged male rats, potentially due to tumor resistance to glucocorticoid inhibition of glucose transport [69]. In prostate cancer cell lines, glucocorticoids can promote androgen-independent growth [70]. Depression alone has been associated with increased CD4/CD8 ratios, decreased percentages of lymphocytes including T and B cells, and decreased circulating NK cells [71, 72]. However, the clinical literature regarding the impact of pre-existing depression on cancer risk and progression remains divided and poorly controlled.
Given the sex-specific susceptibility to certain cancer types, and the bidirectional relationship between cancer and depression, how then does sex affect cancer-related depression? Several studies have attempted to answer this question with mixed results (reviewed in Massie [11]). In a recent Canadian study with over 10,000 patients at two major cancer centers, women were twice as likely as men to report clinical levels of depression as determined using the Psychosocial Screen for Cancer questionnaire [73]. Reported depression varied by cancer type, with the greatest disparity in lung cancer and an absence of a sex difference in neuroendocrine cancers. However, another study examining depression rates using the Hospital Anxiety and Depression Scale in patients with inoperable small cell and non-small cell lung cancer reported that rates of depression in the non-small-cell lung cancer patients differed by sex (31% female vs. 19% male) but not among the small-cell lung cancer patients (46% female vs. 41% male) [74]. Secondary analyses demonstrated that this sex difference or lack thereof correlated with prognosis, such that it existed for the good prognosis patients (with women reporting higher depression) but disappeared among the poor prognosis patients [75]. Miller and colleagues [76] reported a similar finding among patients with stage III or stage IV lung or stage IV gastrointestinal cancer (including colon rectum, pancreas, appendix, and hepatobiliary cancers), using the Beck Depression Inventory II to assess depression. In this study, there were no significant differences between gender and depression or depressive symptom severity [76]. In another study focusing on the role of fatigue, pain, and/or depression in predicting quality of life (QoL) in patients with varying cancer types, women had lower scores for quality of life in the Multidimensional QoL Scale-Cancer and reported greater pain, but equal depressive symptoms on the Center for Epidemiological Studies –Depression Scale [77]. Pain and depression predicted female QoL, while depression alone predicted male QoL, indicating depression as an important factor for both male and female cancer patients. Of course, it is critical to remember that the tools used to assess depression in these studies vary widely, and may account for some disparities seen, making between-study comparisons difficult. However, the trend across assessment tools for a relative leveling of the susceptibility to depression in severe cancer cases seems consistent.
The mechanisms underlying this alteration in sex-specific vulnerability to depression in cancer remain unclear. Some studies have reported increased depression in women associated with estrogen deficiency due to tamoxifen, an estrogen receptor antagonist, or other chemotherapeutic treatment [78, 79]. In contrast, one larger study of tamoxifen treatment vs. placebo in non-cancer patients did not find any statistical difference in rates of depression [80, 81]. It is however possible that the combination of the cancer stressor combined with the estrogen deficiency may be necessary for induction of the depressive state.
Cardiovascular disease, sex, and depression
Aging positively correlates to the incidence and prevalence of cardiovascular-related disease, as age remains one of the greatest risk factors for the emergence of the disease in both men and women [82, 83]. Women, however, begin to outnumber men in cardiovascular disease prevalence starting around age 45, and cardiovascular disease is the leading killer of women over the age of 50 [82, 84]. Similar to other disease states, the change in demographics of cardiovascular disease and the increased burden on women is attributed to the negative pathologic consequences (loss of antioxidant, anti-inflammatory, and neuroprotective properties) associated with the cessation of ovarian function [85, 86].
The manifestation of depression occurs in approximately 15-20% of patients with cardiovascular diseases, a rate three times what is observed in the general population [12] (see Table 1). The links between depression and cardiovascular disease are strong; with evidence supporting the bidirectionality of onset. Low heart rate variability, an indicator of poor autonomic control (i.e. inflexibility), among depressed patients is associated with dramatic effects on vascular function, including increased peripheral resistance, arterial hypertension, and organ damage [87, 88]. Depressive events are reported to precede cardiovascular events, and the manifestation of depressive episodes early in life confers a greater risk of the development of cardiovascular disease [4, 87]. Similarly, cellular mechanisms associated with mood disorders and stress give rise to vascular damage observed in cardiovascular diseases while the co-expression of the two disease states is associated with the greater risk of cardiovascular events as well as a poorer prognosis following these events [12, 87]. Conversely, depression is a common consequence of stroke and other vascular events, occurring in an estimated one third of people [89, 90]. Furthermore, patients who have suffered a stroke are at a six-fold greater risk of developing depression for up to two years following the event [91]. Though direct causality is still a mystery, vascular-induced changes to endocrine status [92], neurotransmission [93], and neuroinflammation [94, 95] have all been associated with the development of depression.
The linkage of cardiovascular disease to depression, especially late-life depression, is thought to be the product of reduced blood flow [96]. The hippocampus and frontal cortices are susceptible to reduced blood flow and have been shown to exhibit reduced blood flow in depressed patients [97–99]. Moreover, in addition to proposed reduced blood flow, white matter hyperintensities, a sign of hypertension [100], are evident in depression, especially in old age [101, 102], and appear to positively correlate with the progress of depressive symptoms [103]. Further, the locations of the white matter intensities correlate with some aspects of the scope of the depressive disorder, though this remains controversial. For instance, when white matter intensities are evident in subcortical areas, a greater percentage of anger attacks are reported in those depressed persons [104]. Similarly, when gray matter hyperintensities have been examined, an apparent positive correlation exists between the presence of those abnormal signals in the basal ganglia and suicide attempts [105], although that study was 85% populated by women.
Sex steroids influence the onset of depressive symptoms as well as the susceptibility to cardiovascular events; however, the interaction of the two disease states is unclear. Though the overall rates of depression are greater among patients with cardiovascular disease, women are still at a double risk of developing depression, a rate that is consistent with what is observed in the general population [12]. Beginning at menopause, a woman’s risk for cardiovascular diseases increases as a result of decreased estrogen activity and parallels a decreased risk for depressive disorders [82, 86]. These data suggest that hormonal status can influence mood and cardiovascular disease independently, even when co-expressed. Because men have higher circulating levels of androgens for a greater proportion of their lives, high levels of androgens have often been associated with the development of cardiovascular disease and implicated in the earlier onset of the condition in men relative to women [85, 86]. Interestingly, however, circulating levels of testosterone are typically lower in men with cardiovascular disease compared to an age-matched healthy population suggesting either an alternative mechanism or a dynamic role for testosterone [82, 86]. Furthermore, low levels of total and bioavailable testosterone in men are associated with depressed mood, although data on the topic are controversial [86, 106, 107]. The role androgens play in depressive behaviors and cardiovascular disease in men is less clear, and the coexpression of the two disease states does little to elucidate the relationship. In men, other factors may weigh more heavily than androgen status for the emergence of cardiovascular disorders, such as cholesterol [85] and alterations to the HPA axis [84], which both are implicated in depressive disorders [108, 109]. Together, these data and others suggest that there is a biological basis of sex-influenced vulnerability for depressive symptoms through a cardiovascular mechanism and that such clinical and preclinical studies should be explored.
Conclusions
In conclusion, we assert that not all depression is created equal. The evidence presented here indicates that sex interacts with disease to precipitate depression whether the disease is neural (as in epilepsy and AD) or somatic (e.g., cancer and cardiovascular disease) in origin. The relationships described in this review highlight the complex disease-depression interaction as well as the multitude of factors that influence the expression and severity of each condition. In each of the diseases discussed herein, the prevalence of comordbid mood disorders is much higher than in the general population (see Table 1), though in each case the direction of causality is unclear. It appears as though the directionality of the depression-disease relationship is two-fold, such that each state lowers the threshold for the other, depending on the genetic and environmental susceptibilities of each.
The typical 2:1 female predominance in the presentation of mood disorders is tempered in at least some comorbid disease states. Baseline changes in neurochemical signaling due to the disease state, as in epilepsy [32], altered neurobiology secondary to inflammatory mechanisms associated with disease, as in cancer [53, 59], or a combination of these factors seem to interact with sex to alter vulnerability to mood disorders. The exact role of sex steroids and whether fluctuations (eg. androgens in cardiovascular disease and estrogen in AD) or absolute levels of hormone may mitigate some degree of disease progression and susceptibility to depression remains unclear. A review by Cahill [110] of “His brain, her brain” similarly addresses the neurobiology of sex differences in disease and advocates for appropriate treatment in light of these differences. Indeed differential coping styles of men and women [111] may contribute at least in part to depressive symptoms; however, several studies have cited not only the bidirectional nature of depression and disease [14, 53, 87], but the biological basis for depression within disease [95, 112, 113].
The precise systems that are dysregulated in the manifestation of depression likely vary by the root cause of the disorder. The originating biological cause of depression will depend on the sex, age, and disease state of the individual. For instance, the point of origin is likely to vary substantially between a healthy adolescent girl abused as a child, an adult woman with epilepsy, and an elderly man with cardiovascular disease and a history of cerebral ischemia. Once it is understood that depression is not a singular disorder but rather a common phenotype for a variety of underlying biological disruptions, both central and peripheral in origin, preclinical research will be better designed to identify and target potential therapeutic mechanisms, leading to more specific and effective clinical treatments.
Abbreviations
- 3α:
-
5αTHP: 3α-hydroxy-5α-pregnan-20one
- Aβ:
-
Amyloid beta
- AD:
-
Alzheimer’s disease
- GABA:
-
Gamma-Aminobutyric acid
- GEPR:
-
Genetically epilepsy prone rat
- HPA:
-
Hypothalamic-adrenal-pituitary
- HPG:
-
Hypothalamic-adrenal-gonadal
- NK:
-
Natural killer
- NMDA:
-
N-methyl-D-aspartate
- QoL:
-
Quality of Life
- SWD:
-
Spike-wave discharge
- TLE:
-
Temporal lobe epilepsy.
References
WHO: The Global Burden of Disease. 2004 Update. Geneva: World Health Organization; 2008.
Pagura J, Cox BJ, Sareen J, Enns MW: Factors associated with multiple versus single episode suicide attempts in the 1990–1992 and 2001–2003 United States national comorbidity surveys. J Nerv Ment Dis 2008, 196: 806–813. 10.1097/NMD.0b013e31818b6a77
Rockett IR, Regier MD, Kapusta ND, Coben JH, Miller TR, Hanzlick RL, Todd KH, Sattin RW, Kennedy LW, Kleinig J, Smith GS: Leading causes of unintentional and intentional injury mortality: united states, 2000–2009. Am J Public Health 2012, 102: e84-e92. 10.2105/AJPH.2012.300960
Steiner M, Dunn E, Born L: Hormones and mood: from menarche to menopause and beyond. J Affect Disord 2003, 74: 67–83. 10.1016/S0165-0327(02)00432-9
Piccinelli M, Wilkinson G: Gender differences in depression. Critical review. Br J Psychiatry 2000, 177: 486–492. 10.1192/bjp.177.6.486
Naninck EF, Lucassen PJ, Bakker J: Sex differences in adolescent depression: do sex hormones determine vulnerability? J Neuroendocrinol 2011, 23: 383–392. 10.1111/j.1365-2826.2011.02125.x
Pitychoutis PM, Nakamura K, Tsonis PA, Papadopoulou-Daifoti Z: Neurochemical and behavioral alterations in an inflammatory model of depression: sex differences exposed. Neuroscience 2009, 159: 1216–1232. 10.1016/j.neuroscience.2009.01.072
Pinkerton JV, Guico-Pabia CJ, Taylor HS: Menstrual cycle-related exacerbation of disease. Am J Obstet Gynecol 2010, 202: 221–231. 10.1016/j.ajog.2009.07.061
Kanner AM: Depression in epilepsy: prevalence, clinical semiology, pathogenic mechanisms, and treatment. Biol Psychiatry 2003, 54: 388–398. 10.1016/S0006-3223(03)00469-4
Dal Forno G, Palermo MT, Donohue JE, Karagiozis H, Zonderman AB, Kawas CH: Depressive symptoms, sex, and risk for Alzheimer’s disease. Ann Neurol 2005, 57: 381–387. 10.1002/ana.20405
Massie MJ: Prevalence of depression in patients with cancer. J Natl Cancer Inst Monogr 2004, 2004: 57–71. 10.1093/jncimonographs/lgh014
Shanmugasegaram S, Russell KL, Kovacs AH, Stewart DE, Grace SL: Gender and sex differences in prevalence of major depression in coronary artery disease patients: A meta-analysis. Maturitas 2012, 73: 305–311. 10.1016/j.maturitas.2012.09.005
Gilliam FG, Santos J, Vahle V, Carter J, Brown K, Hecimovic H: Depression in epilepsy: ignoring clinical expression of neuronal network dysfunction? Epilepsia 2004,45(Suppl 2):28–33.
Kanner AM: Can neurobiological pathogenic mechanisms of depression facilitate the development of seizure disorders? Lancet Neurol 2012, 11: 1093–1102. 10.1016/S1474-4422(12)70201-6
Epilepsy Fast Facts. http://www.cdc.gov/epilepsy/basics/fast_facts.htm
Ropper A, Samuels M: Epilepsy and Other Seizure Disorders. In Adams and Victor's Principles of Neurology. 9th edition. New York: McGraw-Hill; 2009.
Kotsopoulos IA, van Merode T, Kessels FG, de Krom MC, Knottnerus JA: Systematic review and meta-analysis of incidence studies of epilepsy and unprovoked seizures. Epilepsia 2002, 43: 1402–1409. 10.1046/j.1528-1157.2002.t01-1-26901.x
Mula M: Epilepsy: Bidirectional link between epilepsy and psychiatric disorders. Nat Rev Neurol 2012, 8: 252–253. 10.1038/nrneurol.2012.69
Barraclough BM: The suicide rate of epilepsy. Acta Psychiatr Scand 1987, 76: 339–345. 10.1111/j.1600-0447.1987.tb05616.x
Altshuler LL, Devinsky O, Post RM, Theodore W: Depression, anxiety, and temporal lobe epilepsy. Laterality of focus and symptoms. Arch Neurol 1990, 47: 284–288. 10.1001/archneur.1990.00530030050016
Piazzini A, Canevini MP, Maggiori G, Canger R: Depression and anxiety in patients with epilepsy. Epilepsy Behav 2001, 2: 481–489. 10.1006/ebeh.2001.0247
Strauss E, Wada J, Moll A: Depression in male and female subjects with complex partial seizures. Arch Neurol 1992, 49: 391–392. 10.1001/archneur.1992.00530280079026
Kohler C, Norstrand JA, Baltuch G, O’Connor MJ, Gur RE, French JA, Sperling MR: Depression in temporal lobe epilepsy before epilepsy surgery. Epilepsia 1999, 40: 336–340. 10.1111/j.1528-1157.1999.tb00714.x
Mazarati A, Siddarth P, Baldwin RA, Shin D, Caplan R, Sankar R: Depression after status epilepticus: behavioural and biochemical deficits and effects of fluoxetine. Brain 2008, 131: 2071–2083. 10.1093/brain/awn117
Sarkisova K, van Luijtelaar G: The WAG/Rij strain: a genetic animal model of absence epilepsy with comorbidity of depression [corrected]. Prog Neuropsychopharmacol Biol Psychiatry 2011, 35: 854–876. 10.1016/j.pnpbp.2010.11.010
Russo E, Citraro R, Scicchitano F, Urzino A, Marra R, Rispoli V, De Sarro G: Vigabatrin has antiepileptogenic and antidepressant effects in an animal model of epilepsy and depression comorbidity. Behav Brain Res 2011, 225: 373–376. 10.1016/j.bbr.2011.07.030
Jobe PC, Browning RA: The serotonergic and noradrenergic effects of antidepressant drugs are anticonvulsant, not proconvulsant. Epilepsy Behav 2005, 7: 602–619. 10.1016/j.yebeh.2005.07.014
Sarkisova KY, Kuznetsova GD, Kulikov MA, van Luijtelaar G: Spike-wave discharges are necessary for the expression of behavioral depression-like symptoms. Epilepsia 2010, 51: 146–160. 10.1111/j.1528-1167.2009.02260.x
Russo E, Citraro R, Scicchitano F, De Fazio S, Perrotta I, Di Paola ED, Constanti A, De Sarro G: Effects of early long-term treatment with antiepileptic drugs on development of seizures and depressive-like behavior in a rat genetic absence epilepsy model. Epilepsia 2011, 52: 1341–1350. 10.1111/j.1528-1167.2011.03112.x
Tellez-Zenteno JF, Patten SB, Jette N, Williams J, Wiebe S: Psychiatric comorbidity in epilepsy: a population-based analysis. Epilepsia 2007, 48: 2336–2344.
Denning DG, Conwell Y, King D, Cox C: Method choice, intent, and gender in completed suicide. Suicide Life Threat Behav 2000, 30: 282–288.
Frye CA: Hormonal influences on seizures: basic neurobiology. Int Rev Neurobiol 2008, 83: 27–77.
Genazzani AR, Petraglia F, Bernardi F, Casarosa E, Salvestroni C, Tonetti A, Nappi RE, Luisi S, Palumbo M, Purdy RH, Luisi M: Circulating levels of allopregnanolone in humans: gender, age, and endocrine influences. J Clin Endocrinol Metab 1998, 83: 2099–2103. 10.1210/jc.83.6.2099
Veliskova J, Claudio OI, Galanopoulou AS, Lado FA, Ravizza T, Velisek L, Moshe SL: Seizures in the developing brain. Epilepsia 2004,45(Suppl 8):6–12.
Giorgi FS, Veliskova J, Chudomel O, Kyrozis A, Moshe SL: The role of substantia nigra pars reticulata in modulating clonic seizures is determined by testosterone levels during the immediate postnatal period. Neurobiol Dis 2007, 25: 73–79. 10.1016/j.nbd.2006.08.009
Molsa PK, Marttila RJ, Rinne UK: Long-term survival and predictors of mortality in Alzheimer’s disease and multi-infarct dementia. Acta Neurol Scand 1995, 91: 159–164.
Barron AM, Pike CJ: Sex hormones, aging, and Alzheimer’s disease. Front Biosci (Elite Ed) 2012, 4: 976–997.
Lopez OL, Becker JT, Kuller LH: Patterns of Compensation and Vulnerability in Normal Subjects at Risk of Alzheimer’s Disease. J Alzheimers Dis 2012,33(Suppl 1):427–38.
Lopez OL: The growing burden of Alzheimer’s disease. Am J Manag Care 2011,17(Suppl 13):S339-S345.
Dye RV, Miller KJ, Singer EJ, Levine AJ: Hormone replacement therapy and risk for neurodegenerative diseases. Int J Alzheimers Dis 2012, 2012: 258454.
Callahan MJ, Lipinski WJ, Bian F, Durham RA, Pack A, Walker LC: Augmented senile plaque load in aged female beta-amyloid precursor protein-transgenic mice. Am J Pathol 2001, 158: 1173–1177. 10.1016/S0002-9440(10)64064-3
Lee HB, Lyketsos CG: Depression in Alzheimer’s disease: heterogeneity and related issues. Biol Psychiatry 2003, 54: 353–362. 10.1016/S0006-3223(03)00543-2
Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky S: Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: results from the cardiovascular health study. JAMA 2002, 288: 1475–1483. 10.1001/jama.288.12.1475
Luchetti S, Bossers K, Van de Bilt S, Agrapart V, Morales RR, Frajese GV, Swaab DF: Neurosteroid biosynthetic pathways changes in prefrontal cortex in Alzheimer’s disease. Neurobiol Aging 2011, 32: 1964–1976. 10.1016/j.neurobiolaging.2009.12.014
Carlson LE, Sherwin BB, Chertkow HM: Relationships between dehydroepiandrosterone sulfate (DHEAS) and cortisol (CRT) plasma levels and everyday memory in Alzheimer’s disease patients compared to healthy controls. Horm Behav 1999, 35: 254–263. 10.1006/hbeh.1999.1518
Clinton LK, Billings LM, Green KN, Caccamo A, Ngo J, Oddo S, McGaugh JL, LaFerla FM: Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol Dis 2007, 28: 76–82. 10.1016/j.nbd.2007.06.013
Long J, He P, Shen Y, Li R: New evidence of mitochondria dysfunction in the female Alzheimer’s disease brain: deficiency of estrogen receptor-beta. J Alzheimers Dis 2012, 30: 545–558.
Mayberg HS: Clinical correlates of PET- and SPECT-identified defects in dementia. J Clin Psychiatry 1994,55(Suppl):12–21.
Caroli A, Geroldi C, Nobili F, Barnden LR, Guerra UP, Bonetti M, Frisoni GB: Functional compensation in incipient Alzheimer’s disease. Neurobiol Aging 2010, 31: 387–397. 10.1016/j.neurobiolaging.2008.05.001
Seigel R, Naishadham D, Jemal A: Cancer Statistics 2012. CA Cancer J Clin 2012, 62: 10–29. 10.3322/caac.20138
Cousins JP, Harper G: Neurobiochemical changes from Taxol/Neupogen chemotherapy for metastatic breast carcinoma corresponds with suicidal depression. Cancer Lett 1996, 110: 163–167. 10.1016/S0304-3835(96)04486-2
Gawrysiak MJ, Carvalho JP, Rogers BP, Nicholas CR, Dougherty JH, Hopko DR: Neural Changes following Behavioral Activation for a Depressed Breast Cancer Patient: A Functional MRI Case Study. Case reports in psychiatry 2012, 2012: 152916.
Spiegel D, Giese-Davis J: Depression and cancer: mechanisms and disease progression. Biol Psychiatry 2003, 54: 269–282. 10.1016/S0006-3223(03)00566-3
Evans DL, McCartney CF, Nemeroff CB, Raft D, Quade D, Golden RN, Haggerty JJ Jr, Holmes V, Simon JS, Droba M: Depression in women treated for gynecological cancer: clinical and neuroendocrine assessment. Am J Psychiatry 1986, 143: 447–452.
Soygur H, Palaoglu O, Akarsu ES, Cankurtaran ES, Ozalp E, Turhan L, Ayhan IH: Interleukin-6 levels and HPA axis activation in breast cancer patients with major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry 2007, 31: 1242–1247. 10.1016/j.pnpbp.2007.05.001
Giese-Davis J, Wilhelm FH, Conrad A, Abercrombie HC, Sephton S, Yutsis M, Neri E, Taylor CB, Kraemer HC, Spiegel D: Depression and stress reactivity in metastatic breast cancer. Psychosom Med 2006, 68: 675–683. 10.1097/01.psy.0000238216.88515.e5
Musselman DL, Miller AH, Porter MR, Manatunga A, Gao F, Penna S, Pearce BD, Landry J, Glover S, McDaniel JS, Nemeroff CB: Higher than normal plasma interleukin-6 concentrations in cancer patients with depression: preliminary findings. Am J Psychiatry 2001, 158: 1252–1257. 10.1176/appi.ajp.158.8.1252
Andersen BL, Farrar WB, Golden-Kreutz D, Kutz LA, MacCallum R, Courtney ME, Glaser R: Stress and immune responses after surgical treatment for regional breast cancer. J Natl Cancer Inst 1998, 90: 30–36. 10.1093/jnci/90.1.30
Capuron L, Ravaud A, Neveu PJ, Miller AH, Maes M, Dantzer R: Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol Psychiatry 2002, 7: 468–473. 10.1038/sj.mp.4000995
Capuron L, Ravaud A, Dantzer R: Early depressive symptoms in cancer patients receiving interleukin 2 and/or interferon alfa-2b therapy. J Clin Oncol 2000, 18: 2143–2151.
Raison CL, Demetrashvili M, Capuron L, Miller AH: Neuropsychiatric adverse effects of interferon-alpha: recognition and management. CNS Drugs 2005, 19: 105–23. 10.2165/00023210-200519020-00002
Miller AH, Pariante CM, Pearce BD: Effects of cytokines on glucocorticoid receptor expression and function. Glucocorticoid resistance and relevance to depression. Adv Exp Med Biol 1999, 461: 107–116. 10.1007/978-0-585-37970-8_7
Pariante CM, Pearce BD, Pisell TL, Sanchez CI, Po C, Su C, Miller AH: The proinflammatory cytokine, interleukin-1alpha, reduces glucocorticoid receptor translocation and function. Endocrinology 1999, 140: 4359–4366. 10.1210/en.140.9.4359
Kamata M, Higuchi H, Yoshimoto M, Yoshida K, Shimizu T: Effect of single intracerebroventricular injection of alpha-interferon on monoamine concentrations in the rat brain. Eur Neuropsychopharmacol 2000, 10: 129–132.
Lestage J, Verrier D, Palin K, Dantzer R: The enzyme indoleamine 2,3-dioxygenase is induced in the mouse brain in response to peripheral administration of lipopolysaccharide and superantigen. Brain Behav Immun 2002, 16: 596–601. 10.1016/S0889-1591(02)00014-4
DiMatteo MR, Lepper HS, Croghan TW: Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med 2000, 160: 2101–2107. 10.1001/archinte.160.14.2101
Pinquart M, Duberstein PR: Depression and cancer mortality: a meta-analysis. Psychol Med 2010, 40: 1797–1810. 10.1017/S0033291709992285
Sapolsky RM, Donnelly TM: Vulnerability to stress-induced tumor growth increases with age in rats: role of glucocorticoids. Endocrinology 1985, 117: 662–666. 10.1210/endo-117-2-662
Romero LM, Raley-Susman KM, Redish DM, Brooke SM, Horner HC, Sapolsky RM: Possible mechanism by which stress accelerates growth of virally derived tumors. Proc Natl Acad Sci USA 1992, 89: 11084–11087. 10.1073/pnas.89.22.11084
Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM, Feldman D: Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med 2000, 6: 703–706. 10.1038/76287
Irwin MR, Miller AH: Depressive disorders and immunity: 20 years of progress and discovery. Brain Behav Immun 2007, 21: 374–383. 10.1016/j.bbi.2007.01.010
Zorrilla EP, Luborsky L, McKay JR, Rosenthal R, Houldin A, Tax A, McCorkle R, Seligman DA, Schmidt K: The relationship of depression and stressors to immunological assays: a meta-analytic review. Brain Behav Immun 2001, 15: 199–226. 10.1006/brbi.2000.0597
Linden W, Vodermaier A, Mackenzie R, Greig D: Anxiety and depression after cancer diagnosis: Prevalence rates by cancer type, gender, and age. J Affect Disord 2012, 141: 343–351. 10.1016/j.jad.2012.03.025
Hopwood P, Stephens RJ: Depression in patients with lung cancer: prevalence and risk factors derived from quality-of-life data. J Clin Oncol 2000, 18: 893–903.
Anderson H, Hopwood P, Stephens RJ, Thatcher N, Cottier B, Nicholson M, Milroy R, Maughan TS, Falk SJ, Bond MG: Gemcitabine plus best supportive care (BSC) vs BSC in inoperable non-small cell lung cancer–a randomized trial with quality of life as the primary outcome. UK NSCLC Gemcitabine Group. Non-Small Cell Lung Cancer. Br J Cancer 2000, 83: 447–453. 10.1054/bjoc.2000.1307
Miller S, Lo C, Gagliese L, Hales S, Rydall A, Zimmermann C, Li M, Rodin G: Patterns of depression in cancer patients: an indirect test of gender-specific vulnerabilities to depression. Soc Psychiatry Psychiatr Epidemiol 2011, 46: 767–774. 10.1007/s00127-010-0246-7
Pud D: Gender differences in predicting quality of life in cancer patients with pain. Eur J Oncol Nurs 2011, 15: 486–491. 10.1016/j.ejon.2010.12.005
Duffy LS, Greenberg DB, Younger J, Ferraro MG: Iatrogenic acute estrogen deficiency and psychiatric syndromes in breast cancer patients. Psychosomatics 1999, 40: 304–308. 10.1016/S0033-3182(99)71223-5
Cathcart CK, Jones SE, Pumroy CS, Peters GN, Knox SM, Cheek JH: Clinical recognition and management of depression in node negative breast cancer patients treated with tamoxifen. Breast Cancer Res Treat 1993, 27: 277–281. 10.1007/BF00665698
Day R: Quality of life and tamoxifen in a breast cancer prevention trial: a summary of findings from the NSABP P-1 study. National Surgical Adjuvant Breast and Bowel Project. Ann N Y Acad Sci 2001, 949: 143–150.
Day R, Ganz PA, Costantino JP: Tamoxifen and depression: more evidence from the National Surgical Adjuvant Breast and Bowel Project’s Breast Cancer Prevention (P-1) Randomized Study. J Natl Cancer Inst 2001, 93: 1615–1623. 10.1093/jnci/93.21.1615
Banos G, Guarner V, Perez-Torres I: Sex steroid hormones, cardiovascular diseases and the metabolic syndrome. Cardiovasc Hematol Agents Med Chem 2011, 9: 137–146. 10.2174/187152511797037547
Pavlovic AM, Pekmezovic T, Zidverc-Trajkovic J, Jovanovic Z, Mijajlovic M, Pavlovic D, Tomic G, Sternic N: What are the differences between younger and older patients with symptomatic small vessel disease? Clin Neurol Neurosurg 2011, 113: 762–767. 10.1016/j.clineuro.2011.08.011
Steiner M: Serotonin, depression, and cardiovascular disease: sex-specific issues. Acta Physiol (Oxf) 2011, 203: 253–258. 10.1111/j.1748-1716.2010.02236.x
Vitale C, Miceli M, Rosano GM: Gender-specific characteristics of atherosclerosis in menopausal women: risk factors, clinical course and strategies for prevention. Climacteric 2007,10(Suppl 2):16–20.
Reckelhoff JF: Sex steroids, cardiovascular disease, and hypertension: unanswered questions and some speculations. Hypertension 2005, 45: 170–174. 10.1161/01.HYP.0000151825.36598.36
Plante GE: Depression and cardiovascular disease: a reciprocal relationship. Metabolism 2005, 54: 45–48. 10.1016/j.metabol.2005.01.013
Lombard JH: Depression, psychological stress, vascular dysfunction, and cardiovascular disease: thinking outside the barrel. J Appl Physiol 2010, 108: 1025–1026.
Blazer DG 2nd, Hybels CF: Origins of depression in later life. Psychol Med 2005, 35: 1241–1252. 10.1017/S0033291705004411
Kales HC, Maixner DF, Mellow AM: Cerebrovascular disease and late-life depression. Am J Geriatr Psychiatry 2005, 13: 88–98.
Whyte EM, Mulsant BH, Vanderbilt J, Dodge HH, Ganguli M: Depression after stroke: a prospective epidemiological study. J Am Geriatr Soc 2004, 52: 774–778. 10.1111/j.1532-5415.2004.52217.x
Milot MR, Plamondon H: Changes in HPA reactivity and noradrenergic functions regulate spatial memory impairments at delayed time intervals following cerebral ischemia. Horm Behav 2011, 59: 594–604. 10.1016/j.yhbeh.2011.02.017
Fang J, Cheng Q: Etiological mechanisms of post-stroke depression: a review. Neurol Res 2009, 31: 904–909. 10.1179/174313209X385752
Hakim AM: Depression, strokes and dementia: new biological insights into an unfortunate pathway. Cardiovasc Psychiatry Neurol 2011, 2011: 649629.
Spalletta G, Bossu P, Ciaramella A, Bria P, Caltagirone C, Robinson RG: The etiology of poststroke depression: a review of the literature and a new hypothesis involving inflammatory cytokines. Mol Psychiatry 2006, 11: 984–991. 10.1038/sj.mp.4001879
Tiemeier H, Bakker SL, Hofman A, Koudstaal PJ, Breteler MM: Cerebral haemodynamics and depression in the elderly. J Neurol Neurosurg Psychiatry 2002, 73: 34–39. 10.1136/jnnp.73.1.34
Galynker II, Cai J, Ongseng F, Finestone H, Dutta E, Serseni D: Hypofrontality and negative symptoms in major depressive disorder. Journal of nuclear medicine: official publication, Society of Nuclear Medicine 1998, 39: 608–612.
Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Hua X, Toga AW, Jack CR Jr: Automated 3D mapping of hippocampal atrophy and its clinical correlates in 400 subjects with Alzheimer’s disease, mild cognitive impairment, and elderly controls. Hum Brain Mapp 2009, 30: 2766–2788. 10.1002/hbm.20708
Videbech P: PET measurements of brain glucose metabolism and blood flow in major depressive disorder: a critical review. Acta Psychiatr Scand 2000, 101: 11–20.
Breteler MM, van Swieten JC, Bots ML, Grobbee DE, Claus JJ, van den Hout JH, van Harskamp F, Tanghe HL, de Jong PT, van Gijn J: Cerebral white matter lesions, vascular risk factors, and cognitive function in a population-based study: the Rotterdam Study. Neurology 1994, 44: 1246–1252. 10.1212/WNL.44.7.1246
Herrmann LL, Le Masurier M, Ebmeier KP: White matter hyperintensities in late life depression: a systematic review. J Neurol Neurosurg Psychiatry 2008, 79: 619–624.
Godin O, Dufouil C, Maillard P, Delcroix N, Mazoyer B, Crivello F, Alperovitch A, Tzourio C: White matter lesions as a predictor of depression in the elderly: the 3C-Dijon study. Biol Psychiatry 2008, 63: 663–669. 10.1016/j.biopsych.2007.09.006
Soennesyn H, Oppedal K, Greve OJ, Fritze F, Auestad BH, Nore SP, Beyer MK, Aarsland D: White matter hyperintensities and the course of depressive symptoms in elderly people with mild dementia. Dementia and geriatric cognitive disorders extra 2012, 2: 97–111. 10.1159/000335497
Iosifescu DV, Renshaw PF, Dougherty DD, Lyoo IK, Lee HK, Fraguas R, Cassano P, Nierenberg AA, Fava M: Major depressive disorder with anger attacks and subcortical MRI white matter hyperintensities. J Nerv Ment Dis 2007, 195: 175–178. 10.1097/01.nmd.0000253820.69362.87
Ahearn EP, Jamison KR, Steffens DC, Cassidy F, Provenzale JM, Lehman A, Weisler RH, Carroll BJ, Krishnan KR: MRI correlates of suicide attempt history in unipolar depression. Biol Psychiatry 2001, 50: 266–270. 10.1016/S0006-3223(01)01098-8
Amore M, Innamorati M, Costi S, Sher L, Girardi P, Pompili M: Partial androgen deficiency, depression, and testosterone supplementation in aging men. Int J Endocrinol 2012, 2012: 280724.
Seidman SN, Araujo AB, Roose SP, McKinlay JB: Testosterone level, androgen receptor polymorphism, and depressive symptoms in middle-aged men. Biol Psychiatry 2001, 50: 371–376. 10.1016/S0006-3223(01)01148-9
Ceretta LB, Reus GZ, Abelaira HM, Jornada LK, Schwalm MT, Hoepers NJ, Tomazzi CD, Gulbis KG, Ceretta RA, Quevedo J: Increased prevalence of mood disorders and suicidal ideation in type 2 diabetic patients. Acta Diabetol 2012,49(Suppl 1):S227–34.
Bourke C, Harrell CS, Neigh GN: Stress-induced sex differences: Adaptations mediated by the glucocorticoid receptor. Horm Behav 2012, 62: 210–8. 10.1016/j.yhbeh.2012.02.024
Cahill L: His brain, her brain. Sci Am 2005, 292: 40–47.
Campbell T, Lin S, DeVries C, Lambert K: Coping strategies in male and female rats exposed to multiple stressors. Physiol Behav 2003, 78: 495–504. 10.1016/S0031-9384(03)00033-7
Capuron L, Gumnick JF, Musselman DL, Lawson DH, Reemsnyder A, Nemeroff CB, Miller AH: Neurobehavioral effects of interferon-alpha in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology 2002, 26: 643–652. 10.1016/S0893-133X(01)00407-9
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW: From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 2008, 9: 46–56. 10.1038/nrn2297
Acknowledgements
The authors wish to acknowledge Jillybeth Burgado, Renuka Reddy, and Kaela Singleton for their assistance in the primary literature review and final editing steps. Support for GNN was provided by the Creative and Novel Ideas in HIV Research Program (CNIHR) through a supplement to the University of Alabama at Birmingham (UAB) Center for AIDS Research funding (P30 AI027767-24). This funding was made possible by collaborative efforts of the Office of AIDS Research, the National Institutes of Allergies and Infectious Diseases, and the International AIDS Society. Additional support for GNN was provided by NIH grant MH091312. Support for KDB was provided by the U.S. Department of Veterans Affairs Office of Biomedical Laboratory Research and Development (I01 BX000218). The opinions expressed are those of the authors and do not reflect the official position of the U.S. Department of Veterans Affairs.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
CLN declares that she has no competing interests. Authors CSH, KDB, and GNN are members of the Organization for the Study of Sex Differences, for which the Biology of Sex Differences is the official journal.
Authors’ contributions
CLN and CSH participated equally in the literature review, interpretation of the findings, and drafting of the manuscript. KDB helped conceive of the review and participated in drafting. GNN helped conceive of the review, coordinated the efforts of the literature search and interpretation, and participated in drafting the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Nemeth, C.L., Harrell, C.S., Beck, K.D. et al. Not all depression is created equal: sex interacts with disease to precipitate depression. Biol Sex Differ 4, 8 (2013). https://doi.org/10.1186/2042-6410-4-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2042-6410-4-8