
Abstract
Background
Deletions of the mitochondrial DNA (mtDNA) accumulate to high levels in dopaminergic neurons of the substantia nigra pars compacta (SNc) in normal aging and in patients with Parkinson's disease (PD). Human nigral neurons characteristically contain the pigment neuromelanin (NM), which is believed to alter the cellular redox-status. The impact of neuronal pigmentation, neurotransmitter status and brainstem location on the susceptibility to mtDNA damage remains unclear. We quantified mtDNA deletions (ΔmtDNA) in single pigmented and non-pigmented catecholaminergic, as well as non-catecholaminergic neurons of the human SNc, the ventral tegmental area (VTA) and the locus coeruleus (LC), using laser capture microdissection and single-cell real-time PCR.
Results
In healthy aged individuals, ΔmtDNA levels were highest in pigmented catecholaminergic neurons (25.2 ± 14.9%), followed by non-pigmented catecholamergic (18.0 ± 11.2%) and non-catecholaminergic neurons (12.3 ± 12.3%; p < 0.001). Within the catecholaminergic population, ΔmtDNA levels were highest in dopaminergic neurons of the SNc (33.9 ± 21.6%) followed by dopaminergic neurons of the VTA (21.9 ± 12.3%) and noradrenergic neurons of the LC (11.1 ± 11.4%; p < 0.001). In PD patients, there was a trend to an elevated mutation load in surviving non-pigmented nigral neurons (27.13 ± 16.73) compared to age-matched controls (19.15 ± 11.06; p = 0.052), but levels where similar in pigmented nigral neurons of PD patients (41.62 ± 19.61) and controls (41.80 ± 22.62).
Conclusions
Catecholaminergic brainstem neurons are differentially susceptible to mtDNA damage. Pigmented dopaminergic neurons of the SNc show the highest ΔmtDNA levels, possibly explaining the exceptional vulnerability of the nigro-striatal system in PD and aging. Although loss of pigmented noradrenergic LC neurons also is an early feature of PD pathology, mtDNA levels are not elevated in this nucleus in healthy controls. Thus, ΔmtDNA are neither an inevitable consequence of catecholamine metabolism nor a universal explanation for the regional vulnerability seen in PD.
Similar content being viewed by others


Background
Oxidative stress and mitochondrial dysfunction are believed to have a dominant role in mechanisms of aging and neurodegenerative disorders such as Parkinson disease (PD) [1]. The mitochondrial theory of aging proposes that production of reactive oxygen species (ROS) in mitochondria causes accumulating damage to proteins, lipids, and mitochondrial DNA (mtDNA). As a consequence, mitochondrial dysfunction and ROS production may build up in a vicious cycle that eventually results in cell death [2, 3]. Damage to mtDNA is central to this theory and early studies provide evidence for the accumulation of somatic mtDNA deletions (ΔmtDNA) in aging postmitotic tissues with high energy demand, such as skeletal muscle and the brain [4, 5]. Nevertheless, due to the low abundance of ΔmtDNA detected in crude tissue homogenates, their functional significance remained controversial. By combining laser-microdissection (LMD) with a quantitative real-time PCR (RT-PCR) assay, we demonstrated the age-related accumulation of clonally expanded ΔmtDNA in individual post mortem dopaminergic neurons of the substantia nigra pars compacta (SNc) [6]. Indicative of a resulting functionally relevant biochemical defect, neurons with high levels (~ 60%) ΔmtDNA had mitochondrial cytochrome-c oxidase (COX, complex IV of the mitochondrial respiratoy chain) deficiency on histochemical examination in PD patients and controls. These findings were independently confirmed using a different methodological approach [7]. Our studies into the regional distribution of ΔmtDNA further showed that dopaminergic nigral neurons have a higher propensity to accumulate ΔmtDNA than extranigral populations, e.g. in the putamen, the hippocampus or the frontal cortex [8, 9]. Besides their catecholaminergic neurotransmitter status, a prominent feature of these neurons is their pigmentation, i.e. the intraneuronal accumulation of neuromelanin (NM). NM has long been considered a cellular waste product via the non-enzymatic oxidization of dopamine or other catecholamines, but some evidence points towards a regulated production that might involve alpha-synuclein [10, 11]. Its contribution to neurodegenerative processes is far from understood as there is evidence for both neuroprotective and neurotoxic properties [12–14]. As a possible mechanism, it has been proposed that NM might serve to control iron homeostasis within pigmented neurons [15]. If the iron chelation ability of NM is reduced, increased levels of intra-neuronal free iron may stimulate ROS production. In PD, pigmented neurons contain less NM than in healthy brains, while the optical density of the pigment is increased [16, 17]. These changes of neuronal NM content and composition may cause a loss of protective properties [18]. Indeed, we have previously shown that individual dopaminergic neurons have elevated iron levels in PD [19]. Thus, increased ROS generation, mtDNA mutations and mitochondrial dysfunction might pave the way for neurodegenerative processes in PD [20].
In this context the question arises, if location, neurotransmitter status or the presence of NM determines the vulnerability of dopaminergic nigral neurons to accumulate mtDNA damage. Herein, we investigated the association of these factors with ∆mtDNA levels in single post mortem catecholaminergic neurons that were dissected from the SNc, the ventral tegmental area (VTA) and the locus coeruleus (LC) of post mortem brains of healthy aged individuals and PD patients.
Results
Association of pigmentation and ΔmtDNA levels
In the adult human brain, NM is easily identifiable as a black-brown pigment by light microscopy. NM-containing neurons are distributed throughout the entire brainstem, but the largest clusters are found in the SNc, the VTA and the LC [18]. In a pilot experiment, we randomly collected pigmented neurons from the SNc of five healthy controls (80.8 ± 8.6 y) and compared ΔmtDNA levels to those seen in non-pigmented neurons within the same specimens. We found that non-pigmented neurons had a mean of 31.0 ± 25.1% ΔmtDNA, whereas pigmented neurons had a mean of 49.2% ± 18.3% ΔmtDNA (p = 0.017). This preliminary data suggested that an increased vulnerability is associated with NM-pigmentation of midbrain neurons in healthy aged controls.
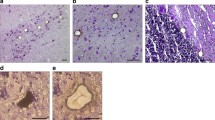
Encouraged by these results, we next established an immunohistochemical staining protocol to facilitate a more specific and extensive analysis of catecholaminergic neurons of the SNc, the VTA and the LC. Using a successive TH/NeuN antibody labeling and DAB/HRP reaction, catecholaminergic (TH+) neurons were positively discriminated from non-catecholaminergic (TH-) neurons (e.g. GABAergic interneurons) and from glia cells. TH+ immunoreactivity results in a brown cytosolic reaction product, while TH- neurons remain unstained. NeuN immunoreactivity results in a grey appearance of TH- neurons due to the successive DAB/Nickel reaction. Pigmented and non-pigmented neurons were further distinguished by the visible presence or absence of NM (Figure 1). For these studies, we extended the analysis with an independent group of 19 controls (68.8 ± 19.4 y) and 14 PD specimens (75.1 ± 7.8 y).

Immunohistochemical identification of target neurons. (a+b) Pigmented vs. non-pigmented catecholaminergic neurons. TH+ neurons were identified by their brown cytosolic reaction product. Pigmented (TH+/NM+; top left) and non-pigmented (TH+/NM-; bottom right, marked for LMD) neurons were further distinguished by the visible presence or absence of NM. (a) TH+/NM- neuron before and (b) after LMD. (c+d) Catecholaminergic vs. non-catecholaminergic neurons. NeuN+ immunoreactivity results in a grey appearance of TH- neurons due to a DAB/Nickel reaction (bottom neuron). (c) TH+/NM+ neuron before and (d) after LMD.
We first asked, whether our initial data could be validated after immunhistochemical identification of catecholaminergic neurons. To this end, we analyzed ΔmtDNA levels of TH+ neurons captured from all three regions (SN, VTA and LC). In a combined analysis of all regions, we confirmed a significant difference of mtDNA levels between non-pigmented (18.0% ± 11.2%) and pigmented neurons (25.2% ± 14.9%; p = 0.003). Thus, in catecholaminergic brainstem neurons, the presence of NM is associated with higher ΔmtDNA levels, independent of location and dopaminergic or noradrenergic neurotransmitter status.
Influence of neurotransmitter status on ΔmtDNA levels
We next asked whether ΔmtDNA levels are generally higher in TH+ (catecholaminergic) compared to TH- neurons, i.e. neurons that contain neither dopamine nor noradrenaline. For this analysis, TH-/NeuN+ neurons were additionally sampled from SN, VTA and LC. RT-PCR data showed that TH-negative midbrain neurons had low mtDNA levels of 12.3% ± 12.3. After combining this and the previous data for statistical analysis, we found a significant difference between TH- neurons and pigmented as well as non-pigmented TH+ neurons (Mann-Whitney-Test; p = 0.005). In these three groups, levels of mtDNA increase in the order (Kruskal-Wallis-Test; p < 0.001): non-catecholaminergic (12.3%) < catecholaminergic/non-pigmented (18.0%) < catecholaminergic/pigmented (25.2%) (Figure 2).

Differential susceptibility of catecholaminergic pigmented neurons to deletions of mitochondrial DNA. Levels of ΔmtDNA raise in the order of non-catecholaminergic (TH-/NeuN+) < catecholaminergic/non-pigmented (TH+/NM-) < catecholaminergic/pigmented neurons (TH+/NM+). Differences were significant at p = 0.005 (**; Mann-Whitney-Test) and p = 0.001 (***; Kruskal-Wallis-Test).
Vulnerability of SNc, VTA and LC neurons
The SNc and VTA both predominately contain dopaminergic neurons, while the LC contains noradrenergic neurons. In the SNc and in the LC the pigment is found in approximately 95% of neurons, whereas in the VTA only about 50% neurons are pigmented [11]. We therefore asked, to what extend the location has an impact on deletion levels independent of neuronal pigmentation. Combined analysis of pigmented and non-pigmented TH+ neurons showed an average of 11.1 ± 11.4% ΔmtDNA in the LC, 21.9 ± 12.3% in the VTA and 33.9 ± 21.6% in the SNc. Interregional differences were significant between all groups at p < 0.001 (Figure 3). Thus, dopaminergic neurons of the SNc are more susceptible to ΔmtDNA than those of the VTA and both are more susceptible than noradrenergic neurons of the LC.

Differential vulnerability of SNc, VTA and LC neurons to deletions of mitochondrial DNA. Lowest levels were seen for noradrenergic neurons of the LC, followed by dopaminergic neurons of the VTA and the SNc. Differences were significant at p = 0.001 (***; Kruskal-Wallis-Test).
ΔmtDNA levels in PD nigral neurons
Lastly, we quantified ΔmtDNA levels in PD cases (n = 14; mean age 75.1 ± 7.8 years). Collection and analysis was restricted to SN dopaminergic neurons due to the paucity of suitable tissue samples. In PD, ΔmtDNA levels of pigmented neurons (41.62 ± 19.61) were again higher than those of non-pigmented neurons (27.13 ± 16.73; p = 9.6E-05), thus independently reproducing the results seen in the control group. We then compared ΔmtDNA levels in PD cases to those seen in age-matched controls (n = 19; mean age 78.7 ± 9.0 years). In non-pigmented neurons there was a trend to higher deletions in PD vs. controls (PD = 27.13 ± 16.73; controls = 19.15 ± 11.06; p = 0.052). No difference was seen for pigmented neurons in PD (41.62 ± 19.61) and controls (41.80 ± 22.62; Figure 4).

Levels of mitochondrial DNA deletions in nigral neurons of PD and controls. Pigmented neurons (TH+/NM+) of the SNc have considerably higher ΔmtDNA levels than non-pigmented neurons (TH+/NM-) in controls (C) and PD (*** p = 0.001). In nonpigmented neurons there was a trend to higher deletions in PD vs. controls (p = 0.052).
Differential vulnerability of catecholaminergic brainstem nuclei in healthy aging
To generate a concise picture of ΔmtDNA levels in the aged human brain, we extracted data coming from all control individuals over 60 years of age (n = 19; mean age 78.7 ± 9.0 years). In our synopsis, individual levels of ΔmtDNA in relation to brainstem location, pigmentation and neurotransmitter status are illustrated (Figure 5). This data underlines the prominent vulnerability of pigmented nigral neurons, followed by pigmented neurons of the VTA and non-pigmented neurons of both nuclei, whereas the LC has overall low deletion levels.

Differential vulnerability of catecholaminergic brainstem neurons in healthy aged controls. 3-D model of the brain showing location of brainstem nuclei and ΔmtDNA levels of pigmented (TH+/NM+) and non-pigmented (TH+/NM-) catecholaminergic neurons, as well as non-catecholaminergic (TH-) neurons in these nuclei. Values represent mean ± standard deviation of data collected from healthy aged controls (78.7 ± 9.0 years). Highest deletion levels are seen in pigmented neurons of the SNc (black). VTA neurons show intermediate ΔmtDNA levels (grey) and LC neurons lowest (light grey). PD pathology is deviating from this pattern, as SNc and LC show heavy degeneration whereas the VTA is relatively spared.
Discussion
Degeneration of pigmented dopaminergic neurons is one of the neuropathological hallmarks of PD [21, 22]. In these neurons, ROS generation may be enhanced by the presence of autooxidizable dopamine, low glutathion and high iron content [23]. Additionally, recent data has revealed the reliance on Ca2+ channels to maintain autonomous activity in aging dopaminergic neurons, causing sustained metabolic stress on mitochondria [24]. Decreased expression of nuclear encoded mitochondrial genes and of genes in energy-sensing pathways might further aggravate mitochondrial dysfunction [25]. The combination of these and other factors likely accelerates cellular aging processes and propagates clonal expansion of mtDNA, which are believed to arise during the repair of oxidatively damaged mtDNA [6, 26, 27]. COX-deficient neurons are also found in hippocampal neurons of aged individuals and Alzheimer's patients, but dopaminergic neurons of the SNc accumulate deletions to considerably higher levels [8, 9]. These neurons are characterized by the age-related appearance and accumulation of NM-pigment. In vitro data provides an intriguing view on the potential protective and harmful properties of NM, which might change depending on the melanin species, its protein component, sulfhydryl residues and the cellular redox-state. Depletion of glutathione and upregulation of glutathione peroxidase activity in response to oxidative stress may further drive the production of neuromelanin [28]. While NM can cause apoptosis of DA neurons through an impact on mitochondrial redox state and S-glutathionylation [29], a protective role was shown in primary mesencephalic neurons under conditions of high oxidative load [30].
Increased ΔmtDNA levels in pigmented neurons
To further elucidate the relationship of pigmentation and ROS damage in the human brain, we determined ΔmtDNA levels by RT-PCR analysis of single human post mortem brainstem neurons that were obtained from control individuals and PD patients by immunohistochemical characterization and LMD. The primary finding of this study is that ΔmtDNA levels are reproducibly elevated in neurons containing NM compared to non-pigmented neurons. To our knowledge, these data establish for the first time an association of neuronal pigmentation and mtDNA damage in human post mortem brain. Due to the limitations of a post mortem study, a causal relationship is difficult to prove or exclude. On the one hand, NM itself might cause increased ROS and mtDNA damage leading to the clonal expansion of ΔmtDNA. On the other hand, an independent pathophysiological mechanism may impose cellular stress on these neurons and cause increase in both, ΔmtDNA levels and NM content.
Only dopaminergic neurons show elevated ΔmtDNA levels
In the literature, several lines of evidence argue against the notion that NM is a simple degradation product of catecholamine transmitter metabolism. On the contrary, NM synthesis and turnover may underlie a yet to determined enzymatic regulatory process, possibly involving alpha-synuclein [11, 17, 31]. While NM is found in most dopaminergic neurons of the SN and noradrenergic neurons of the LC, it does not develop in dopaminergic neurons of the olfactory bulb, some hypothalamic nuclei, nor in medullary adrenergic neurons [32]. Moreover, despite the fact that TH is the rate limiting enzyme in catecholamine synthesis there is no clear correlation between the degree of NM pigmentation and TH immunoreactivity [33, 34]. We therefore asked, whether ΔmtDNA levels are generally higher in catecholaminergic than in non-catecholaminergic neurons. We found that in noradrenergic neurons of the LC, ΔmtDNA were not elevated compared to TH- neurons. In contrast to this, dopaminergic neurons of the SNc and VTA have considerably elevated ΔmtDNA levels (Figure 2) compared to other neuronal populations.
SNc neurons are highly susceptible to ΔmtDNA - LC neurons are not
During PD disease-progression, intraneuronal pathology and neurodegeneration are seen in an 'ascending' pattern throughout the entire brain (i.e. Braak stages) [21]. Within the brainstem, catecholaminergic nuclei display a differential pathology: whereas the LC is affected early in disease and degeneration is reported to exceed that of the SNc [35], neuronal survival in the VTA is considerably higher even in severe cases [36, 37]. We therefore sought to determine mtDNA mutation load in these nuclei in healthy controls. If ΔmtDNA played an integral part in the selective vulnerability in PD, a regional pattern of ΔmtDNA resembling that of Braak stages might be seen. Contrary to these expections, we found that deletion levels were considerably higher in SNc neurons than in the similarly pigmented LC neurons, (Figure 3). This raises doubts about a causative continuity of catecholamine metabolism > NM pigmentation > ROS generation > mtDNA damage. Thus, ΔmtDNA levels are not tightly associated with the differential neuronal vulnerability seen in PD (Figure 5). This finding argues against the notion that PD pathology is an acceleration of molecular events found in healthy aging, which has also been shown in morphometric studies [34]. While the agedependent loss of DA neurons is a linear process and mainly affects the dorsal part of the SNc (6,9% loss per decade), followed by the medial ventral part (5,4% per decade) and nearly sparing the ventral part (2,1% loss per decade), neurodegeneration in PD is rapidly progressive with an overall loss of DA neurons of 45% in the first decade. Furthermore, the ventral part of the SNc is the most affected part in this progress with an average loss of 95%, followed by the medial ventral part (71%) and the dorsal part (56%). It has to be stressed that - due to tissue limitations - we were not able to analyze the regional distribution of ΔmtDNA in PD samples to the extent done for controls. Therefore, we cannot rule out that ΔmtDNA levels increase to significant levels in the LC or other neuronal populations affected in PD.
Elevated ΔmtDNA levels in SNc of PD
We further asked, whether ΔmtDNA levels reflect disease pathology in PD as was suggested by previous studies [6]. Results from PD cases independently confirmed higher ΔmtDNA levels in pigmented vs. non-pigmented neurons. Compared to controls, we found a not significant trend to higher ΔmtDNA levels in non-pigmented SNc neurons of PD brains, but this was not seen for pigmented neurons (Figure 4). The interpretation of this data is complicated by the extensive and differential neuronal loss seen in PD brains. Most severely affected neurons might have already been lost and probably only well protected and 'more resistant' neurons were left for sampling. Furthermore, within the SNc, a regional vulnerability is seen for PD: nigral neurons of the lightly pigmented ventral tier degenerate first, while the heavily pigmented of the dorsal tier are relatively preserved [16, 34, 38]. If pigmented neurons are mainly sampled from the pool of 'resistant' neurons located in the dorsal tier this might result in similar ΔmtDNA levels comparing PD and controls. In this study, the available midbrain samples did not allow for the discrimination of regional differences in PD, but this is planned for future studies.
Following a different line of argumentation, ΔmtDNA may simply play no relevant role in the pathogenesis of PD. Contradicting this notion, high ΔmtDNA levels are associated with a clear biochemical defect in nigral neurons and the percentage of these COX-deficient neurons is increased in the SN of PD patients [6]. Furthermore, gene defects of the mtDNA polymerase γ (POLG) result in the accumulation of ΔmtDNA and can cause parkinsonism [39]. Importantly, breakpoint analysis revealed that the types of ΔmtDNA that have clonally expanded in nigra neurons from PD patients and age-matched controls are similar to those from a patient with POLG mutations who had parkinsonism [40].
This study shows that ΔmtDNA are a common age-related phenomenon in pigmented nigral neurons, both in PD patients and healthy individuals. Thus, accumulation of ΔmtDNA clearly cannot serve as the single explanation for neurodegeneration in PD, but may precipitate dopamine neuron death in combination with other endogenous and exogenous factors. Interestingly, recent data implies that age-related accumulation of NM-pigment might induce α-synuclein expression, another important factor determining PD pathology [41].
Conclusions
NM formation and turnover might constitute a protective system regulating neuronal redox state [11, 18]. On the other hand, NM may contribute to ROS generation through release of redox-active iron under certain pathological conditions [15]. In this study, we found increased ΔmtDNA levels in pigmented midbrain neurons, which is consistent with both theories. Dopaminergic neurons have elevated levels of ΔmtDNA, supporting the role of dopamine metabolism in the generation of ROS and mtDNA damage. Importantly, in noradrenergic neurons, a causative relation of pigmentation, production of ROS and accumulation of ΔmtDNA cannot be established. Since the LC is affected early and severely in PD pathology, different factors must account for the vulnerability of catecholamingergic neurons in this nucleus.
Methods
Ethics statement
Frozen midbrain tissue was requested from the Newcastle Brain Tissue Resource and the German brain bank (Brain-Net®). Written consent was obtained with verification/assent in writing from next of kin who confirmed the wishes at time of death. All procedures were in line with the UK Human Tissue Authority guidance and approved by the Local Research Ethics Committee. The study is in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki.
Patients and controls
Control individuals had no prior history of neurological disease (n = 24, mean age 75.1 ± 7.8 years, post mortem interval (PMI) 24.0 ± 9.0 hours). Patients had a clinical and neuropathological diagnosis of PD (n = 14, mean age 71.3 ± 18.2 years, PMI 32.1 ± 18.7 hours). Neither age (p = 0.24) nor PMI (p = 0.1) were significantly different between groups. Neuropathological examination had demonstrated the presence of Lewy body pathology in the substantia nigra with typical pathological features, including moderate to severe neuronal loss and gliosis. Synuclein immunohistochemistry or ubiquitin immunohistochemistry was used to confirm findings from H&E stained sections and cases were graded according to published criteria for Lewy body disorders (LBD)[21, 42].
Histology and immunohistochemistry
Unfixed human midbrains stored at -80°C were used for analysis. 20 μm sections were cut and mounted onto Leica 2 μm PEN membrane slides (Leica Microsystems, Wetzlar, Germany) prior to staining and microdissection. For pilot experiments (n = 5 controls), sections were stained with cresyl violet (Merck, Darmstadt, Germany) and dehydrated in an ethanol series. For most experiments, a successive Tyrosin-Hydroxylase (TH) and Neuronal Nuclei (NeuN) double-staining was applied. In detail, frozen sections were fixed in ice-chilled acetone for 7 minutes and air-dried for 10 minutes at room temperature (RT). All solutions were prepared in phosphate buffered saline 1% Triton X-100 (PBST) and all incubations were performed at RT. Sections were washed twice in PBST and blocked with 5% normal goat serum (NGS) for 30 min. An anti-mouse poly horseradish peroxidase (HRP) kit was used for detection of primary antibodies according to the manufacturers protocol (Millipore, Billerica, MA, USA). Primary rabbit anti-TH antibody (abcam, Cambridge, UK) was applied at 1:300 in PBST with 5% NGS for 60 min and staining was developed with the DAB Chromogen-Buffer provided with the kit for 5 min. Sections were washed in tap water followed by blocking and incubation with 1:400 mouse IgG1 anti-NeuN (Millipore, Billerica, MA, USA) for 45 min. For successive NeuN detection, nickel chloride was added to the DAB substrate solution and developed for 10 min. The addition of nickel chloride produces a dark grey appearance of NeuN+ neurons, thereby enabling distinction from the light brown of DAB-mediated TH+ immunolabelling. Finally, sections were washed, dehydrated in 100% ethanol and air-dried for 30 min. Membrane sections were used for LMD immediately or frozen at -20°C for later use.
Quantification of ΔmtDNA
UV-Laser-microdissection was performed on a Leica LMD6000 microscope (Leica, Wetzlar, Germany). Single neurons were collected into separate reaction tubes and DNA was extracted with the DNA Micro Kit (Qiagen, Düsseldorf, Germany), according to the manufacturer's protocol. Quantification of ΔmtDNA levels was based on the RT-PCR method previously described, using relative quantification of the mitochondrial ND1 and ND4 genes by means of the delta-delta-CT-method [6]. Differing from the original method, we further optimized the RT-PCR assay to be run as a duplex experiment with quantification of ND1 and ND4 genes within the same reaction. Using this protocol, there is high correlation with deletion quantification by southern blot and by the original method [43]. The following primers (MWG Biotech, Ebersberg, Germany) and TaqMan probes (Life Technologies, Carlsbad, CA, USA) were used: ND1 (forward primer nt 3485-3504, reverse primer nt 3553-3532; VIC-labeled probe nt 3506-3529) and ND4 (forward primer nt 12087-12109, reverse primer nt 12170-12140, FAM-labeled probe nt 12111-12138). Final concentrations were 900nM for primers and 250nM for probes. The Taqman Universal PCR Mastermix (Life Technologies, Carlsbad, CA, USA) was used for the assay in a 25 μl reaction mix per sample. Experiments were performed on an Applied Biosystems StepOnePlusTM system (Life Technologies, Carlsbad, CA, USA). Standard cycling conditions were used as follows: activation for 2 minutes @ 50°C followed by 10 minutes @ 95°C; PCR (40 cycles) for 15 seconds @ 95°C followed by 1 minute at 60°C. Samples were analyzed in triplicates and the resulting mean values were used for statistical analysis.
Statistical analysis and graphic design
All statistical analyses were performed with SPSS 18.0 for Mac (PASW Statistics, IBM). A total of 383 neurons were captured and mtDNA deletion levels were analyzed for every single neuron. Data of all neurons was available for analysis. Since the mtDNA deletion values did not have a normal distribution (Kolmogorov-Smirnov-Test p = 0.007), we performed non-parametric tests for all statistical procedures (Mann-Whitney-Test and Kruskal-Wallis one-way analysis of variance). Values are expressed as mean ± standard deviation. Box plots show median, lower and upper quartile. Whiskers are extended to extreme data points. Figure 5 was generated using Cinema 4D (Maxon, Friedrichsdorf, Germany).
Abbreviations
- mtDNA:
-
mitochondrial DNA
- SNc:
-
substantia nigra pars compacta
- VTA:
-
ventral tegmental area
- LC:
-
locus coeruleus
- PD:
-
Parkinson's disease
- NM:
-
neuromelanin
- mtDNA:
-
mtDNA deletions
- ROS:
-
reactive oxygen species
- LMD:
-
laser-microdissection
- TH:
-
tyrosine hydroxylase
- COX:
-
cytochrome-c oxidase
- PMI:
-
post mortem interval.
References
Schapira AH, Gegg M: Mitochondrial contribution to Parkinson's disease pathogenesis. Parkinsons Dis. 2011, 2011: 159160-
Harman D: The biologic clock: the mitochondria?. J Am Geriatr Soc. 1972, 20: 145-147.
Surmeier DJ, Guzman JN, Sanchez-Padilla J, Goldberg JA: The origins of oxidant stress in Parkinson's disease and therapeutic strategies. Antioxid Redox Signal. 2011, 14: 1289-1301. 10.1089/ars.2010.3521.
Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC: Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet. 1992, 2: 324-329. 10.1038/ng1292-324.
Cortopassi GA, Shibata D, Soong NW, Arnheim N: A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc Natl Acad Sci USA. 1992, 89: 7370-7374. 10.1073/pnas.89.16.7370.
Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM: High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006, 38: 515-517. 10.1038/ng1769.
Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K: Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006, 38: 518-520. 10.1038/ng1778.
Bender A, Schwarzkopf RM, McMillan A, Krishnan KJ, Rieder G, Neumann M, Elstner M, Turnbull DM, Klopstock T: Dopaminergic midbrain neurons are the prime target for mitochondrial DNA deletions. J Neurol. 2008, 255: 1231-1235. 10.1007/s00415-008-0892-9.
Krishnan KJ, Ratnaike TE, Gruyter HL, Jaros E, Turnbull DM: Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer's disease. Neurobiol Aging. 2011
Bisaglia M, Mammi S, Bubacco L: Kinetic and structural analysis of the early oxidation products of dopamine: analysis of the interactions with alphasynuclein. J Biol Chem. 2007, 282: 15597-15605. 10.1074/jbc.M610893200.
Double KL, Dedov VN, Fedorow H, Kettle E, Halliday GM, Garner B, Brunk UT: The comparative biology of neuromelanin and lipofuscin in the human brain. Cell Mol Life Sci. 2008, 65: 1669-1682. 10.1007/s00018-008-7581-9.
Fasano M, Bergamasco B, Lopiano L: Modifications of the iron-neuromelanin system in Parkinson's disease. J Neurochem. 2006, 96: 909-916. 10.1111/j.1471-4159.2005.03638.x.
Zhang W, Phillips K, Wielgus AR, Liu J, Albertini A, Zucca FA, Faust R, Qian SY, Miller DS, Chignell CF, Wilson B, Jackson-Lewis V, Przedborski S, Joset D, Loike J, Hong JS, Sulzer D, Zecca L: Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: implications for progression of Parkinson's disease. Neurotox Res. 2011, 19: 63-72. 10.1007/s12640-009-9140-z.
Zecca L, Casella L, Albertini A, Bellei C, Zucca FA, Engelen M, Zadlo A, Szewczyk G, Zareba M, Sarna T: Neuromelanin can protect against iron-mediated oxidative damage in system modeling iron overload of brain aging and Parkinson's disease. J Neurochem. 2008, 106: 1866-1875.
Gerlach M, Double KL, Ben-Shachar D, Zecca L, Youdim MB, Riederer P: Neuromelanin and its interaction with iron as a potential risk factor for dopaminergic neurodegeneration underlying Parkinson's disease. Neurotox Res. 2003, 5: 35-44. 10.1007/BF03033371.
Kastner A, Hirsch EC, Lejeune O, Javoy-Agid F, Rascol O, Agid Y: Is the vulnerability of neurons in the substantia nigra of patients with Parkinson's disease related to their neuromelanin content?. J Neurochem. 1992, 59: 1080-1089. 10.1111/j.1471-4159.1992.tb08350.x.
Halliday GM, Ophof A, Broe M, Jensen PH, Kettle E, Fedorow H, Cartwright MI, Griffiths FM, Shepherd CE, Double KL: Alpha-synuclein redistributes to neuromelanin lipid in the substantia nigra early in Parkinson's disease. Brain. 2005, 128: 2654-2664. 10.1093/brain/awh584.
Fedorow H, Tribl F, Halliday G, Gerlach M, Riederer P, Double KL: Neuromelanin in human dopamine neurons: comparison with peripheral melanins and relevance to Parkinson's disease. Prog Neurobiol. 2005, 75: 109-124. 10.1016/j.pneurobio.2005.02.001.
Oakley AE, Collingwood JF, Dobson J, Love G, Perrott HR, Edwardson JA, Elstner M, Morris CM: Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology. 2007, 68: 1820-1825. 10.1212/01.wnl.0000262033.01945.9a.
Sulzer D: Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 2007, 30: 244-250. 10.1016/j.tins.2007.03.009.
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E: Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003, 24: 197-211. 10.1016/S0197-4580(02)00065-9.
Hirsch E, Graybiel AM, Agid YA: Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature. 1988, 334: 345-348. 10.1038/334345a0.
Chinta SJ, Andersen JK: Redox imbalance in Parkinson's disease. Biochim Biophys Acta. 2008, 1780: 1362-1367. 10.1016/j.bbagen.2008.02.005.
Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ: Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010, 468: 696-700. 10.1038/nature09536.
Elstner M, Morris CM, Heim K, Bender A, Mehta D, Jaros E, Klopstock T, Meitinger T, Turnbull DM, Prokisch H: Expression analysis of dopaminergic neurons in Parkinson's disease and aging links transcriptional dysregulation of energy metabolism to cell death. Acta Neuropathol. 2011, 122: 75-86. 10.1007/s00401-011-0828-9.
Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, Wanrooij S, Spelbrink JN, Lightowlers RN, Turnbull DM: What causes mitochondrial DNA deletions in human cells?. Nat Genet. 2008, 40: 275-279. 10.1038/ng.f.94.
Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, Krantz DE, Kobayashi K, Edwards RH, Sulzer D: Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009, 62: 218-229. 10.1016/j.neuron.2009.01.033.
Bellinger FP, Bellinger MT, Seale LA, Takemoto AS, Raman AV, Miki T, Manning- Bog AB, Berry MJ, White LR, Ross GW: Glutathione Peroxidase 4 is associated with Neuromelanin in Substantia Nigra and Dystrophic Axons in Putamen of Parkinson's brain. Mol Neurodegener. 2011, 6: 8-10.1186/1750-1326-6-8.
Naoi M, Maruyama W, Yi H, Yamaoka Y, Shamoto-Nagai M, Akao Y, Gerlach M, Tanaka M, Riederer P: Neuromelanin selectively induces apoptosis in dopaminergic SH-SY5Y cells by deglutathionylation in mitochondria: involvement of the protein and melanin component. J Neurochem. 2008, 105: 2489-2500. 10.1111/j.1471-4159.2008.05329.x.
Li J, Scheller C, Koutsilieri E, Griffiths F, Beart PM, Mercer LD, Halliday G, Kettle E, Rowe D, Riederer P, Gerlach M, Rodriguez M, Double KL: Differential effects of human neuromelanin and synthetic dopamine melanin on neuronal and glial cells. J Neurochem. 2005, 95: 599-608. 10.1111/j.1471-4159.2005.03404.x.
Fedorow H, Halliday GM, Rickert CH, Gerlach M, Riederer P, Double KL: Evidence for specific phases in the development of human neuromelanin. Neurobiol Aging. 2006, 27: 506-512. 10.1016/j.neurobiolaging.2005.02.015.
Saper CB, Petito CK: Correspondence of melanin-pigmented neurons in human brain with A1-A14 catecholamine cell groups. Brain. 1982, 105: 87-101. 10.1093/brain/105.1.87.
Gaspar P, Berger B, Gay M, Hamon M, Cesselin F, Vigny A, Javoy-Agid F, Agid Y: Tyrosine hydroxylase and methionine-enkephalin in the human mesencephalon. Immunocytochemical localization and relationships. J Neurol Sci. 1983, 58: 247-267. 10.1016/0022-510X(83)90221-6.
Fearnley JM, Lees AJ: Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain. 1991, 114 (Pt 5): 2283-2301.
Zarow C, Lyness SA, Mortimer JA, Chui HC: Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003, 60: 337-341. 10.1001/archneur.60.3.337.
McRitchie DA, Cartwright HR, Halliday GM: Specific A10 dopaminergic nuclei in the midbrain degenerate in Parkinson's disease. Exp Neurol. 1997, 144: 202-213. 10.1006/exnr.1997.6418.
Damier P, Hirsch EC, Agid Y, Graybiel AM: The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain. 1999, 122 (Pt 8): 1437-1448.
Gibb WR, Lees AJ: Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson's disease. J Neurol Neurosurg Psychiatry. 1991, 54: 388-396. 10.1136/jnnp.54.5.388.
Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, Oldfors A, Rautakorpi I, Peltonen L, Majamaa K, Somer H, Suomalainen A: Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004, 364: 875-882. 10.1016/S0140-6736(04)16983-3.
Reeve AK, Krishnan KJ, Elson JL, Morris CM, Bender A, Lightowlers RN, Turnbull DM: Nature of mitochondrial DNA deletions in substantia nigra neurons. Am J Hum Genet. 2008, 82: 228-235. 10.1016/j.ajhg.2007.09.018.
Xuan Q, Xu SL, Lu DH, Yu S, Zhou M, Ueda K, Cui YQ, Zhang BY, Chan P: Increase expression of alpha-synuclein in aged human brain associated with neuromelanin accumulation. J Neural Transm. 2011, 118: 1575-1583. 10.1007/s00702-011-0636-3.
McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, et al: Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005, 65: 1863-1872. 10.1212/01.wnl.0000187889.17253.b1.
Krishnan KJ, Bender A, Taylor RW, Turnbull DM: A multiplex real-time PCR method to detect and quantify mitochondrial DNA deletions in individual cells. Anal Biochem. 2007, 370: 127-129. 10.1016/j.ab.2007.06.024.
Acknowledgements
This work has been supported by the Else-Kröner-Fresenius-Stiftung (P65/06//EKMS 06/13; AB), the Deutsche Forschungsgemeinschaft (DFG; BE 4185/1-1; AB), the BMBF (NGFN, 01GS08134; BL), the German Federal Ministry of Education and Research (grant No 01EO0901 to the IFBLMU; ME, CL) and the Wellcome Trust and Newcastle University Centre for Brain Ageing and Vitality supported by the Biotechnology and Biological Sciences Research Council, Engineering and Physical Sciences Research Council, Economic and Social Research Council and Medical Research Council (G0700718; DMT). Tissue for this study was provided by the Newcastle Brain Tissue Resource, which is funded in part by grants from the UK Medical Research Council (G0400074; DMT, CM), and the Alzheimer's Society and Alzheimer's Research Trust as part of the Brains for Dementia Research Project. We would like to thank Sebastian Krieg of '3D-bewegungswerkstatt' for the graphic design of figure 5.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AB, TK and ME designed the study. HP, EM, DMT aided with design details and contributed essential interpretations of findings. CM, BL and FS contributed, characterized and prepared tissue samples for the experiments. SKM, LL, CL and LK performed histochemistry and LMD. ME directly supervised experiments and wrote the paper with assistance of all authors, who have read and approved the final manuscript.
Thomas Klopstock and Andreas Bender contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Elstner, M., Müller, S.K., Leidolt, L. et al. Neuromelanin, neurotransmitter status and brainstem location determine the differential vulnerability of catecholaminergic neurons to mitochondrial DNA deletions. Mol Brain 4, 43 (2011). https://doi.org/10.1186/1756-6606-4-43
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-6606-4-43