
Abstract
Background
Bicuspid aortic valve (BAV) is the most common type of congenital heart disease with a population prevalence of 1-2%. While BAV is known to be highly heritable, mutations in single genes (such as GATA5 and NOTCH1) have been reported in few human BAV cases. Traditional gene sequencing methods are time and labor intensive, while next-generation high throughput sequencing remains costly for large patient cohorts and requires extensive bioinformatics processing. Here we describe an approach to targeted multi-gene sequencing with combinatorial pooling of samples from BAV patients.
Methods
We studied a previously described cohort of 78 unrelated subjects with echocardiogram-identified BAV. Subjects were identified as having isolated BAV or BAV associated with coarctation of aorta (BAV-CoA). BAV cusp fusion morphology was defined as right-left cusp fusion, right non-coronary cusp fusion, or left non-coronary cusp fusion. Samples were combined into 19 pools using a uniquely overlapping combinatorial design; a given mutation could be attributed to a single individual on the basis of which pools contained the mutation. A custom gene capture of 97 candidate genes was sequenced on the Illumina HiSeq 2000. Multistep bioinformatics processing was performed for base calling, variant identification, and in-silico analysis of putative disease-causing variants.
Results
Targeted capture identified 42 rare, non-synonymous, exonic variants involving 35 of the 97 candidate genes. Among these variants, in-silico analysis classified 33 of these variants as putative disease-causing changes. Sanger sequencing confirmed thirty-one of these variants, found among 16 individuals. There were no significant differences in variant burden among BAV fusion phenotypes or isolated BAV versus BAV-CoA. Pathway analysis suggests a role for the WNT signaling pathway in human BAV.
Conclusion
We successfully developed a pooling and targeted capture strategy that enabled rapid and cost effective next generation sequencing of target genes in a large patient cohort. This approach identified a large number of putative disease-causing variants in a cohort of patients with BAV, including variants in 26 genes not previously associated with human BAV. The data suggest that BAV heritability is complex and polygenic. Our pooling approach saved over $39,350 compared to an unpooled, targeted capture sequencing strategy.
Similar content being viewed by others

Background
Congenital bicuspid aortic valve (BAV) is the most common type of cardiac malformation, with an estimated prevalence of 1-2% in the general population [1]. BAV, in which two of the three normal aortic cusps are fused together, encompasses a wide spectrum of clinical phenotypes. The valve abnormality may be isolated in some cases, whereas in others the aortic valve abnormality is present in conjunction with other cardiac malformations [2]. BAV may also be associated with varying degrees of aortic valve stenosis and/or insufficiency as well as with aortopathy. Among BAV patients, there is variability in cusp fusion phenotypes. Right coronary and left coronary (R-L) cusp fusion is more common than right coronary and non-coronary (R-NC) cusp fusion. Moreover, R-L cusp fusion is more often associated with additional cardiac malformations, whereas R-NC cusp fusion is more likely to be associated with aortic valve dysfunction [3]. The etiologies of these associations are unknown.
While multiple studies have demonstrated the high heritability of BAV, the underlying genetic causes remain poorly understood [4–7]. NOTCH1 and GATA5 are the only genes that have been linked to bicuspid aortic valve in humans, yet variants in these genes are present in only a minority of individuals with BAV [8–14]. Mice lacking Gata5 have partially penetrant BAV of the R-NC subtype, but human studies have not yet demonstrated a specific association between GATA5 variants and the R-NC subtype of BAV. Animal models of R-L BAV demonstrate excess fusion of the septal and parietal ridges of the outflow tract, whereas R-NC BAVs result from fusion of the septal ridge and posterior intercalated cushions [15]. These studies suggest that these two cusp fusion phenotypes may arise from distinct genetic perturbations in humans.
Despite tremendous advances in gene sequencing technology, the genetic etiology of many common human conditions, including BAV, remains poorly understood. Candidate gene studies have long been used to detect variants in individual genes; such studies are easy to perform but require selection of genes with a proposed role in the disease process of interest. Genome-wide association studies allow investigators to compare multiple individuals with a given condition and identify common variants in a non-candidate driven approach [16]. However, because genome-wide association studies are predicated upon the common disease-common variant hypothesis, this approach is not ideal for the study of rare variants, particularly in complex conditions in which rare variants at multiple loci may be needed to produce a clinically recognizable phenotype [17, 18].
Next-generation sequencing (NGS) provides an opportunity for rapid, high-throughput sequencing of entire patient genomes and may overcome the limitation of genome-wide association studies in exploring the role of rare variants in complex diseases [19]. Whole genome sequencing remains at this time a costly technology, thus limiting its application to the sequencing of large cohorts of patients. It also produces a vast amount of data necessitating extensive bioinformatics processing. One option to overcome this issue is the design of targeted capture kits that allow for the rapid and accurate sequencing of only the genetic regions of interest. The two most common approaches to this technique have distinct limitations. Sequencing of a targeted set of genes can be done on individual samples, but this approach is very costly in larger cohorts. Alternatively, sequencing can be performed on pools of individual samples, wherein each sample is labeled with a unique genetic “barcode”; this approach is cost saving, but is quite labor intensive [20]. Combinatorial pooling schemes, wherein individuals are sampled in multiple pools, have been utilized to overcome these pitfalls and still permit identification of the individual sample contributing a given rare variant [21, 22].
Here, we present an approach using combinatorial pooling and targeted multi-gene sequencing to study a well-phenotyped cohort of individuals with BAV. We hypothesize that rare variants will be identified amongst a large proportion of the candidate genes, that multiple rare variants will be found in individual probands, and that such variants will segregate by cusp fusion phenotype.
Results
Identification of sequence variants
We studied a previously described cohort of 78 patients with echocardiogram-identified BAV [8]. Using a targeted capture approach, we sequenced 97 candidate genes selected by reviewing the literature for genes relevant to heart valve development.
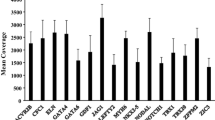
The average depth of coverage for the targeted regions was 268X. Greater than 50X coverage was obtained for 99.04% of the bases sequenced (range: 94.19-99.62), with greater than 100X for 96.11% of bases covered. The percentage of sequencing on target was 71.81%.
Targeted capture identified 42 rare, non-synonymous, exonic variants involving 35 of the candidate genes (Additional file 1: Table S1). Among these variants, in-silico analysis classified 33 of these 42 variants as putative disease-causing changes; Sanger sequencing did not validate two of these 33 variants. The remaining 31 changes were identified in 16 individuals and involved 28 genes (Table 1). Each variant was identified in only one proband. There were no significant differences in variant burden among BAV fusion phenotypes or isolated BAV versus BAV-CoA, with p = 0.78 and p = 0.77, respectively (Additional file 2: Table S2). Only 2 of these variants (rs72541816 at APC and rs116164480 at GATA5) were de novo changes not present in either parent of the affected probands. These two variants were identified in the same individual with a family history of coarctation of the aorta. Of the 16 individuals in whom putative disease-causing variants were identified, two had variants in genes previously known to be involved in human BAV (NOTCH1, GATA5), one of whom we previously described [8]. Four of these 16 individuals had a family history of a left ventricular outflow tract malformation.
Pathway analysis
Pathway analysis was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID). Pathway analysis was used to draw comparisons between the background of only those genes included in the targeted capture and the subset of genes in which rare, non-synonymous exonic variants predicted damaging by in silico analysis were identified. The pathway analysis revealed significant enrichment in genes involved in the WNT signaling pathway (p = 0.035).
Pooling design validation
All samples in the cohort underwent Sanger sequencing of the coding regions of GATA5 as previously reported by our group, used here as a test of the pooling design as well as the sensitivity and specificity of the variant calling algorithm. Four rare variants in GATA5 (each present in 1/78 individuals) were discovered by Sanger sequencing, of which three were identified by NGS [8]. All of the rare GATA5 variants identified by our NGS pooling design were attributed to the correct individual as confirmed by Sanger sequencing.
Sanger sequencing of GATA5 found a variant, p.Q3R, in one individual that was not identified through the pooling design [8]. No pool had this variant above our cut-off threshold of 2.5% (four pools had allele frequencies over 1% with a range of 1.06-1.36%). Coverage of this base was good, with average read depth of 460X.
Discussion
This NGS design utilizing targeted sequencing of pooled BAV patient samples identified 33 rare, non-synonymous exonic variants predicted damaging by in silico analysis. Traditional Sanger sequencing methods confirmed 31 of these 33 changes (94%). Analysis of the GATA5 comparison dataset indicated that the pooling scheme allowed for accurate subject identification. This investigation identified rare variants in 26 genes not previously known to be involved in human BAV; such variants are considered hypothesis-generating and merit further testing in replication cohorts.
Animal models of BAV suggest a possible genotype-phenotype correlation related to cusp fusion phenotypes. However, our data does not support such a correlation in regards to cusp fusion, nor was there a correlation for isolated BAV versus BAV associated with coarctation of the aorta. Sample size and low incidence of familial BAV may limit our ability to detect such an association, but other groups have had similar findings. Rare, non-synonymous exonic variants in GATA5 have not been shown to correlate with cusp fusion [8, 13]. Investigations of familial BAV in large cohorts have demonstrated that cusp fusion morphologies were inherited interchangeably within families [23, 24]. Taken together, these studies suggest that differing BAV phenotypes may derive from a common genetic pathway influenced by downstream modifying elements. Thorough testing of genotype-phenotype correlations would require larger cohorts with significant representation of cusp fusion phenotypes, associated congenital cardiac malformations, aortopathy, and aortic valve insufficiency/stenosis.
Prior to this study, only GATA5 and NOTCH1 variants had been associated with isolated human BAV. Our data identified variants in 26 additional genes not previously identified in human BAV patients. Interestingly, all of these variants are reported in less than 1% of the Exome Variant Server controls and half are absent in this control population. Nonetheless, only 2 of the 31 putative disease-causing changes confirmed by traditional sequencing methods were de novo, in that they were not identified in either parent of the affected proband. We speculate that these 31 variants may be susceptibility alleles, with additional factors (genetic or environmental) required for full phenotype expression [25]. Our finding of multiple variants in the same proband further supports this hypothesis. Among the 16 individuals in whom putative disease-causing variants were identified, the mean variant burden was 1.8 with a range of 1 to 5.
Pathway analysis provides an opportunity to ascribe further meaning to the large number of candidate genes that may be identified in high-throughput approaches such as the one described here. Bioinformatics analysis via DAVID identified significant enrichment of WNT pathway genes including WNT4, PPP3CA, NFATC1, APC, AXIN1 and AXIN 2. DAVID pathway analysis can compare a subset of variants to any background of an investigator’s choosing; by utilizing a background of only the genes included in the targeted capture as opposed to the whole genome, the pathway analysis is not biased by overrepresentation of WNT pathway genes in the targeted capture design. WNT pathway genes display variable expression at various stages in valvulogenesis and have also been implicated in calcific valvular degeneration [26, 27]. Coupling of NGS with pathway analysis allows for the development of more targeted sequencing approaches for subsequent studies. Further investigation into this and similar BAV cohorts could include an enhanced focus on the WNT signaling pathway. A more narrow scope of investigation would then facilitate advanced functional investigations of identified variants.
Several methods are now available for combining multiple individuals into a single sequencing run. Sample-specific indexing uses a short barcode sequence that is unique to each individual in a pool. This barcode is attached to the adapter sequence during library preparation. Commercially available kits now allow up to 96 individuals to be combined in a single run, with deconvolution allowing identification of the individual. Some problems remain in identifying correctly which sequence reads belong to the individual tagged, particularly if single (one end) indexing is used. The pooling method used here does not allow direct deconvolution, but it is not difficult to identify the individual possessing the identified variant. However, the pooling method offers the advantage of error mitigation through use of biological replicates, reducing the false positive rate due to the high frequency of sequencing errors in NGS [28]. Pooling will also overcome problems inherent in the indexing technique itself (including double indexing) that lead to sequencing errors [29].
More precise estimates of the pooling strategy false negative rates and investigation into the causes of these false negatives are necessary to improve the technique. The GATA5 p.Q3R variant may have been missed for a variety of reasons including, but not limited to: error in DNA concentration measurement of the individual possessing the variant, volume measurement variability during pooling, or stochastic events during sequencing. One potential solution may be utilizing different DNA quantification methods for more accurate concentration prior to pooling. Additionally, a combinatorial design wherein each individual is represented in exactly three rather than two pools would potentially reduce false positive and negative rates.
A cost analysis of our approach showed significant savings. Targeted capture used in conjunction with the pooling scheme herein described resulted in a total sequencing cost of $15,950 for the entire 78 proband cohort. Targeted capture without pooling would have a total cost of $54,300 for a cohort of the same sample number, representing a cost savings of $39,350 from pooling alone. Moreover, assuming a cost of $1200 per sample for whole exome sequencing, the pooled and targeted approach would produce a relative cost saving of $77,650 for this cohort as compared to whole exome sequencing without pooling. Compared to whole genome sequencing without pooling (assumed to cost $5950 per sample), the pooled and targeted technique would realize a savings of $448,150.
Conclusions
This unique approach to targeted gene sequencing identified a large number of putative disease-causing variants in a cohort of patients with BAV, including variants in 26 genes not previously associated with human BAV. Pathway analysis supported a role for WNT pathway genes in human BAV. The data as a whole further underscore the complex, polygenic nature of BAV. This technique provides a method for sample multiplexing that lowers costs and reduces sequencing errors.
Methods
Study population
The study cohort, previously described by our group, included 78 unrelated individuals (59 male, 19 female) with BAV [8]. Subjects were prospectively recruited from June 2004 to June 2011 as part of a larger study involving genetic testing in patients with congenital left ventricular tract outflow defects. Informed consent was obtained from study subjects or parents of subjects less than 18 years of age (assent was obtained from subjects 9–17 years of age) under protocols approved by the Institutional Review Board (IRB) at Nationwide Children’s Hospital. Subjects with known chromosomal abnormalities were excluded from the analysis. The majority of individuals were of Caucasian ethnicity, with 1 African-American, 1 Asian, and 3 Hispanic individuals. Each subject had undergone clinical echocardiography with images sufficient to identify associated cardiac malformations and aortic valve cusp fusion morphology (Table 2). Fifty of the 78 subjects (64%) had isolated BAV while the remainder had BAV-CoA. Forty-six subjects (59%) had R-L cusp fusion, 39% had R-NC fusion, and 2% had L-NC fusion. Eighteen of the 78 subjects had a family history of a left ventricular outflow tract defect. For the majority of subjects, parent samples were also obtained under the same IRB protocol. Genomic DNA was isolated from blood or saliva samples using the 5 PRIME DNA extraction kit (Thermo Fisher Scientific, Pittsburgh, PA).
Pooling scheme
Proband genomic DNA was combined into 19 unique pools each representing 9 or 10 individuals. The pools were constructed using overlapping design such that each individual was represented in exactly two pools, and a given rare variant could be uniquely attributed to a single individual on the basis of which two pools contained the variant. Individual genomic DNA samples were quantified by Nanodrop (Thermo Fisher Scientific), diluted to a concentration of 200 ng/microliter, and then requantified by Qubit fluorometer (Invitrogen Life Technologies, Carlsbad, CA). Quality of the DNA was assessed by SYBR Gold agarose gel (Life Technologies). Samples were then pooled, with the total amount of DNA for each pool consisting of 5 micrograms in 50 microliters (i.e. 500 ng per sample for a pool of 10 individuals and 550 ng per sample for a pool of 9 individuals).
Targeted capture
A custom, targeted gene capture was designed using the Agilent SureSelect Target Enrichment kit (Table 3). Candidate genes were selected on the basis of relevance to cardiac development and/or congenital heart defects in humans and animal models. Reference sequences were obtained from the Ensembl database. Probes were designed using paired, double-end, 75 base pair reads with centered design and 2x tiling frequency. A total of 97 candidate genes were probed using a whole gene interval approach, representing 7.6 Mb of DNA. Analysis was subsequently confined to exonic regions.
Sequencing
Sequencing of the pooled target captured proband genomic DNA was performed on the Illumina HiSeq 2000. Variants considered potentially pathogenic identified by NGS were subsequently confirmed by Sanger sequencing. Where available, parent samples were also sequenced for these potentially pathogenic variants. Sequencing primers are available upon request.
Bioinformatics algorithms
Bioinformatics analysis was performed using Churchill, our laboratory’s pipeline for the discovery of human genetic variation. Churchill utilizes the Burrows Wheeler Aligner (BWA) for the alignment of sequence data to the reference genome, hg19. Further refinement steps were performed on the aligned sequence data using Genome Analysis ToolKit (GATK) following the Broad Institute’s guidelines for best practices (https://www.broadinstitute.org/gatk/guide/best-practices). We utilized the GATK’s (version 2.4-9) UnifiedGenotyper (UG) to call variants in the pooled samples. In order to properly handle the pooled data, we amended the recommended UG settings by including the –sample_ploidy configuration parameter and giving it a value of 20, reflecting the potential for 20 individual alleles in a pooled sample of 10 individuals. The threshold for calling was set to 2.5% alternate allele frequency on the basis of the pooling scheme.
In-silico analysis
Rare, non-synonymous, exonic variants were analyzed using the Polyphen 2 and SIFT algorithms. Reference populations from the 1000 Genomes Project and Exome Variant Server were utilized as control populations [30, 31]. Pathway analysis was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) with cutoffs of p-value less than 0.05 [32, 33].
Availability of supporting data
This project has been registered with the National Center for Biotechnology Information (NCBI) BioProject database, identifier PRJNA260036, and can be accessed at: http://www.ncbi.nlm.nih.gov/bioproject/260036.
Supporting sequence data for this project has been deposited with the NCBI Sequence Read Archive. The study accession is SRP045998, available at the following link: http://www.ncbi.nlm.nih.gov/sra/?term=SRP045998 Biosample IDs for the pools, with their corresponding URLs are:
3015266: http://www.ncbi.nlm.nih.gov/biosample/3015266
3015267: http://www.ncbi.nlm.nih.gov/biosample/3015267
3015268: http://www.ncbi.nlm.nih.gov/biosample/3015268
3015269: http://www.ncbi.nlm.nih.gov/biosample/3015269
3015270: http://www.ncbi.nlm.nih.gov/biosample/3015270
3015271: http://www.ncbi.nlm.nih.gov/biosample/3015271
3015272: http://www.ncbi.nlm.nih.gov/biosample/3015272
3015273: http://www.ncbi.nlm.nih.gov/biosample/3015273
3015274: http://www.ncbi.nlm.nih.gov/biosample/3015274
3015275: http://www.ncbi.nlm.nih.gov/biosample/3015275
3015276: http://www.ncbi.nlm.nih.gov/biosample/3015276
3015277: http://www.ncbi.nlm.nih.gov/biosample/3015277
3015278: http://www.ncbi.nlm.nih.gov/biosample/3015278
3015279: http://www.ncbi.nlm.nih.gov/biosample/3015279
3015280: http://www.ncbi.nlm.nih.gov/biosample/3015280
3015281: http://www.ncbi.nlm.nih.gov/biosample/3015281
3015282: http://www.ncbi.nlm.nih.gov/biosample/3015282
3015283: http://www.ncbi.nlm.nih.gov/biosample/3015283
Abbreviations
- BAV:
-
Bicuspid aortic valve
- BAV-CoA:
-
Bicuspid aortic valve associated with coarctation of the aorta
- R-L:
-
Right coronary and left coronary
- R-NC:
-
Right coronary and non-coronary
- NGS:
-
Next-generation sequencing
- DAVID:
-
Database for annotation, visualization and integrated discovery
- GATK:
-
Genome analysis toolkit
- UG:
-
UnifiedGenotyper.
References
Ward C: Clinical significance of the bicuspid aortic valve. Heart. 2000, 83 (1): 81-85. 10.1136/heart.83.1.81.
Duran AC, Frescura C, Sans-Coma V, Angelini A, Basso C, Thiene G: Bicuspid aortic valves in hearts with other congenital heart disease. J Heart Valve Dis. 1995, 4 (6): 581-590.
Fernandes SM, Sanders SP, Khairy P, Jenkins KJ, Gauvreau K, Lang P, Simonds H, Colan SD: Morphology of bicuspid aortic valve in children and adolescents. J Am Coll Cardiol. 2004, 44 (8): 1648-1651. 10.1016/j.jacc.2004.05.063.
Clementi M, Notari L, Borghi A, Tenconi R: Familial congenital bicuspid aortic valve: a disorder of uncertain inheritance. Am J Med Genet. 1996, 62 (4): 336-338. 10.1002/(SICI)1096-8628(19960424)62:4<336::AID-AJMG2>3.0.CO;2-P.
Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW: Bicuspid aortic valve is heritable. J Am Coll Cardiol. 2004, 44 (1): 138-143. 10.1016/j.jacc.2004.03.050.
Glick BN, Roberts WC: Congenitally bicuspid aortic valve in multiple family members. Am J Cardiol. 1994, 73 (5): 400-404. 10.1016/0002-9149(94)90018-3.
Huntington K, Hunter AG, Chan KL: A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J Am Coll Cardiol. 1997, 30 (7): 1809-1812. 10.1016/S0735-1097(97)00372-0.
Bonachea EM, Chang SW, Zender G, Lahaye S, Fitzgerald-Butt S, McBride KL, Garg V: GATA5 Sequence Variants Identified in Individuals with Bicuspid Aortic Valve. Pediatr Res. 2014, 76 (2): 211-6. 10.1038/pr.2014.67.
Foffa I, Ait Ali L, Panesi P, Mariani M, Festa P, Botto N, Vecoli C, Andreassi MG: Sequencing of NOTCH1, GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic valve. BMC Med Genet. 2013, 14: 44.
Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D: Mutations in NOTCH1 cause aortic valve disease. Nature. 2005, 437 (7056): 270-274. 10.1038/nature03940.
McBride KL, Riley MF, Zender GA, Fitzgerald-Butt SM, Towbin JA, Belmont JW, Cole SE: NOTCH1 mutations in individuals with left ventricular outflow tract malformations reduce ligand-induced signaling. Hum Mol Genet. 2008, 17 (18): 2886-2893. 10.1093/hmg/ddn187.
Mohamed SA, Aherrahrou Z, Liptau H, Erasmi AW, Hagemann C, Wrobel S, Borzym K, Schunkert H, Sievers HH, Erdmann J: Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochem Biophys Res Comm. 2006, 345 (4): 1460-1465. 10.1016/j.bbrc.2006.05.046.
Padang R, Bagnall RD, Richmond DR, Bannon PG, Semsarian C: Rare non-synonymous variations in the transcriptional activation domains of GATA5 in bicuspid aortic valve disease. J Mol Cell Cardiol. 2012, 53 (2): 277-281. 10.1016/j.yjmcc.2012.05.009.
Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu L, Liu H, Li RG, Xu YJ, Wang Q, Zheng HZ, Li X, Wang XZ, Zhang M, Qu XK, Yang YQ: GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int J Mol Med. 2014, 33 (5): 1219-1226.
Fernandez B, Duran AC, Fernandez-Gallego T, Fernandez MC, Such M, Arque JM, Sans-Coma V: Bicuspid aortic valves with different spatial orientations of the leaflets are distinct etiological entities. J Am Coll Cardiol. 2009, 54 (24): 2312-2318. 10.1016/j.jacc.2009.07.044.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM: Finding the missing heritability of complex diseases. Nature. 2009, 461 (7265): 747-753. 10.1038/nature08494.
Iyengar SK, Elston RC: The genetic basis of complex traits: rare variants or “common gene, common disease”?. Methods Mol Biol. 2007, 376: 71-84. 10.1007/978-1-59745-389-9_6.
Visscher PM, Brown MA, McCarthy MI, Yang J: Five years of GWAS discovery. Am J Hum Genet. 2012, 90 (1): 7-24. 10.1016/j.ajhg.2011.11.029.
Metzker ML: Sequencing technologies - the next generation. Nat Rev Genet. 2010, 11 (1): 31-46. 10.1038/nrg2626.
Kim SY, Li Y, Guo Y, Li R, Holmkvist J, Hansen T, Pedersen O, Wang J, Nielsen R: Design of association studies with pooled or un-pooled next-generation sequencing data. Genet Epidemiol. 2010, 34 (5): 479-491. 10.1002/gepi.20501.
Erlich Y, Chang K, Gordon A, Ronen R, Navon O, Rooks M, Hannon GJ: DNA Sudoku–harnessing high-throughput sequencing for multiplexed specimen analysis. Genome Res. 2009, 19 (7): 1243-1253. 10.1101/gr.092957.109.
Prabhu S, Pe'er I: Overlapping pools for high-throughput targeted resequencing. Genome Res. 2009, 19 (7): 1254-1261. 10.1101/gr.088559.108.
Calloway TJ, Martin LJ, Zhang X, Tandon A, Benson DW, Hinton RB: Risk factors for aortic valve disease in bicuspid aortic valve: a family-based study. Am J Med Genet A. 2011, 155A (5): 1015-1020.
Robledo-Carmona J, Rodriguez-Bailon I, Carrasco-Chinchilla F, Fernandez B, Jimenez-Navarro M, Porras-Martin C, Montiel-Trujillo A, Garcia-Pinilla JM, Such-Martinez M, De Teresa-Galvan E: Hereditary patterns of bicuspid aortic valve in a hundred families. Int J Cardiol. 2013, 168 (4): 3443-3449. 10.1016/j.ijcard.2013.04.180.
McBride KL, Ware SM: Modifying Mendel: approaches for identification of susceptibility alleles for human cardiovascular malformations. Circ Cardiovasc Genet. 2012, 5 (3): 274-276. 10.1161/CIRCGENETICS.112.963579.
Alfieri CM, Cheek J, Chakraborty S, Yutzey KE: Wnt signaling in heart valve development and osteogenic gene induction. Dev Biol. 2010, 338 (2): 127-135. 10.1016/j.ydbio.2009.11.030.
Tzahor E: Wnt/beta-catenin signaling and cardiogenesis: timing does matter. Dev Cell. 2007, 13 (1): 10-13. 10.1016/j.devcel.2007.06.006.
Robasky K, Lewis NE, Church GM: The role of replicates for error mitigation in next-generation sequencing. Nat Rev Genet. 2014, 15 (1): 56-62.
Kircher M, Sawyer S, Meyer M: Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012, 40 (1): e3-10.1093/nar/gkr771.
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA: An integrated map of genetic variation from 1,092 human genomes. Nature. 2012, 491 (7422): 56-65. 10.1038/nature11632.
NHLBI Exome Sequencing Project (ESP) Exome Variant Server. [http://evs.gs.washington.edu/EVS/]
da Huang W, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocol. 2009, 4 (1): 44-57.
da Huang W, Sherman BT, Lempicki RA: Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37 (1): 1-13. 10.1093/nar/gkn923.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1755-8794/7/56/prepub
Acknowledgements
This work was supported by funding from the National Institutes of Health/National Heart, Lung, and Blood Institute and The Research Institute at Nationwide Children’s Hospital (grant R01HL109758). Recruitment was conducted under approved IRB protocol #0405HS134. We thank the participants and their families for their involvement in this study. The authors would like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EB participated in study conception and design, sample preparation, statistical analysis, and drafted the manuscript. GZ participated in sample preparation and sequencing. PW, DN, and DC participated in sequencing, bioinformatics processing, and data interpretation. SFB participated in study conception and study recruitment. VG and KM participated in study conception and design, study coordination, and helped to draft the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12920_2014_512_MOESM2_ESM.doc
Additional file 2: Table S2: Clinical characteristics of probands with rare, non-synonymous, exonic variants predicted damaging by in-silico analysis and confirmed by Sanger sequencing. (DOC 61 KB)
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Bonachea, E.M., Zender, G., White, P. et al. Use of a targeted, combinatorial next-generation sequencing approach for the study of bicuspid aortic valve. BMC Med Genomics 7, 56 (2014). https://doi.org/10.1186/1755-8794-7-56
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8794-7-56