
Abstract
Here we report on a healthy and fertile 30 years old man, who was carrier of a small supernumerary marker chromosome (sSMC). The application of molecular techniques such as fluorescence in situ hybridisation (FISH), microdissection and reverse painting, helped to characterize the sSMC which resulted to be derived from chromosome 16. In fact, the presence of euchromatin material from the long arm (16q) in the sSMC was demonstrated, and the karyotype can be written as mos 47, XY,+min(16)(:p11.1->q12.1:)[20]/46, XY [10].
Similar content being viewed by others

Brief report
Small supernumerary marker chromosomes (sSMC) have been recently defined by Liehr et al. [1] as "structurally abnormal chromosomes that cannot be identified or characterized unambiguously by conventional banding cytogenetics alone, and generally are equal in size or smaller than a chromosome 20 of the same metaphase spread". These sSMC have been described from all human chromosomes although most of them are derivatives of acrocentric chromosomes [1, 2], and approximately 30% of them were parentally inherited [3, 4].
The individuals carrying a sSMC, have a wide range of clinical variability, which may be related with the different sizes of the sSMC, the presence and/or absence of euchromatic material, the degree of mosaicism and/or uniparental disomy (UPD) [2]. Indeed, 70% of non-acrocentric sSMC do not have phenotypic repercussion, while the remaining 30% have different clinical manifestations [5]. Therefore, it is very important to characterize the content and the structure of the sSMC, in order to establish an adequate genotype-phenotype correlation.
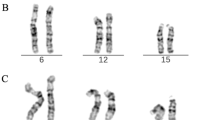
The present possibilities of the new molecular techniques such as fluorescence in situ hybridisation (FISH), microdissection and reverse painting, are enhancing our knowledge about sSMC. Here we present the result of using these techniques to characterize a sSMC, which was found in a blood sample of a healthy male. He is a healthy and fertile 30 years old man, father of a malformed newborn (NB) infant who was cytogenetically studied according to standard procedures, because of dysmorphic features at birth and resulted to have on her blood sample high resolution G-band karyotype, a tiny interstitial deletion on a chromosome 2 [46, XX, del(2)(q36.3)] (no other tissues were studied on this patient). A high resolution G-band karyotype was performed on her parents blood samples, and the 2q chromosome deletion of the NB was diagnosed to be "de novo". Nevertheless, unexpectedly, the father karyotype showed two different cellular lines. The first one, with 47 chromosomes was found in 20 cells (66.6%) having a sSMC (Fig. 1), and the second one with 46 chromosomes was observed in the 33.3% (10 cells). The application of centromere-specific multicolour fluorescence in situ hybridization (FISH) together with subcentromere specific multicolor FISH (subcenM-FISH) [6] and glass-needle based microdissection [7], revealed that the sSMC was from a chromosome 16 origin, containing not only centromeric and heterochromatic material but also euchromatin from the long arm of chromosome 16 (16q)(Fig. 2). Consequently, his karyotype resulted to be: mos 47, XY, +min(16)(:p11.1->q12.1:)[20]/46, XY[10]. This alteration was produced "de novo" since his parents blood karyotypes were normal, and seems to have no relation with her daughter chromosome interstitial deletion, which was also produced "de novo". An EBV-transformed, immortalized cell line has been established and included in a cell bank [8].

High resolution G-band chromosomes karyotype showing the sSMC.

Centromere-specific multicolour fluorescence in situ hybridization (FISH) and subcentromere specific multicolor FISH (subcenM-FISH) showing that the sSMC was from a chromosome 16 origin, containing centromeric, heterochromatic and euchromatic material from the long arm of chromosome 16 (16q).
The chromosome 16 long arm (16q) is characterized by the presence of a block of heterochromatin located between its centromere and the euchromatin region. Trisomy of the heterochromatin block is not related with any clinical manifestations, in fact there are five reported cases with sSMC (16), whose content is limited to the centromeric D16Z and heterochromatin regions without clinical manifestations. Among these five cases, the first one, had an abnormal phenotype attributed to an obstetric trauma [9]. The second case ended in a termination of pregnancy (TOP) with no clinical information [10]. And finally, the other three cases were ascertained in the amniotic fluid of three pregnant woman being normal the prenatal outcome and/or baby follow-up [11–13].
Trisomy of the next euchromatic region of the long arm of chromosome 16 (involving the q12 band) has been reported in seven case reports and seven members of two families [14–20] and after a recent review of the clinical data from all those patients, Barber et al. [20] concluded that "duplications of proximal 16q do not have characteristic facies but frequently have short stature, developmental delay, speech delay, learning difficulties and behavioural problems which range from mild to severe". A few genes has been located at 16q12-13 band, such as the Hereditary cylindromatosis gene (CYLD1), which has been defined as a tumor suppressor gene [21], the beta subunit gene of the Phosphorylase kinase (PHKB) which is an enzyme that activates glycogen phosphorylases in muscle, liver, and other tissues [22] and the human smooth muscle myosin heavy chain (MHC) which corresponds to MYH11 gene, and is expressed in the human umbilical artery, bladder, esophagus and trachea [23].
The case we present here, has a partial trisomy of the long arm of chromosome 16, involving the heterochromatin block and the proximal euchromatin region at 16q12 band, and as far as we know is the first one reported in a phenotypically normal patient, with no developmental delay neither mental retardation. It was produced "de novo", and consequently we could not disregard that it could be due to low repetitive elements present in the pericentromeric region of chromosome 16, as recently proposed by different authors for chromosomes 2, 10 and 12 [24–26]. And it was found as a mosaicism, although other tissues were not studied, where different degrees of mosaicism may be present influencing the patient phenotype.
In conclusion, the advantage of molecular techniques have allowed to know the real euchromatic/heterochromatic content of the sSMC(16) in our patient. In fact, they helped to show that not all the euchromatic trisomies are associated with clinical repercussion, as previously reported by Barber [27]. Nevertheless, more affected and non-affected patients are needed to be described in order to elucidate the phenotype-genotype correlations of different sSMC, that we consider is essential for a right genetic prenatal counselling.
References
Liehr T, Claussen U, Starke H: Small supernumerary marker chromosomes (sSMC) in humans. Cytogenet Genome Res 2004, 107: 55–67. 10.1159/000079572
Liehr T, Mrasek K, Weise A, Dufke A, Rodriguez L, Martinez Guardia N, Sanchis A, Vermeesch JR, Ramel C, Polityko A, Haas OA, Anderson J, Claussen U, von Eggeling F, Starke H: Small supernumerary marker chromosomes – progress towards a genotype-phenotype correlation. Cytogenet Genome Res 2006, 112: 23–34. 10.1159/000087510
Dennis NR, Veltman MW, Thompson R, Craig E, Bolton PF, Thomas NS: Clinical findings in 33 subjects with large supernumerary marker(15) chromosomes and 3 subjects with triplication of 15q11-q13. Am J Med Genet A 2006, 140: 434–41.
Liehr T, Weise : A Frequency of small supernumerary marker chromosomes in prenatal, newborn, developmentally retarded and infertility diagnostics. Int J Mol Med 2007, 19: 719–31.
Crolla JA, Dennis NR, Jacobs PA: A non-isotopic in situ hybridisation study of the chromosomal origin of 15 supernumerary marker chromosomes in man. Journal of Medical Genetics 1992, 29: 699–703.
Starke H, Nietzel A, Weise A, Heller A, Mrasek K, Belitz B, Kelbova C, Volleth M, Albrecht B, Mitulla B, Trappe R, Bartels I, Adolph S, Dufke A, Singer S, Stumm M, Wegner RD, Seidel J, Schmidt A, Kuechler A, Schreyer I, Claussen U, von Eggeling F, Liehr T: Small supernumerary marker chromosomes (SMC): genotype-phenotype correlation and classification. Hum Genet 2003, 114: 51–67. 10.1007/s00439-003-1016-3
Starke H, Raida M, Trifonov V, Clement JH, Loncarevic IF, Heller A, Bleck C, Nietzel A, Rubtsov N, Claussen U, Liehr T: Molecular cytogenetic characterization of an acquired minute supernumerary marker chromosome as the sole abnormality in a case clinically diagnosed as atypical Philadelphia-negative chronic myelogenous leukaemia. Br J Haematol 2001, 113: 435–8. 10.1046/j.1365-2141.2001.02787.x
Tönnies H, Pietrzak J, Bocian E, MacDermont K, Kuechler A, Belitz B, Trautmann U, Schmidt A, Schulze B, Rodriguez L, Binkert F, Yardin C, Kosyakova N, Volleth M, Mkrtchyan H, Schreyer I, von Eggeling F, Weise A, Mrasek K, Liehr T: New immortalized cell lines of patients with small supernumerary marker chromosome: towards the establishment of a cell bank. J Histochem Cytochem 2007, 55(6):651–60. 10.1369/jhc.6A7161.2007
Callen DF, Ringenbergs ML, Fowler JC, Freemantle CJ, Haan E: A Small marker chromosomes in man: origin from pericentric heterochromatin of chromosomes 1, 9, and 16. J-Med-Genet 1990, 27: 155–9.
Callen DF, Eyre HJ, Ringenbergs ML, Freemantle CJ, Woodroffe P, Haan EA: Chromosomal origin of small ring marker chromosomes in man: characterization by molecular genetics. Am J Hum Genet 1991, 48: 769–782.
Crolla JA: FISH and molecular studies of autosomal supernumerary marker chromosomes excluding those derived from chromosome 15: II. Review of the literature. Am J Med Genet 1998, 75: 367–81. 10.1002/(SICI)1096-8628(19980203)75:4<367::AID-AJMG5>3.0.CO;2-N
Paoloni-Giacobino A, Morris MA, Dahoun SP: Prenatal supernumerary r(16) chromosome characterized by multiprobe FISH with normal pregnancy outcome. Prenat Diagn 18(7):751–2. 10.1002/(SICI)1097-0223(199807)18:7<751::AID-PD312>3.0.CO;2-5
Sanz R, Anabitarte MA, Querejeta ME, Lorda-Sanchez I, Ibanez MA, Rodriguez de Alba MR, Ayuso C, Ramos C: Rapid identification of a small dicentric supernumerary marker derived from chromosome 16 with a modified FISH technique on amniotic fluid. Prenat Diagn 2000, 20: 63–5. 10.1002/(SICI)1097-0223(200001)20:1<63::AID-PD741>3.0.CO;2-W
Hirai S, Ujüe J, Suzuki J, Ishiyama S, Tsukanishi A, Muramoto J, Kano H, Suzuki H: Duplication of the long arm of chromosome 16. Jpn J Pédiatr 1981, 34: 1963–1967.
Romain DR, Frazer AG, Columbano-Green LM, Parfitt RG, Smythe RH, Chapman CJ: Direct intrachromosomal duplication of 16q and heritable fragile site fra (10) (q25) in the same patient. Am J Med Genet 1984, 19: 507–13. 10.1002/ajmg.1320190312
Fryns JP, Kleczkowska A, Decock P, Van den Berghe H: Direct duplication 16q11.1–16q13 is not associated with a typical dysmorphic syndrome. Ann Genet 1990, 33: 46–8.
Mascarello JT, Hubbard V: Routine use of methods for improved G-band resolution in a population of patients with malformations and developmental delay. Am J Med Genet 1991, 38: 37–42. 10.1002/ajmg.1320380110
Engelen JJ, De Die-Smulders CE, Vos PT, Meers LE, Albrechts JC, Hamers AJ: Characterization of a partial trisomy 16q with FISH. Report of a patient and review of the literature. Ann Genet 1999, 42: 101–4.
Trimborn M, Wegner RD, Tönnies H, Sarioglu N, Albig M, Neitzel H: Prenatal diagnosis and molecular cytogenetic characterisation of a small de novo interstitial duplication 16q11.2-q13. Prenat Diagn 2006, 26: 273–6. 10.1002/pd.1396
Barber JC, Zhang S, Friend N, Collins AL, Maloney VK, Hastings R, Farren B, Barnicoat A, Polityko AD, Rumyantseva NV, Starke H, Ye S: Duplications of proximal 16q flanked by heterochromatin are not euchromatic variants and show no evidence of heterochromatic position effect. Cytogenet Genome Res 2006, 114: 351–8. 10.1159/000094225
Thomson SA, Rasmussen SA, Zhang J, Wallace MR: A new hereditary cylindromatosis family associated with CYLD1 on chromosome 16. Hum Genet 1999, 105: 171–3. 10.1007/s004390051083
Francke U, Darras BT, Zander NF, Kilimann MW: Assignment of human genes for phosphorylase kinase subunits alpha (PHKA) to Xq12-q13 and beta (PHKB) to 16q12-q13. Am J Hum Genet 1989, 45: 276–82.
Matsuoka R, Yoshida MC, Furutani Y, Imamura S, Kanda N, Yanagisawa , Masaki T, Takao A: Human smooth muscle myosin heavy chain gene mapped to chromosomal region 16q12. Am J Med Genet 1993, 46: 61–7. 10.1002/ajmg.1320460110
Jackson MS, Rocchi M, Thompson G, Hearn T, Crosier M, Guy J, Kirk D, Mulligan L, Ricco A, Piccininni S, Marzella R, Viggiano L, Archidiacono N: Sequences flanking the centromere of human chromosome 10 are a complex patchwork of arm-specific sequences, stable duplications and unstable sequences with homologies to telomeric and other centromeric locations. Hum Mol Genet 1999, 2: 205–15. 10.1093/hmg/8.2.205
Vermeesch JR, Duhamel H, Raeymaekers P, Van Zand K, Verhasselt P, Fryns JP, Marynen P: A physical map of the chromosome 12 centromere. Cytogenet Genome Res 2003, 103: 63–73. 10.1159/000076291
Mrasek K, Starke H, Liehr T: Another small supernumerary marker chromosome (sSMC) derived from chromosome 2: towards a genotype/phenotype correlation. J Histochem Cytochem 2005, 53: 367–70. 10.1369/jhc.4B6414.2005
Barber J: Directly transmitted unbalanced chromosome abnormalities and euchromatic variants. J Med Genet 2005, 42: 609–629. 10.1136/jmg.2004.026955
Acknowledgements
We thank the family and their physicians for their collaboration in this study. This work was supported by a Grant (PI020028) from the Fondo de Investigaciones Sanitarias (FIS), Instituto de Salud Carlos III. Ministerio de Sanidad y Consumo. Spain.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
LR, carried out the high resolution G-band cytogenetic studies from the patient and his family as well as the preliminary FISH techniques and drafted the manuscript. TL carried out the application of centromere-specific multicolour FISH, the subcentromere specific multicolor FISH (subcenM-FISH) [6] and glass-needle based microdissection techniques. MLMF, help with the high resolution G-band cytogenetic studies. AL and AT are the pediatricians who follow the child and sent us the clinical data and the blood samples to perform the citogenetic and molecular studies. MLMartínez-Frías participated in the design of the manuscript. All the authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rodríguez, L., Liehr, T., Martínez-Fernández, M.L. et al. A new small supernumerary marker chromosome, generating mosaic pure trisomy 16q11.1–q12.1 in a healthy man. Mol Cytogenet 1, 4 (2008). https://doi.org/10.1186/1755-8166-1-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-1-4