
Abstract
The idea of renewable and regenerative resources has inspired research for more than a hundred years. Ideally, the only spent energy will replenish itself, like plant material, sunlight, thermal energy or wind. Biodiesel or ethanol are examples, since their production relies mainly on plant material. However, it has become apparent that crop derived biofuels will not be sufficient to satisfy future energy demands. Thus, especially in the last decade a lot of research has focused on the production of next generation biofuels. A major subject of these investigations has been the microbial fatty acid biosynthesis with the aim to produce fatty acids or derivatives for substitution of diesel. As an industrially important organism and with the best studied microbial fatty acid biosynthesis, Escherichia coli has been chosen as producer in many of these studies and several reviews have been published in the fields of E. coli fatty acid biosynthesis or biofuels. However, most reviews discuss only one of these topics in detail, despite the fact, that a profound understanding of the involved enzymes and their regulation is necessary for efficient genetic engineering of the entire pathway. The first part of this review aims at summarizing the knowledge about fatty acid biosynthesis of E. coli and its regulation, and it provides the connection towards the production of fatty acids and related biofuels. The second part gives an overview about the achievements by genetic engineering of the fatty acid biosynthesis towards the production of next generation biofuels. Finally, the actual importance and potential of fatty acid-based biofuels will be discussed.
Similar content being viewed by others


Introduction
During the recent decades it has become evident that the world’s fossil fuel reserves are decreasing and will be most probably depleted rather soon. However, until 2016 the global demand for crude oil will increase by more than 1 million barrels per day [1], and also for the time after 2018 no dramatic change in energy need is expected. As a result governments, companies and scientists work on the development of sustainable ways to produce energy. Concerning biofuels there are actually two major products of great commercial importance: ethanol and biodiesel (fatty acid alkyl ester, FAAE), which currently account for roughly 90% of the biofuel market [2, 3].
The main producers of bioethanol are the USA and especially Brazil, where the gasoline must be blended with at least 25% ethanol [4, 5]. The production of ethanol is based on fermentation of Saccharomyces cerevisiae and the most often used substrates are carbohydrates obtained from sugarcane, corn, wheat, sugar beet and some other plants [6]. In comparison to petrol, bioethanol and bioethanol blends have a higher cetane number, but a roughly 30% lower energy density [7]. Another problem is the hygroscopicity of ethanol, which makes storage and transportation challenging [8]. In theory, bioethanol can be carbon-neutral as plant material is used for its production. But due to the kind of fermentation process, forest clearance, intensive use of fertilizers and the energy consumption during distillation, the overall emissions of CO2 exceed its consumption. In addition, other pollutants such as mono-nitrogen oxides (NOx) or carbon monoxide (CO) are produced, which result in an environmental impact that might even be worse than from the use of fossil fuels [9, 10].
Biodiesel is produced by the transesterification of mostly plant-derived triacylglycerols (TAG), yielding glycerol and FAAEs. Methanol is being used as alcohol moiety, due to its low price [11]. In contrast to ethanol, biodiesel has very similar properties to petrol and can therefore be used in the same engines and distributed through the same infrastructure. Like ethanol, it has some environmentally friendly aspects because it is degradable, its biosynthesis consumes carbon dioxide (CO2), and it has low sulfur content when compared to crude oils. However, most studies conclude that the overall environmental impact of biodiesel is also negative [12], due to the use of fertilizers for growing the oil plants and the transesterification process, which is energy consuming and relies on the use of toxic methanol [11].
Another drawback of currently used biofuels is that to date all economically feasible processes are based on the utilization of cereal crops (for example, wheat, and maize), oil crops (for example rape, palm oil and soya) or sugar crops (for example, sugar beet and sugar cane) [6, 13]. In the last years this has led to intensive political debate concerning the social and environmental consequences of the use of food and agricultural land for biofuel production [9, 14–16]. Owing to these drawbacks one speaks of first-generation biofuels in contrast to second-generation biofuels (Figure 1) which rely on the use of lignocellulose or other feedstocks that do not directly compete with food, and reduce the need for agricultural land [13].

Overview of the production of first- and second-generation biofuels.
As an alternative to the use of plant-derived TAGs, the microbial production of free fatty acids (FFA) or FFA-derived biofuels offers a great potential due to short production times and very low land-use. To reduce the competition with food, the use of cellulose, lignin, hemicellulose, CO2 or other non-food carbon sources needs further optimization, although many strategies have already been established [17–19] and their suitability for biofuel production has been shown [20–23]. Furthermore, direct microbial production of FAAE has also been published, which makes the subsequent transesterification unnecessary and thus saves energy costs and reduces the use of methanol [24–26]. Some recent investigations lead to the production of fatty acid-derived alkanes, alcohols, methylketones or 3-hydroxyalkanoates. All mentioned compounds are suitable as diesel replacement. The production of TAG in Escherichia coli (E. coli) may in future become an alternative for TAG production by native producers (like species of the genera Rhodococcus, Mycobacterium or Streptomyces) that mostly exhibit a rather slow growth rate and are not as easy to genetically modify as E. coli.
To date, promising results for microbial production of free fatty acids and derived products have been obtained mainly by metabolic engineering, but for the production of large amounts of cheap biofuels much effort still needs to be undertaken. For this a detailed knowledge about the participating enzymes and their regulation is crucial. The objective of this review is to focus on the biosynthesis of fatty acids in the fast growing and industrially important microorganism E. coli. Especially, we will sum up possibilities to genetically modify this bacterium towards an overproduction of fatty acids or fatty acid-derived biofuels.
Fatty acid biosynthesis
Synthesis of fatty acids is one of the most ubiquitous pathways in organisms. In eukaryotic and prokaryotic cells fatty acids are precursors for a variety of important building blocks such as phospholipids, sphingolipids, sterols, as secondary metabolites and signaling molecules, or they are attached to proteins. By changing the grade of saturation of the phospholipids in cellular membranes, their fluidity can be altered, which makes an adaption to temperature changes possible. Because the degradation of fatty acids yields a high amount of ATP and reducing equivalents, they also represent a suitable storage compound for energy and carbon. Especially in multicellular organisms, but also in unicellular eukaryotes and prokaryotes, fatty acids are stored as TAG or wax esters, whereas the storage of hydroxyfatty acids as polyhydroxyalkanoates is limited to bacterial species. In Archaea, fatty acids play a minor role, owing to their differing membrane, which mainly consists of fatty alcohol-glycerol diethers instead of fatty acid-glycerol diesters. Nevertheless, fatty acid biosynthesis is also performed by Archaea, and the products can be used to acylate membrane proteins [27, 28].
Despite the early development of fatty acid biosynthesis during evolution of life but due to its importance, the pathway is highly conserved within the kingdoms of life. At the first step, malonyl-CoA is formed by carboxylation of acetyl-CoA with hydrogencarbonate, by the expense of ATP. Coenzyme A is then exchanged by the acyl carrier protein (ACP) resulting in malonyl-ACP. The ACP prevents the growing fatty acid chain from degradation and from being used for anabolic reactions. With malonyl-ACP, the first turn of the fatty acid biosynthesis cycle starts by an initial condensation of malonyl-ACP with acetyl-CoA, yielding hydrogencarbonate, free coenzyme A and acetoacetyl-ACP. The latter is then reduced to 3-hydroxybutyryl-ACP, dehydrated to 2-butenoyl-ACP and further reduced to butyryl-ACP. Butyryl-ACP enters the next turn of the cycle again by a condensation with malonyl-ACP. Fatty acid synthesis stops when a certain chain length is reached, and the acyl-ACP is used for membrane synthesis. As both reduction steps require two reduction equivalents, derived from nicotinamide adenine dinucleotide (NADPH), the following equation for the elongation of a fatty acid by a two-carbon unit applies:
Despite high similarities in the general pathway, different enzymes and different genetic organizations have evolved. In animals and fungi, the type-I fatty acid synthase caries out all steps of fatty acid biosynthesis as one multifunctional protein complex. This type is further divided into the fungal typeIa, in which the fatty acid synthase is encoded by two genes and is assembled to a α6β6 heterododecamer of about 2.6 MDa. Fatty acid synthase typeIb is found in animals. Here, all required proteins are encoded by a single gene, and the translated peptide chains form an α2 homodimer of about 540 kDa [29, 30].
Type-II fatty acid synthase is predominant in prokaryotes as well as in the plastids of plants, in which de novo synthesis of plant fatty acids takes place [31–34]. An exception are Gram-positive, mycolic acid-producing bacteria, which contain a type-I fatty acid synthase as one polypeptide chain [35–37] and additionally a type-II fatty acid synthase, which is only involved in the elongation of fatty acids with medium chain length but cannot start de novo fatty acid biosynthesis [38, 39]. The main difference of FAS type-II is that it consists of a set of enzymes that are not organized as one single gene or operon. In any case, in many bacteria such as E. coli, a number of the genes are organized in the fab cluster. The bacterial acetyl-CoA carboxylase represents an example for a bacterial enzyme complex that is involved in fatty acid biosynthesis.
Studies of the fatty acid synthase of Archaea are rare, because the de novo synthesis of fatty acids seems not to be comparably important as in other organisms. The use of fatty acids as anchor molecules for membrane proteins has been proven [28], and additionally they have been found as parts of phospholipids [40] and as free fatty acids [41, 42]. Due to similarities with the bacterial fatty acid profile [43, 44] and within the inhibition pattern at high salt-concentration of archaeal and bacterial FAS, as well as in the isolation procedure, it has been suggested that Archaea contain a type-II FAS [45].
The E. coli type-II FAS enzymes
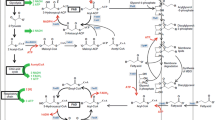
This section deals with the set of enzymes that perform the fatty acid biosynthesis of E. coli. Transcriptional and biochemical regulation is emphasized, and studies concerning overexpression or deletion of the respective genes are discussed with special interest in the impact on overproduction of fatty acids. The enzymes and regulation of the membrane synthesis and fatty acid degradation are also of great interest, as they represent the main competing pathways with a FFA overproduction. An overview of the involved pathways is shown in Figure 2.

Biosynthesis and degradation of fatty acids and membrane lipids. ATP and reduction equivalents are colored red if consumed and green if gained. Enzymes are colored blue. FAB, fatty acid biosynthesis; FAD, fatty acid degradation.
AccABCD: acetyl-CoA carboxylase
Acetyl-CoA carboxylase represents the starting enzyme and directs acetyl-CoA towards de-novo fatty acid biosynthesis and chain elongation. In E. coli, the four subunits form a very unstable complex that could be purified as two subcomplexes: 1) the biotin carboxylase-biotin carboxyl carrier protein (BC-BCCP), which is a homodimer of AccC (biotin carboxylase), interacting with four molecules of AccB (biotin carboxyl carrier protein) [46], and 2) the carboxyl transferase, which is a heterotetramer, consisting of two subunits of AccA and two subunits of AccD [47]. The reaction can be divided into two half-reactions with (i) the carboxylation of biotin, by the expense of ATP and with Mg2+-ions as cofactor and (ii) the subsequent transfer of the carboxyl group to acetyl-CoA, yielding malonyl-CoA [47].
Transcription of accABCD is strictly coordinated and regulated, as the subunits have to be synthesized in equimolar amounts. In addition, the carboxylation of acetyl-CoA is driven by cleavage of ATP and thus consumes energy. The genes accB and accC build one mRNA, which serves as a template for the translation of both AccB and AccC [48]. Their transcription, which positively correlates with the growth rate, is further autoregulated by the N-terminal domain of AccB [49, 50]. Overexpression of accB inhibits the transcription of accB and accC, whereas in an accB-deletion mutant the transcription of the accBC operon is not altered [50]. Additionally, the excess of AccB deregulates the biotin synthetic operon and thus, the cells are stressed due to the strong biotin-synthesis [51, 52]. Enhanced production of AccC also affects the biotin operon, as AccC forms complexes with AccB, which is more efficiently biotinylated in its unbound form. If AccC is more abundant, almost no biotin is attached to AccB, and transcription of the biotin operon is shut down by BirA, which has a dual function as biotin protein ligase and as repressor for the bio-operon [51]. The resulting reduction of fatty acid biosynthesis by AccB or AccC overexpression however, is not seen in a strain with overexpression of both proteins in equimolar amounts [53].
In contrast to accB and accC, the genes coding for the carboxyl transferase are not part of an operon, and no transcriptional regulation has been found by sequence analysis [49]. Instead, the translation of the respective mRNAs is controlled by the β-subunit (AccD) of the mature carboxyl transferase [54]. This subunit forms a zinc-finger motif, which binds to accA and accD mRNA, but is also required for the catalytic activity [55]. Since the binding of mRNA is preferred in comparison to the binding of acetyl-CoA, high levels of acetyl-CoA (as in growing cells) are required to resolve the complex of carboxyl transferase and its mRNA and thus, to promote both, synthesis of malonyl-CoA and translation of AccA and AccD [54]. As the zinc-finger motif is found in E. coli and Staphylococcus aureus AccD [56], as well as in the chloroplast-encoded β-subunit of the carboxyltransferase of pea, tobacco, rice, liverwort [57, 58] and wheat but not in type-I fatty acid synthase, this leads to the assumption that this type of regulation might be common in type-II fatty acid synthases [56].
For the E. coli acetyl-CoA carboxylase it has been shown that the enzyme activity is inhibited by acyl-ACP with chain lengths of C6 to C20. Thus, an accumulation of fatty acids that are not used for membrane lipid synthesis is prevented [59].
Overexpression of different combinations of the four subunits of the acetyl-CoA carboxylase in equimolar amounts has been extensively studied by Davis et al. [60]. The normally very weak enzyme activity in cellular crude extracts could be enhanced 50-fold. Interestingly the overexpression of accBCD led to an 11-times enhanced activity, whereas all tested combinations of one or two subunits did not result in enhanced activity. Comparing this with the more recent results for the translational regulation of AccA and AccD [54], it seems reasonable that a higher copy-number of the mRNA of AccA or AccD, or both, lead to an overall higher level of translation of accA and accD mRNA. Translation will be higher than in the wild-type until equilibrium between the copy number of AccAD and the respective mRNAs is reached.
However, overproduction of AccABCD was found to result only in enhanced production of free fatty acids if a cytosolic form of thioesterase I (‘tesA) was coexpressed and thus a metabolic sink for fatty acids was provided. Nearly the same amount of free fatty acids was achieved upon coexpression of accBCD and ‘tesA, and the expression of ‘tesA alone still resulted in high amounts of free fatty acids. Additional coexpression of birA, to attach the biotin cofactor to AccB, did not enhance the enzyme activity significantly; therefore, biotin availability should not be a limiting factor [60]. In the study of Zha et al. [61], the level of malonyl-CoA in E. coli was increased 15-fold. They overexpressed the acetyl-CoA carboxylase, and additionally improved acetate assimilation and deleted the formation of ethanol and acetate.
FabD: malonyl-CoA:ACP transacylase
Malonyl-CoA:ACP transacylase catalyzes the transfer of the malonyl-moiety to ACP, which directs it towards fatty acid neogenesis and fatty acid chain elongation. Kinetic studies have shown that the E. coli FabD cannot use acetyl-CoA as substrate but the latter shows a weak competitive inhibition of the reaction. Also the binding of ACP is inhibited by high CoA-SH concentrations. It was further shown that in presence of equal concentrations of substrates and products the formation of malonyl-CoA is favored over the production of malonyl-ACP [62].
Deletion of fab D has been shown to be lethal [63, 64], and overexpression of fab D in E. coli leads to a slightly altered fatty acid composition. The proportion of palmitoleic acid was shown to decrease, whereas the proportion of cis-vaccenic acid increases [65]. The authors suggest that the different composition is caused by an enhanced malonyl-ACP pool and thus, enhanced activity of the 3-ketoacyl-ACP synthase II (FabF), which is the enzyme responsible for chain elongation of C16:1 to C18:1 [66]. Overexpression of the E. coli fabD, together with ‘tesA, increases the amount of free fatty acids by about 11% when compared to overexpression of the thioesterase gene alone [67]. The fab D gene is transcribed as part of the fab-cluster, which is described in more detail in the regulation section.
FabB, FabF and FabH: 3-ketoacyl-ACP synthase I, II and III
The 3-ketoacyl-ACP synthase catalyzes the formation of 3-ketoacyl-ACP by condensation of fatty acyl-ACP with malonyl-ACP. In the case of FabH, the substrates are malonyl-ACP and acetyl-CoA, initiating the first cycle of chain elongation during fatty acid biosynthesis. Subsequent elongation steps are performed by FabF and FabB.
The activity of FabH with propionyl-CoA is as good as with acetyl-CoA, leading to the formation of fatty acids with an uneven number of carbon atoms. The activity with butyryl-CoA is much lower, and hexanoyl-CoA is no natural substrate of FabH [68]. The E. coli FabH exhibits no activity with branched-chain acyl-CoA esters, but heterologous expression of the Bacillus subtilis fabH gene leads to the formation of branched-chain fatty acids in E. coli[69].
The activities of FabF and FabB differ only in the catalysis of some reactions. Both show activity with C6 to C14 saturated fatty acyl-ACP esters; however, C14:0 is a weak substrate of both enzymes and only FabF exhibits low activity with C16:0 [70]. However, the condensation reaction with butyryl-ACP was not tested [70], but as activity of both enzymes with acetyl-ACP was found [71] it seems likely that butyryl-ACP is also a suitable substrate.
In the synthesis of unsaturated fatty acids, FabB catalyzes the condensation of cis-3-decenoyl-ACP (formed by the FabA catalyzed reaction, see the section about FabA and FabZ), cis-5-dodecenoyl-ACP and cis-7-tetradecenoyl-ACP each with malonyl-ACP [72]. Only the last elongation step that leads to the formation of vaccenic acid is catalyzed by FabF [70].
Deletion of the fabH gene has been thought to be lethal [73]. However, a recent study has shown that this is only true in a mutant strain that is also SpoT1-negative [74] and only a significant reduction in growth rate and cell size upon fabH deletion was reported. Thus, the activity of FabH can only partly be substituted by other enzymes. Regarding the fatty acid profile, deletion of fabH leads to enhanced production of C18 species, whereas the amount of C16 species is reduced [74]. Overexpression of fabH leads to the opposite effect - at the expense of C18:1, C14 and C16 fatty acids are more abundant than in wild-type cells [75].
Deletion of fabF leads to a temperature-sensitive mutant, because the elongation of C14:1 to C16:1 is performed mainly by FabF [66]. Overexpression has been shown to be lethal and a strong increase of malonyl-CoA intermediates has been observed [76]. The lethal effect could partly be resolved by coexpression of fabD, and thus, it has been proposed that FabD (which catalyzes the transacylation of malonyl-CoA to malonyl-ACP) forms complexes with FabF or FabH and eventually also with FabB. Overexpression of fabF would leave substantially no free FabD proteins and thus the FabD-FabH complex would not be formed. This could hinder the correct FabH activity and block the synthesis of new fatty acids. Malonyl-ACP would easily be converted to malonyl-CoA, which accumulates, as has been shown [76].
Deletion of fabB in E. coli leads to auxotrophy for unsaturated fatty acids [77]. Overexpression is only suitable to enhance the unsaturated fatty acid proportion if fabA is also overexpressed [78]. However, a significant increase in total fatty acid content is not achieved by fabA and fabB overexpression and also the co-production of ‘TesA increases the total fatty acid content only by 50% [78].
The enzyme FabH catalyzes the first condensation reaction and its activity is inhibited by high levels of long-chain acyl-ACP esters [79], which accumulate when the rate of membrane synthesis slows down. Furthermore, the accumulation of long-chain acyl-ACP esters redirects the activities of FabF and FabB towards the formation of acetyl-ACP on the expense of malonyl-ACP. Transesterification of acetyl-ACP to acetyl-CoA is catalyzed by FabH, when the enzyme is bound to long-chain acyl-ACP [80]. As fab F and fab H are cotranscribed with the E. coli fab-cluster, their transcriptional regulation will be discussed in Section ‘Promoters of the fab-operon’. Transcription of fabB is modulated by the repressors FadR and FabR (detailed in ‘Transcriptional regulation by FadR and FabR’).
FabG: 3-ketoacyl-ACP reductase
Following the condensation reaction, the resulting 3-ketoacyl-ACP is reduced to 3-hydroxyacyl-ACP at the concomittant expense of NADPH and H+. This reversible reaction is catalyzed by FabG. FabG of E. coli was first purified and analyzed by Toomey and Wakil [81], who found that it is active over a wide range of different 3-ketoacyl-ACPs. Activity with acetoacetyl-CoA has also been demonstrated, although the rate of reduction was only 2% of the rate with the corresponding ACP ester. NADH is not used as a coenzyme [81]. Also 3-ketoacyl-CoA with longer chains are suitable substrates [82]; thereby, the FabG protein is also involved in the engineered biosynthesis of polyhydroxyalkanoates consisting of medium chain-length constituents [82–84]. In E. coli the FabG activity in fatty acid biosynthesis cannot be substituted by any other enzyme [64, 85]. Homologous expression of fabG in E. coli increases the content of C16:0 and C18:0 acids by two and three times [86]. The transcriptional regulation of fabG is discussed in Section ‘Promoters of the fab-operon’.
FabA and FabZ: 3-hydroxyacyl-ACP dehydrase
The enzymes encoded by fab Z and fab A in E. coli perform the dehydration of 3-hydroxyacyl-ACP. Moreover, FabA subsequently isomerizes trans-2-decenoyl-ACP into cis-3-decenoyl-ACP [87], which is the first reaction towards the synthesis of unsaturated fatty acids. Under physiological conditions, these reactions can be catalyzed in both directions depending on the substrate and product concentrations.
In earlier studies FabA was thought to only act in the synthesis of unsaturated fatty acids, because a defect in fab A led to auxotrophy of unsaturated fatty acids [88–90]. However, further investigations showed that fab A overexpression increases the amount of saturated fatty acids [91] and that FabA also catalyzes the dehydration of saturated 3-hydroxyacyl-ACPs with different chain lengths [92]. The explanation for the saturated fatty acid accumulation in a FabA-overproducing strain is, that cis-3-decenoyl-ACP is not further reduced to decenoyl-ACP at an appropriate rate and thus, reacts in the reversible reaction and re-enters the cycle for the synthesis of saturated fatty acids [91]. Only a strain that co-overexpresses fabA and fabB will produce an enhanced proportion of unsaturated fatty acids [78]. FabA can also dehydrate 3-hydroxydecanoyl-CoA with an activity of 11% in comparison to 3-hydroxydecanoyl-ACP [93].
The second enzyme of E. coli that catalyzes the dehydration of 3-hydroxyacyl-ACP intermediates was detected in 1994 [94] and was designated as FabZ. It was found that the dehydration of 3-hydroxymyristoyl-ACP is mainly performed by FabZ, and thus it was suggested that disruption of fabZ leads to an enhanced pool of 3-hydroxymyristoyl-ACP [79, 94]. Besides this reaction, FabZ exhibits activity for all 3-hydroxyacyl-ACP with shorter chain-length [79]. Homologous overexpression of fabZ results in an about 2-fold increase in palmitic acid and stearic acid levels [86]. In E. coli, fab Z is part of the lipid A cluster, whereas fab A is not transcribed together with other fatty acid or lipid-metabolism genes. However, fabA is under control of the regulators FadR and FabR (detailed in ‘Transcriptional regulation by FadR and FabR’).
FabI: enoyl-ACP reductase
In E. coli there is a single enoyl-ACP reductase that performs the last step in each fatty acid biosynthesis cycle, which makes the gene essential if no external fatty acids are fed [95, 96]. The enzyme, which is encoded by fabI[97], catalyzes the reduction of 2-enoyl-ACP to fatty acyl-ACP at the expense of NADPH + H+ or NADH + H+.
As the two preceding steps in fatty acid biosynthesis are reversible under physiological conditions, FabI plays a determinant role in completing each elongation cycle [98]. This important role makes the enzyme a suitable candidate for posttranslational regulation. The enzyme is severely inhibited by low concentrations of palmitoyl-CoA (with a Ki of 3.3 μM in an NADH-dependent enzyme assay and 1.6 μM with NADPH), presumably to prevent the energy-expensive biosynthesis of fatty acids, when they are already available [95]. The enoyl-ACP reductase is also inhibited by its product palmitoyl-ACP, but at about 50 times higher concentrations [79, 98]. The fab I gene in E. coli is not part of any cluster that concerns lipid synthesis. Overexpression of the fabI gene does not result in any growth defect [99] but also does not increase the cellular lipid, palmitic acid or stearic acid content [86].
ACP, ACP synthase and ACP phosphodiesterase
The acyl carrier protein is encoded by acpP, which in E. coli is part of the fab-cluster. To get the physiologically active form, a phosphopantethein group is attached to a serine of the translated apo-ACP by the action of the ACP synthase (AcpS). In E. coli an ACP phosphodiesterase also exists (AcpH) that cleaves the phosphopantethein residue of [100, 101]. The physiological role of ACP is to differentiate fatty acid biosynthesis where all intermediates are bound to ACP from fatty acid catabolism, where all intermediates are CoA esters. In E. coli, ACP represents 0.25% of all soluble proteins [102] and thus, belongs to the most abundant proteins. Its absolute requirement has been demonstrated by Goh et al. [103], who stopped cell growth by inducible gene-silencing of acpP.
Overexpression of acpS leads to cessation of cellular growth [104], which is due to strong inhibition of the glycerol-3-phosphate acyltransferase [104, 105]. This effect can be slightly resolved by coexpression of acpH, because apo-ACP is a stronger inhibitor than holo-ACP. However, the inhibition of fatty acid biosynthesis has additionally been shown in vitro by the supplementation of holo- or apo-ACP to cell-free extracts of a FFA-producing E. coli strain [106]. Coproduction of ‘TesA and apo-ACP stops cell growth completely and leads to 5-fold stronger formation of free fatty acids compared to the expression solely of ‘tesA[104]. Also the heterologous expression of acpP offers some potential, as it has been shown that the expression of acpP from Azospirillum brasilense alters the E. coli fatty acid profile and the content of C18:1 is increased 2-fold at 30°C [107].
A recent study of Battesti and Bouveret [108] has shown evidence for an interaction between ACP and SpoT, a protein that can synthesize or degrade the signal molecule (p) ppGpp (guanosine 5-triphosphate, 3-diphosphate). They suggested a model in which SpoT senses the functionality of the fatty acid biosynthesis and the consumption of acyl-ACP, by the interaction with ACP and mediates the stringent response if fatty acid biosynthesis is impaired. Additionally, they showed that SpoT cannot interact with apo-ACP [108].
Membrane synthesis
In bacteria two systems exist for the formation of 1-acyl-glycerol-3-phosphate; the genes for both are present in E. coli[109]. The end product of fatty acid biosynthesis, acyl-ACP, can be activated with an inorganic phosphate group by the action of PlsX, leading to acyl-phosphate, which is subsequently added to glycerol-3-phosphate by the action of PlsY. Alternatively the fatty acid moiety of acyl-ACP can directly be condensed with glycerol-3-phosphate by PlsB. The following steps to synthesize diacylglycerol-3-phosphate and cytosine diphosphate diacylglycerol are performed by PlsC and CdsA. The latter intermediate is then used for the formation of phosphatidylethanolamine, phosphatidylglycerol, cardiolipin or other membrane lipids [110, 111].
PlsB of E. coli is active with both acyl-CoA and acyl-ACP with similar affinity. The Km values for palmitoyl-CoA and palmitoyl-ACP have been determined to be 50 μM and 70 μM [112]. Elevated (p)ppGpp levels, as in the stringent response, seriously reduce the in vivo activity of PlsB, whereas the in vitro activity is not altered. Accordingly posttranslational inhibition of PlsB by (p)ppGpp has been suggested [113]. As a result, the intracellular level of acyl-ACP increases, which inhibits the fatty acid biosynthesis at several points [79] (see Section ‘Regulation by the stringent response’). Contrary to the data from Heath and coworkers [113], the inhibition of PlsB by (p)ppGpp has been shown in vitro by Ray and Cronan [112] with the substrate palmitoyl-CoA. The reaction with palmitoyl-ACP was not affected under the conditions of the enzyme test. Overexpression of the plsB gene resolves the inhibition of PlsB by (p)ppGpp and thus, the fatty acid biosynthesis remains active, which leads to the formation of longer cells because the cell division is still affected [113].
Transcription of plsB has been found to be antagonistic to transcription of the gene for diacylglycerol kinase (dgkA) [114], so that the cell can react to different stresses with either the biosynthesis of new membrane lipids or the modification of the headgroups via degradation to diacylglycerol, phosphorylation and addition of an alternative headgroup. Transcription of plsB is inhibited in response to high levels of BasR [114], a regulator protein that mediates iron or zinc stress [115]. If E. coli is in an environment that leads to degradation of the lipopolysaccharides or otherwise stresses the cell envelope, the σE regulon is activated, which leads to induction of plsB transcription [114]. During the multiple changes in the transcriptome that are mediated by the stringent response (due to amino-acid starvation) the plsB transcription is downregulated [116].
Despite the existence of both systems, PlsX and PlsY or PlsB cannot substitute for a complete loss of the other system in E. coli[117]. However, no significant increase in glycerol-3-phosphate acyltransferase activity has been found upon overexpression of plsX and plsY[118]. Transcription of plsX and plsB are both inhibited during the stringent response [116, 119]. Thus, it was suggested that PlsX and PlsY determine the concentration of acyl-phosphate, which might have a further regulatory function [117].
Fatty acid degradation
To metabolize fatty acids, they must be activated to acyl-CoA esters. If fatty acids are the exogenous carbon source, they bind to the transporter protein FadL. By a conformational change, a pore is opened and the diffusion of fatty acids into the periplasm is enabled [120]. Disruption of fadL impaires growth on oleate [121]. Transport from the periplasm to the cytosol is performed by FadD and is coupled to the acyl-CoA ester formation at the expense of ATP [122]. As has been shown recently, FadD also uses free fatty acids that are cleaved from membrane lipids and the formed acyl-CoA is consumed via the β-oxidation pathway. Consequently, a fadD- disruption mutant accumulates free fatty acids in the cytosol [123] and apart from this, is unable to grow on oleate as a sole carbon source [121]. Homologous overexpression of fadD enables E. coli strains to grow on fatty acids with medium chain-length and enhances the transcription of the fadE and fadBA genes [124]. The affinity of FadD for medium chain-length fatty acids can be enhanced by directed mutagenesis [125].
The degradation of acyl-CoA compounds proceeds in a cycle that reverses the steps of the fatty acid biosynthesis, resulting in the release of one unit of acetyl-CoA in each cycle. FadE catalyzes the oxidation of acyl-CoA to enoyl-CoA with a concomitant reduction of FAD to FADH2. Disruption of fadE disables the ability of E. coli to grow on dodecanoate or oleic acid as the sole carbon source [121]. The enzyme FadB performs the hydration of enoyl-CoA to 3-hydroxyacyl-CoA and further oxidizes this intermediate to 3-ketoacyl-CoA [126]. The β-ketothiolase FadA catalyzes the last step in the cycle in which acetyl-CoA and an acyl-CoA (reduced by two carbon atoms) are formed [127]. The final cleavage of acetoacetyl-CoA is performed by YquF [127, 128]. The catabolism of unsaturated fatty acids additionally involves the proteins FadH (2,4-dienoyl-CoA reductase) [129] and probably also FadM (thioesterase III) [130]. However, the exact role of FadM is still not clear, as its transcription is quite strong during growth on glucose. This suggests that the protein function is not limited to fatty acid degradation [131].
In anaerobic growth on fatty acids E. coli possesses some alternative proteins, namely YfcYX (homologs of FadBA) and YdiD (FadD homolog). An alternative FadE protein (YdiO) is also suggested. These two sets of proteins are not completely selective for aerobic or anaerobic conditions, for example, YfcYX can partially compensate for a loss of FadB and FadA [132–134]. In the anaerobic fatty acid degradation pathway, nitrate is used as the final electron acceptor. Repression of the transcription of the genes for the aerobic cycle is mediated by the ArcA/ArcB two-component system [135]. For more detailed information about the bacterial β-oxidation, the reader is referred to recent review articles [136, 137].
All genes of the aerobic fatty acid degradation cycle are under transcriptional control of the repressor protein FadR (for details see Section ‘Transcriptional regulation by FadR and FabR’), which releases the DNA upon binding of long-chain acyl-CoA, and thus enables transcription of the β-oxidation genes [138, 139]. Furthermore, the fatty acid degradation cycle is under positive control by the cyclic adenosine-monophosphate (cAMP) receptor protein-cAMP complex (CRP-cAMP), so that at least fadL, fadD and fadH are upregulated when glucose is limited [140]. In addition, the upregulation of several genes of the β-oxidation involves the sigma factor σS (RpoS) [141], as detailed in Section ‘Regulation by the stringent response’.
As discussed in the Section about free fatty acid production, fadL, fadD or fadE have been deleted to prevent product uptake or utilization in fatty acid or related biofuel-producing strains [26, 142, 143]. All of them have been shown to increase the yields of the desired product, at least in some of the studies.
Regulation in E. coli
In E. coli several genes for fatty acid biosynthesis or degradation are controlled at the transcriptional level. The main transcription factors are the proteins FabR and FadR, but also other regulators, notably the signal molecule (p)ppGpp are involved (Figure 3).

Regulation of the lipid metabolism. Activating compounds are colored green, and repressing compounds are shown in red. FAB, fatty acid biosynthesis; FAD, fatty acid degradation.
Transcriptional regulation by FadR and FabR
The regulator FadR represses the transcription of all genes that code for proteins of the β-oxidation cycle [136, 140] and via activation of the repressor protein IclR, also of the glyoxylate shunt [144, 145]. FadR repression is due to binding to the promotor sites, which is released by interaction with long-chain acyl-CoA esters that accumulate only if external fatty acids are taken up or if phospholipids from the membrane are degraded [138, 139, 146]. Since the uptake of fatty acids is mediated by FadL and FadD, the corresponding genes are only partially repressed by FadR [140] and additionally activated by CRP-cAMP, under glucose limiting conditions. In the presence of glucose, transcription of the fad-genes occurs only at a low level, even when fatty acids are also available [147]. Besides the negative control of the fad-regulon, FadR also acts as a positive regulator for the transcription of fabA, fabB and the operon fabHDG[138, 139, 148, 149], and thus, is the activator for the formation of both unsaturated and saturated fatty acids.
In a mutant strain of E. coli that synthesizes no functional FadR, all fad-genes are derepressed in addition to the activation under glucose limiting conditions. The resulting phenotype is such that the strain can grow on fatty acids with medium chain-length, whereas the wild-type can use them only if long-chain fatty acids are also present and sequester FadR [150]. Due to the lacking induction of the transcription of fabA and fabB, a fadR mutant strain has a roughly 30% lower concentration of unsaturated fatty acids [140, 151]. On the contrary, the homologous expression of fadR in a fatty acid-producing strain of E. coli can increase the yield of saturated and unsaturated fatty acids significantly [152].
In the study of Farewell et al. [153] a FadR mutant was investigated that cannot be derepressed by long-chain fatty acids. Probably owing to the impairment of unsaturated fatty acid synthesis, this mutant exhibited a low survival rate during long-time cultivations.
Besides their regulation by FadR, fabA and fabB are further under control of the fatty acid biosynthesis regulator FabR [154, 155]. More recently it has been shown that FabR senses the composition of the cytosolic fatty acid pool. In complex with unsaturated fatty acyl-ACP the binding and thus, the repression of fabA and fabB is strengthened, whereas the binding is weakened when FabR is bound to saturated fatty acyl-ACP [156]. According to the study of Feng and Cronan [157], FabR is the main regulator for fabB, whereas the transcription of fabA is more strongly influenced by the action of FadR.
Promoters of the fab-operon
Many of the genes coding for enzymes of fatty acid biosynthesis in E. coli are organized in a cluster and under control of different promoters. This so called fab-cluster comprises the genes plsX, fabH, fabD, fabG, acpP and fabF. PlsX is the first gene of this cluster; however, its own promoter is rather weak so that about 60% of all transcripts containing plsX-mRNA result from promoters located further upstream [64, 158]. This longer mRNA contains transcripts of yceD (coding for an uncharacterized protein) and rpmF (coding for the 50S ribosomal subunit protein L32) so that coordinated regulation of these proteins and fatty acid biosynthesis seems possible [159].
The genes fabH, fabD and fabG encode for proteins that catalyze subsequent steps in the initiation of fatty acid biosynthesis. They are transcribed by a strong promoter within the upstream region of plsX and a weak promoter upstream of fabD[64, 160]. The strong promoter has been investigated unravelling a 4-fold downregulation of transcription upon amino-acid starvation [160], which is known to induce the synthesis of (p)ppGpp. Furthermore, the transcription of fabHDG at a normal level requires induction by FadR [149]. The weaker promoter is thought to complement for polar effects [64]. Lacking a promoter directly upstream of the coding sequence, fabG mRNA seems to be formed by processing of the longer transcripts [64]. The last two genes of the fab-cluster, acpP and fabF, each possess a strong promoter and for the former, no control by the growth rate or by FadR could be found [64].
Regulation by the stringent response
Many studies have shown that fatty acid biosynthesis in E. coli correlates with the growth rate [74, 161, 162], but the underlying mechanism was only partially investigated. Of great importance is the concentration of the global regulator (p)ppGpp, which in E. coli can be synthesized by the action of RelA and either hydrolyzed or synthesized by SpoT. Elevated concentrations of (p)ppGpp, in combination with the regulator protein DksA [163, 164], influence the stability of the RNA polymerase complex. As a result, the respective genes are activated or inactivated. As enhanced (p)ppGpp concentrations tend to destabilize σ70 promoters, the use of sigma factors other than σ70 is facilitated, thereby further extending the alteration of gene expression [116, 119, 165, 166]. The resulting changes of the cellular processes are referred to as the stringent response [167].
One sigma factor that is upregulated by elevated concentrations of (p)ppGpp in the stationary phase is RpoS [168]. RpoS is involved in multiple stress responses, including UV, acid, heat, oxidative stress or starvation [169–172]. With respect to the fatty acid metabolism, the induction of fadA, fadB, fadE and fadH expression by RpoS is of interest [141]. For additional information about the regulation and influences of RpoS, the reader is referred to a recent review article [173].
In cells growing under optimal conditions the concentration of (p)ppGpp is very low; however, it can be increased by amino-acid starvation [174], carbon-source depletion [175], phosphate limitation [176, 177], iron limitation [178] or inhibition of fatty acid biosynthesis [108, 179]. In E. coli, RelA is associated with the ribosomes and senses the binding of uncharged tRNAs (during amino-acid limitation), upon which the synthase activity of RelA is induced [174, 180, 181]. Furthermore, the degradation of (p)ppGpp by SpoT is reduced 5-fold, leading to a more than 20-fold increase in (p)ppGpp concentration. Glucose starvation triggers an approximately 5-fold increase of the (p)ppGpp concentration [176, 182], which is achieved by a strong inhibition of the hydrolase activity and a decrease of the synthesis of (p)ppGpp to 30%. As SpoT permanently interacts with the acyl carrier protein [108], it is likely that the degradation or synthase activities of SpoT are influenced by the difference in the charge of ACP, similar to RelA. The hereby provided link between fatty acid biosynthesis and (p)ppGpp concentration might be responsible for the increase in (p)ppGpp concentration under carbon limitation.
Elevated (p)ppGpp concentrations have direct and indirect effects on the biosynthesis of fatty acids. Fastest is the inhibition of enzymes, as shown in vivo for PlsB [113] and in vitro for FabZ [183]. On the level of transcription, a 4-fold decrease of the promoter of fabH, which controls fabH, fabD and fabG, is triggered by amino-acid starvation [160]. Additionally, the transcription of plsB, accBC and fadR is downregulated [116, 119, 149], whereas fabA, fabB, cfa and ybhO appear to be upregulated. The respective gene products are involved in the synthesis of stearic acid, unsaturated and cyclopropane fatty acids and cardiolipin [119]. These processes are typically for E. coli cells entering the stationary phase [153, 184, 185]. However, the enhanced expression of cfa is due to its σS-dependent promoter [168]. The same or a similar mechanism might also explain the upregulation of fabA and fabB, which stands in contrast to the decreased synthesis of their activator, FadR, during the stationary growth phase.
The inhibition of PlsB leads to an increase in the amount of long-chain fatty acyl-ACP, which in turn inhibit FabH, FabI [79] and the acetyl-CoA carboxylase [59]. An additional result of acyl-ACP accumulation is the modified activity of FabH, FabF and FabB, which leads to the degradation of malonyl-ACP to acetyl-CoA via the intermediate, acetyl-ACP. Overexpression of plsB partially relieves the inhibition of the fatty acid synthesis, leading to very long cells in the stationary phase [113].
As for the genes coding for enzymes of the β-oxidation pathway, the expression of yfcX and fadE is enhanced during the stringent response. On the contrary, the expression of fadD during stringent response of the wild-type was lower than in the mutant control strain that was not able to accumulate (p)ppGpp upon amino-acid starvation [166]. In this context one should keep in mind that the expression of the enzymes of the fatty acid degradation cycle is regulated by FadR [138, 139], whereas the expression of fadD can be induced by CRP-cAMP [140] (see also Section ‘Fatty acid degradation’).
Further impacts of the high (p)ppGpp concentration during the stringent response are the inhibition of the genes for tRNAs, rRNAs and ribosomal proteins [186, 187], as well as of the initiation factor IF2 [188]. A recent study by Edwards et al. [189] also found interactions between the carbon-storage regulators CsrA, CsrB and CsrC with the regulators of the stringent response.
Regulation according to the growth conditions
Long-chain fatty acids can be metabolized by E. coli; however, carbohydrates are the preferred carbon sources [140]. Aerobic growth on glucose is accompanied by a missing transcriptional induction of the β-oxidation genes, by CRP-cAMP [140, 147], and by the repression by FadR [136, 140]. Since the repression of fadD and fadL is less stringent, fatty acids can be taken up in small amounts [140] and relieve the binding of FadR to the respective promoters [138, 139]. Due to the similar affinity of PlsB towards fatty acyl-CoA, as towards fatty acyl-ACP, the former can directly be used for the membrane biosynthesis [112]. If glucose (and any other suitable carbohydrate carbon source) is also missing, the concentration of cAMP increases and the complex CRP-cAMP is formed, which binds to the promoters of the β-oxidation genes and induces their transcription [140].
Under anaerobic conditions, the situation is somewhat different: Transcription of fadA, fadB, fadE, fadD and, to a lesser extend, fadL is inhibited by the regulator ArcA [135], and the proteins FadK, YfcY, YfcX, YdiO and YdiD are responsible for the anaerobic fatty acid degradation, if a more suitable carbon source is not available [132]. Under these conditions, uptake of long-chain fatty acids by FadL is very slow and activation by FadD is not possible. However, FadK, which replaces FadD under anaerobic conditions, has a low activity towards long-chain fatty acids. Consequently, anaerobic growth on oleic acid is possible but is very slow [133]. In contrast to long-chain fatty acids, the transport of fatty acids with a medium or short chain-length does not require the activity of FadL [132, 151]. Also the activity of the other proteins of the anaerobic β-oxidation, for short and medium chain-length fatty acids, is sufficient to enable a robust growth on these substrates under anaerobic conditions [132].
Production of free fatty acids
The use of E. coli and other microorganisms for the production of free fatty acids was initiated by the discovery that the periplasmic enzyme thioesterase I (TesA) deregulates the tight product inhibition of fatty acid synthesis, when expressed as a cytosolic enzyme (‘TesA) [190]. This enzyme cleaves the fatty acyl-ACP, and with a considerably lower activity the fatty acyl-CoA thioester bond also. The resulting free fatty acids accumulate in the late exponential and in the stationary phase and are mostly released to the culture medium [190].
Since fatty acids are very energy-dense, produced in relatively large amounts and in every organism, they represent a suitable target for the development of single-cell oils. Also the use of alternative carbon sources has been demonstrated in a variety of microorganisms. With increasing attention towards the search of sustainable energy sources, many studies have been performed in the last 15 years with the aim of utilizing fatty acid biosynthesis for biofuel production. However, due to the strict regulation of this pathway much basic research is still needed to improve the yields of free fatty acids or related products. Besides of E. coli, fatty acid overproduction has been established in cyanobacteria [23, 191] and yeast [192].
Thioesterase expression and physiological consequences
As shown in Table 1, every strategy that yielded a concentration of more than 0.2 g l-1 fatty acids used a cytosolic thioesterase from E. coli or from a different organism. By the use of different thioesterases, the product can be considerably altered with respect to yield, fatty acid chain-length and degree of unsaturation [143, 193–195]. However, the expression level of any thioesterase must be tuned carefully, because already low levels increase the fatty acid titer significantly, and too strong a thioesterase activity has been shown to impair FFA production, both in in vitro and in vivo experiments [106, 196]. In addition, a high titer of FFA in the culture medium can also cause severe defects in the cellular viability. Desbois and Smith [197] summarized the antibacterial actions of FFA, ranging from membrane lysis and interruption of the electron transport chain to possible interferences with membrane proteins or nutrient uptake. Concerning the physiological effects of endogenous FFA overproduction, it has been shown that thioesterase overexpression can alter the degree of saturation of the membrane lipids in E. coli[193] and in Synechococcus elongatus[191]. Additional effects are the induction of stress responses and reduced membrane integrity and viability of the production strains [193, 198]. In the study of Lennen and coworkers [199], an improved fatty acid export system has been suggested to improve viability, and several components of the E. coli system have been identified and investigated. Deletion of the gene of the fatty acid transporter FadL has already been tested in combination with tesA overexpression [142] and gave promising results.
Of importance for the microbial production of biofuels are strategies to enhance the tolerance of E. coli towards organic solvents, as performed by Oh and coworkers [200]. Deletion of fadR resulted in an enhanced proportion of saturated fatty acids in the membrane of E. coli, as has been observed in previous studies [140, 151]. The higher grade of saturation made the membrane less permeable for organic solvents. By deletion of marR, the repressor of marA expression, the multidrug resistance of E. coli was permanently induced. Besides others this led to the constitutive expression of tolC, acrA and acrB. The gene products build an efflux system for organic solvents and thus enhance the survival of E. coli in presence of high concentrations of organic solvents. Interestingly, these genes are exactly the same genes that had been proposed by Lennen and coworkers [199] for improved fatty acid export. A combination of both deletions (fadR and marR) led to an even higher tolerance of organic solvents, compared to the single deletions [200]. However, fadR deletion may not be ideal, if one aims at the production of fatty acids. Hence, a combination of marR deletion and the improved synthesis of saturated fatty acids, for example, by overexpression of fabA[91] or fabZ[86], appears to be promising.
Deletion of β-oxidation genes
To prevent product degradation, many studies have been performed in a strain that was inhibited in fatty acid β-oxidation. The main target for deletion was fadD[26, 106, 143, 190, 194, 201–203], whereas deletion of fadE was mainly done when the activation of the FFA to fatty acyl-CoA esters was necessary for further product processing [26, 142, 152]. Although most studies found FFA levels enhanced upon (partial) deletion of the β-oxidation pathway or did not control the success of this deletion, Cho and Cronan [190], as well as Liu and coworkers [142] did not detect a positive effect when thioesterase overexpression was combined with the deletions of fadD, fadE or fadL (to impair re-uptake of FFA). In these studies it was suggested that the β-oxidation pathway has not the capacity to cope with the strong FFA production. An alterative explanation might be that the positive control of fadL, fadD and fadH by the cAMP receptor protein-cAMP complex [140] was limiting in some of the performed studies, which might be caused by different cultivation conditions. In contrast, it seems unlikely that the negative control via the FadR repressor (released upon acyl-CoA binding) differed in the studies where the fadD or fadL genes were not deleted.
Investigation and remodeling of the whole pathway
In order to improve FFA production on a broad scale, a computational model of the E. coli metabolism has been used, and several deletions in the glycolysis or tricarboxylic acid cycle have been investigated along with the overexpression of genes of fatty acid biosynthesis [204]. Deletion of the genes responsible for acetate formation has been tested to improve malonyl-CoA titers [61] or FFA productivity [194, 201, 205]. This strategy clearly reduced acetate formation; however, in the two latter studies the reduction of acetate formation did not enhance FFA yields. Instead, Zhang and coworkers [194] state that the acetate formation is already diminished in efficient FFA producers. This is also interesting with respect to the pH of the medium, as E. coli production strains tend to slightly increase the pH, instead of decreasing it as wild-type cells [194].
An alternative way to investigate the production of FFA as a whole is the reconstruction of the pathway under controlled conditions in vitro. In the study of Liu et a l. [106] cell extracts of E. coli production strains were used to determine the concentrations of NADPH and malonyl-CoA, which enabled half-maximal reaction velocity. For NADPH a Km value of 34.7 μM was calculated, and based on an estimated concentration of NADPH in the cytosol of 100 to 200 μM [206–208] it was concluded that this cofactor should not be limiting. In contrast, the calculated Km value for malonyl-CoA of 15.7 μM exceeds the estimates of Davis et al. [60] and Bennet et al. [206], who give a cellular concentration of malonyl-CoA of less than 5 μM. Consequently, the addition of malonyl-CoA or acetyl-CoA-carboxylase to cell free extracts enhanced the in vitro reaction velocity of fatty acid biosynthesis [106]. In addition, the overexpression of the acetyl-CoA carboxylase was also investigated several times in combination with other genetic modifications, and successfully enhanced FFA production [60, 106, 143]. However, Acc overexpression alone was not sufficient for FFA production in a ΔfadD background [202]. A detailed study of how to improve the malonyl-CoA concentration in the cytosol has been performed by Zha and coworkers [61]. By deletion of acetate formation pathways, heterologous expression of Acc from Corynebacterium glutamicum and the E. coli acetyl-CoA synthase, 16-fold higher malonyl-CoA content was achieved.
For those who wish to improve the substrate or cofactor supply, Yu et al. [196] have tested higher concentrations of acetyl-CoA (0.5 mM), malonyl-CoA (1.5 mM), NADH (1 mM), NADPH (1.5 mM), NAD+ (5 mM) and NADP+ (5 mM) in an in vitro reconstitution experiment of fatty acid biosynthesis. With these concentrations, no inhibition was observed. In contrast, Liu and coworkers [106] found a strong inhibitory effect of high concentrations of apo- and holo-ACP on in vitro fatty acid synthesis. The limit for a beneficial effect has been determined to be 32 μM for both apo- or holo-ACP [196]. This finding is in agreement with the study of Keating et al. [104], who found that overexpression of tesA and acpP strongly inhibits the growth of E. coli. Coexpression of acpS relieved this phenotype only slightly, suggesting that both apo- and holo-ACP are also inhibitory in vivo.
With respect to the other enzymes of fatty acid biosynthesis, it was found that FabA, FabB, FabD, FabF, FabG, FabH, FabI and FabZ occur in about equal concentrations in wild-type E. coli cells, whereas TesA and holo-ACP proteins are considerably more abundant [196]. To get an even better understanding of potential candidates for overexpression, an in vitro assay was performed using purified enzymes, with fixed concentrations. When all enzymes were used in a concentration of 1 μM and 10 μM for ACP and TesA respectively, a further enhancement of the concentrations of FabA, FabB, FabD or FabG did not result in an increase of activity. FabF and FabH (the latter less pronounced) inhibited the enzymes of FAB at higher concentrations, whereas FabI and FabZ enhanced the FAB activity 2- and 6-fold, respectively, when added at a concentration of 10 μM [196]. Some of these data were confirmed in vivo (see Table 1), for example, the lack of enhanced FFA production upon (co-)overexpression of FabA [78, 152], whereas coexpression of FabB doubled the FFA production in an E. coli strain with a fadD-deletion and overexpression of tesA[152]. The role of FabF remains even more controversial. Due to the strong inhibition of the in vitro assay, Yu et al. [196] tested coexpression of the fabF gene in their production strain and could also detect a strong decrease in FFA titer. On the contrary, Zhang et al. [152] enhanced the FFA yield of their production strain nearly 3-fold upon fabF overexpression. However, they observed a higher FFA yield, when fabF was expressed at a lower rate. Both studies were performed using a fadE deletion mutant of E. coli and strong tesA and fabF overexpression. Apart from the use of different plasmid systems, Yu and coworkers [196] coexpressed an additional thioesterase from Cinnamomum camphorum in both in vitro and in vivo experiments. This thioesterase has activity towards fatty acids of a chain length ranging from 12 to 18 carbon atoms [209]. It is also noteworthy, that overexpression of fabF has a lethal effect in a strain that does not overproduce FFA [76] (compare to the Section about FabB, FabF and FabH).
Similar discrepancies can be found in the literature on fabZ overexpression. Upon co-overexpression of fabZ, the FFA titer was enhanced nearly 3-fold in a fadD deletion mutant with expression of a thioesterase from Ricinus communis[76]. In contrast, in the study of Yu and coworkers [196], enhanced levels of FabZ improved the rate of fatty acid biosynthesis only in vitro, whereas in vivo the combined co-overexpression of fabZ, fabI and fabG was necessary to outperform the control strain with fadE deletion and overexpression of tesA and the thioesterase from C. camphorum. Taken together these results indicate that overexpression of more than one enzyme of the FAB are much more likely to improve FFA production of already existing production strains.
Altering the regulation or pathway direction
A promising addition to the overexpression of genes that are involved in fatty acid biosynthesis offers the use of regulatory mutants. A possible target to improve the FFA yields is the carbon-storage regulator, which consists of the CsrA protein and the non-coding RNAs CsrB and CsrC [210]. CsrA acts as a posttranscriptional inhibitor or activator by binding to the 5’-untranslated sequence of target mRNAs [211]. This binding can be prevented by the interaction with CsrB or CsrC that consist of several CsrA binding sites and sequester this protein [212]. In the study of Edwards et al. [189] 721 transcripts have been identified that copurify with CsrA, which regulates cellular processes such as glycolysis, glycogen formation or the stringent response. McKee et al. [213] have used csrB overexpression to enhance the productivity of a tesA expression strain. Besides the nearly doubled FFA production, a concomitant reduction in acetate formation was observed.
A more obvious candidate to alter the regulatory network is the repressor of fatty acid degradation, FadR. In an E. coli strain with a fadE deletion and with tesA overexpression, coexpression of fadR resulted in a more than seven-fold enhanced FFA production [152]. Due to the induction of fabA and fabB, the coexpression of fadR leads to an increase of the unsaturated fatty acid (UFA) content from 13% to 43% in the production strain.
Deletion or disruption of the fadR gene leads to a constant expression of the β-oxidation genes. This enables E. coli to grow aerobically on fatty acids with medium chain-length [214]. The expression can further be enhanced by a mutation in the cAMP receptor protein (crp*) that leads to a deregulated catabolite repression [215]. By the deletion of arcA[135] and a mutation in the regulatory gene atoC, aerobic growth of E. coli on fatty acids with short chain-length is possible [216, 217]. The aforementioned mutations in fadR, crp, arcA and atoC have been used in combination with the deletion of fermentative pathways and overexpression of the genes fadB, fadA and fadM for a functional reversal of the fatty acid degradation cycle with the aim to produce FFA from non-related carbon sources [201]. As this way does not need the energy-consuming conversion of acetyl-CoA to malonyl-CoA, the theoretical yield for FFA production from glucose can be increased from about 36% (g g-1) to 43% (g g-1) [218].
Process optimization
Although many studies have focused on the engineering of an efficient production strain, they were mostly performed in a batch mode. If investigated, fed-batch fermentations have significantly increased the productivity, and titers of 2.5 g l-1 to 7 g l-1 have been achieved [106, 143, 201]. Due to the growth inhibitory effect of high concentrations of fatty acids in the culture medium and to the fact that the product must be somehow purified, Liu and coworkers [142] have applied an extraction unit to their fed-batch fermentation system. Beginning 10 hours after induction, the culture volume was pumped through a tributylphosphate phase at a rate of 0.8% per minute (volume for extraction per volume cultivation medium). After passage of the tributylphosphate phase, the culture medium was pumped back into the fermenter vessel. By this process, a total fatty acid production of roughly 9 g l-1 was achieved [142].
Although fed-batch fermentations have advantages over batch cultures, continuous fermentations offer an even higher potential, because the cells can be kept under optimal conditions and in the most suitable growth phase. With the aim of FFA production, continuous cultivations of an E. coli strain with replacements of fadD, fadE and fadAB, each by one copy of the thioesterse gene from Umbellularia californica, have been performed [203, 204]. Limitation of carbon, nitrogen or phosphate source has been applied with phosphate limitation enabling the best results. With respect to the carbon source (glucose) a conversion rate to FFA of 0.1 (g g-1) has been achieved, and the highest biomass-specific productivity was 0.068 g FFA per g cell dry weight per hour [203].
Production of FAAE
The production of FAAE has been the focus of several studies in recent years. It relies on the microbial production of fatty acids and of a short chain-length alcohol. After the fatty acids are activated to acyl-CoA the formation of an ester bond with an alcohol is performed enzymatically. For this reaction, nearly all studies performed so far used the promiscuous wax ester synthase/acyl-CoA:diacylglycerol acetyl transferase (WS/DGAT; AtfA) of Acinetobacter baylyi ADP1. This enzyme has been shown to exhibit activity with an extraordinary wide range of alcohols and fatty acids [219, 220]. As alcohol moiety ethanol is preferred due to its low toxicity for the production organism, compared to methanol or butanol and the ease of its microbial production. The activity of the AtfA towards ethanol is considerably lower than towards the natural substrates diacylglycerol or long chain-length alcohols [219]. An attempt to overcome this possible bottleneck has been the comparison of five different wax ester synthases towards FAEE production in S. cerevisiae[221].
The first study to produce FAEE-based (fatty acid ethyl ester) microdieselin E. coli has been performed by Kalscheuer et al. [24], yielding 1.3 g l-1 FAEE by fed-batch fermentation (Table 2). They expressed the Zymomonas mobilis pyruvate decarboxylase (pdc) and alcohol dehydrogenase B (adhB) to produce ethanol. By coexpression of atfA, the cells were enabled to form an ester consisting of ethanol and a fatty acid. This study provided the blue print for most subsequent studies to produce FAEE. Optimization of the fed-batch process by Elbahloul and Steinbüchel [222] yielded a maximum FAEE concentration of 11 g l-1. However, these two studies used oleic acid that was externally added to the culture medium and did not rely on endogenously produced fatty acids. To produce FAEE from non-related carbon sources, at least a thioesterase (for cleavage of acyl-ACP) and a fatty acid CoA ligase have to be combined with ethanol production and atfA expression [26, 223]. These studies have also shown a significant increase in FAEE production upon deletion of the fadE gene. Fed-batch fermentations to produce FAEE from glucose have been optimized with respect to the medium, time of induction, temperature and feeding rate, by Duan and co-workers [223], yielding a maximal FAEE content of 0.9 g l-1.
Despite the advances in FAEE production derived from non-related carbon sources, the yields are still far too low for commercial purposes. One problem is the instability of production strains that probably results from ethanol accumulation [26, 224]. Cultivations of the production strain have been performed at 25 or 30°C [26, 223], and the latter study reports that at 37°C no FAEE production was achieved. A more recent study approached the problem of ethanol toxicity by finetuning the expression of all overexpressed genes [152]. This is achieved by coexpression of fadR and introduction of FadR binding sites in the strong promoters of all overexpressed genes, despite tesA. When FFAs accumulate, the repression of fadD, pdc, adhB and atfA by FadR is relieved depending on the amount of FFA. As a result, the formation of ethanol, as well as of acetate could be clearly reduced.
With respect to the use of FAEE as an alternative and renewable energy source, carbon sources must be used that are even cheaper than glucose and do not compete with food and feed production. Attempts have already been made that prove the possibility of using hemicelluloses or pretreated switchgrass for FAEE production by fermentation of E. coli[26, 225].
Besides FAEE production, the biosynthesis of fatty acid methyl esters has also been performed in E. coli, by the action of a fatty acid methyltransferase that uses S-adenosylmethionine as the donor for the methyl group [25]. However, the yield in this study was very low at roughly 15 μM FAME . Alternative products in the class of FAAE are wax esters, which are formed naturally by some plant species. Microbial production of wax esters in E. coli was first achieved by Kalscheuer and coworkers [226], who used the acyl-CoA reductase from the jojoba plant to reduce fatty acyl-CoA to fatty alcohol. The latter was esterified with fatty acyl-CoA, yielding wax esters. In this study, the maximal concentration of wax esters reached 1% of the cell dry weight. Steen et al. [26] demonstrated wax ester formation in E. coli ΔfadE upon overexpression of the genes for an acyl-CoA reductase (from Mus musculus), tesA, fadD and atfA. More recently wax ester synthesis was also established in the cyanobacterium S. elongatus by the overexpression of atfA, an acyl-ACP reductase and alcohol dehydrogenase [227].
The production of large amounts of fatty acid ethyl esters in microorganisms other than E. coli is so far restricted to S. cerevisiae[221, 228], which is known to naturally synthesize small amounts of FAEE with medium chain-length [229]. FAEE production could potentially benefit from the endogenous ethanol production and tolerance of high ethanol concentrations of S. cerevisiae. However, the study of Yu et al. [228] shows that the endogenous ethanol production has to be boosted in order to achieve higher amounts of FAEE. The best result in this study was a concentration of 0.52 g l-1 after 72 hours of batch cultivation.
Production of other fatty acid-derived biofuels
Besides free fatty acids and fatty acid alkyl esters some research has focused on the production of alternative biofuels that are derived from E. coli fatty acid biosynthesis. The native and engineered pathways of E. coli are summarized in Figure 4. Examples for this are triacylglycerols that also occur naturally in a variety of eukaryotic and prokaryotic cells, methyl ketones, alcohols and alkanes and polyhydroxyalkanoates with a medium chain-length.

Metabolic pathways for the production of fatty acids and derived compounds. Enzymes and arrows are shown in green, if the pathway to which they belong departs from the wild-type fatty acid metabolism. The end products are shown in gray boxes. Appropriate deletions or homologous overexpressions within the wild-type pathway are not highlighted in this figure. For details, the reader is referred to the text. FAB, fatty acid biosynthesis; FAD, fatty -acid degradation; Aar, acyl-ACP reductase; Aco, acyl-CoA oxidase; Acr1, acyl-CoA reductase; Adc, aldehyde decarbonylase; Ahr, aldehyde recuctase; AtfA, acyltransferase; Cad, carboxylic acid decarboxylase; Car, carboxylic acid reductase; FadM, thioesterase; PgpB, phosphatidylglycerol phosphate phosphatase; PhaC2, PHA synthase 2; PhaG, (R)-3-hydroxydecanoyl-ACP transacylase; PhaJ, (R)-specific enoyl-CoA hydratase; SHMKS1, methylketone synthase 1; SHMKS2, methylketone synthase 2; TesA, thioesterase.
TAG
The formation of TAG is native to only a few bacterial genera, such as Rhodococcus, Mycobacterium, Streptomyces, Nocardia, Acinetobacter or Alcanivorax[230]. Of these the organisms Rhodococcus opacus[38, 231–234], Streptomyces coelicolor[235–237] and species of the genus Mycobacterium[238–240] are the best studied. R. opacus has been shown to accumulate TAG up to 86% of the cellular dry weight [231] and was subject to pilot-scale fermentation and optimization [241–243].
Attempts to establish TAG formation in E. coli have been very rare. In 2008 Arabolaza and coworkers [235] transformed an E. coli dgkA (diacylglycerol kinase A) mutant with plasmids containing three different WS/DGAT enzymes from S. coelicolor. The dgkA mutant of E. coli has earlier been found to accumulate high levels of diacylglycerol, due to an impaired membrane lipid-recycling following to the synthesis of membrane-derived oligosaccharides [111, 244, 245]. Expression of one of the investigated WS/DGAT enzymes (Sco0958) led to TAG formation in the mutant strain [235].
An alternative way has been investigated more recently [236, 246], which employs enzymes that catalyze the dephosphorylation of phosphatidic acid, yielding diacylglycerol. The first study used the overexpression of the atfA and E. coli pgpB (phosphatidate phosphatase) genes, by which the synthesis of 1.1 mg l-1 TAG was achieved. The study of Comba et al. [236] used the same biosynthetic route, but with the genes Sco0958 and lppα or lppβ (phosphatidate phosphatases) from S. coelicolor.
Apart from E. coli, heterologous TAG formation has also been performed in the cyanobacterium S. elongatus, by overexpression of atfA[227]. As S. elongatus uses an acyl-ACP synthetase instead of an acyl-CoA ligase for fatty acid activation [247], this raises the question as to whether AtfA can also use acyl-ACP as substrate. Optimization of the native TAG production in algae by metabolic engineering also offers great potential for the production of next-generation biofuels [248, 249].
Methyl ketones
Methyl ketones are formed by the hydrolysis of an acyl-ACP intermediate and the subsequent decarboxylation of the 3-keto acid. These volatile substances were first found in rue (Ruta graveolens) [250] but are widespread among plant, animal and microbial species [251]. Wild-type E. coli cells do not produce significant amounts of methyl ketones, but the ability can be established by metabolic engineering. In the first study small amounts of methyl ketones were obtained by overexpression of the genes shmks1 and shmks2 (methylketone synthases 1 and 2) from wild tomato (Solanum habrochaites) [252]. Park et al. [253] applied overexpression of these genes in an E. coli strain that was blocked in four pathways of the fermentation metabolism by deletion of the genes adhE, ldhA, poxB and pta. This strain procuced 450 mg l-1 methyl ketones. Shortly before, a methyl ketone titer of 380 mg l-1 was published upon overexpression of the genes fadB, fadM and Mlut11700 (an acyl-CoA oxidase of Micrococcus luteus) in an E. coli strain with deleted fadE and fadA genes [254]. The combination of the genes fadB, fadM and Mlut11700 was also sufficient for chemolithoautotrophic production of up to 180 mg l-1 methyl ketones in a strain of Ralstonia eutropha with both β-oxidation operons deleted [255].
Fatty alcohols and alkanes
Another way to process fatty acids for fuel production is the reduction to long-chain alcohols, alkanes and alkenes. First attempts have been performed by Keasling and coworkers [256], who showed that an E. coli fadE mutant produced small amounts of fatty alcohols. Additional overexpression of fadD, acr1 (acyl-CoA reductase 1 from A. calcoaceticus BD413) and tesA enhanced the fatty alcohol production to 60 mg l-1[26]. Another study has shown that the overexpression of only a fatty acyl-CoA reductase (in this case from Arabidopsis thaliana) is sufficient for fatty alcohol production in otherwise unmodified wild-type E. coli[257]. By reversal of the β-oxidation pathway [201] (see Section ‘Altering the regulation or pathway direction’ for details) and overexpression of an alcohol dehydrogenase, 330 mg l-1 n-alcohols with chain lengths of 5 to 10 carbon atoms were produced. A slightly higher concentration of 350 mg l-1 fatty alcohols was achieved by expression of the carboxylic acid reductase (Mycobacterium marinum), ahr (aldehyde reductase of E. coli) and tesA genes [258].
The biosynthesis of alkanes and alkenes is not done by further reduction of a fatty alcohol, but by a decarboxylation or decarbonylation of a fatty acid or aldehyde. In 2010 Lennen et al. reported on the production of alkanes by the conversion of fatty acids, extracted from an overproducing strain of E. coli[202]. Complete biosynthesis of alkanes was achieved by overexpression of acyl-ACP reductase and aldehyde decarbonylase (both from S. elongatus) in E. coli[259]. As these enzymes can use acyl-ACP, the coexpression of fadD does not lead to higher productivity. Presumably, a thioesterase overexpression would also rather diminish fatty alkane production. By coexpression of fabH2 (β-ketoacyl-ACP synthase of B. subtilis) up to 80 mg l-1 alkanes with even and uneven chain length could be produced [260].
A similar system based on FFA took advantage of the genes luxCED (fatty acid reductase complex from Photorhabdus luminescens) and NpAD (aldehyde decarbonylase from Nostoc punctiforme) for the production of fatty alkanes [261]. By coexpression of fabH2, branched-chain fatty acids were also produced and processed to the respective alkanes. The coexpression of fatB (thioesterase of C. camphora, specific for tetradecanoyl-ACP) resulted mainly in the synthesis of tridecane. However, the yields of the alkane production were lower than 10 mg l-1, also with fatB overexpression [261]. In the study of Akhtar and coworkers [258] a carboxylic acid reductase of M. marinum and an aldehyde decarboxylase of Prochlorococcus marinus were used in combination with tesA expression. The exact yields were not given, but it was stated that the yields were considerably lower than for fatty acids or alcohols.
The production of 1-alkenes by the decarboxylation of FFA has been studied in the Gram-positive bacterium Jeotgalicoccus sp. ATCC 8456. Identification of the responsible gene (a fatty acid decarboxylase) revealed that heterologous expression is sufficient for one-step production of 1-alkenes in E. coli[262].
PHAmcl
Polyhydroxyalkanoates (PHA) are polymers that can be used as biodegradable plastics. However, their physical properties and thus their usability depend on the kind of the monomer (s) [263]. Whereas the well-investigated and first-discovered polyhydroxybutyrate (PHB) is synthesized by the condensation of two molecules of acetyl-CoA (reaction of PhaA) and the subsequent reduction of acetaldehyde (by PhaB) and polymerization of 3-hydroxybutyrate (by PhaC) [264], the monomers of PHA with longer carbon chains typically are taken from fatty acid biosynthesis [265] or degradation [266]. Biosynthesis of these PHAmcl (with medium chain-length) also occurs in many prokaryotes and is well studied in species of the genus Pseudomonas[267] that were also genetically modified [268–270]. The monomers of PHA with medium or long alkyl chains are 3-hydroxy fatty acids and in addition to their use as bioplastics may be considered as potential biofuels.
Aiming to establish the production of PHAmcl in E. coli, the cells are made to overproduce or grow on FFA that enter the β-oxidation, and the intermediate product 3-hydroxyacyl-CoA is then polymerized by a suitable PhaC (PHA synthase) enzyme. In the first studies concerning PHAmcl production in E. coli, this was achieved by expression of the PHA synthases 1 and 2 of Pseudomonas aeruginosa in E. coli mutants, impaired in the β-oxidation and with acrylic acid as the inhibitor [271, 272].
Klinke et al. [273] reported the synthesis of PHAmcl up to 2.3% of the cell dry weight by overexpression of tesA and phaC mcl from P. aeruginosa. With the same PHA polymerase and the thioesterase of U. californica, coexpressed in an E. coli fadB mutant 6% of the cell dry weight could be achieved [274]. The effect of fadA, fadB or fadAB mutant strains was further studied by Park and Lee [275]. By the coexpression of phaA, phaB (both of R. eutropha) and phaC2 and with decanoate feeding, the authors showed the production of PHB-PHAmcl copolymers with different ratios of PHB to PHAmcl, depending on the deletion mutant used.
A step towards the production of PHA homopolymers with medium chain-length-3-hydroxyalkanoates as constituents has been the production of 3-hydroxydecanoate, up to 46% of the cell dry weight by the combined expression of tesB (thioesterase II from E. coli) and phaG ((R)-3-hydroxydecanoyl-ACP transacylase from P. putida) [276]. However, a polymerization of 3-hydroxydecanoate has not been reported in this study. Further attempts towards the production of copolymers with short and medium chain-lengths have been the coexpression of several mutant genes of fabH and phaC1 (from Pseudomonas sp. 61–3) with phaA and phaB (from R. eutropha) [277] and additionally with fabG[82]. These studies succeeded in the production of PHA with an enhanced proportion of monomers with 6, 8, 10 and 12 carbon atoms. By application of an E. coli fadR and atoC mutant, the polymer content could be further enhanced [83]. To date, the highest amount of PHA copolymer (15% of the cell dry weight) from non-related carbon sources has been achieved by Agnew et al. [278]. They expressed the tesA gene for FFA production in combination with the P. aeruginosa genes phaC, phaJ ((R)-specific enoyl-CoA hydratase) and PP_0763 (putative acyl-CoA synthetase), in an E. coli strain with deletions in fadR, fadI, fadJ (also named yfcX), fadA and fadB.
Feedstocks
If one wishes to produce microbial biofuels (or bulk chemicals) in a cost-competitive way with petrochemical or oil plant-based production processes, there are two key factors to consider. First, it is important to achieve high product-concentrations, as that will influence the productivity of a fermentation plant and maximize the yields in product recovery and refinement. The second key factor is the cost for the carbon source [279]. In far-developed processes, the carbon source can account for 30 to 60% of the production costs, as is the case for the production of polyhydroxybutyrate from glucose [280], propanediol from glycerol [281] or ethanol from sugar cane molasses [282].
Glucose is the most common substrate for bacterial growth and is used in most of the studies that were performed in the field of biofuel production with engineered E. coli. However, glucose is rather expensive, whereas sucrose from sugar cane was described as the cheapest carbon source available for industrial fermentations in 2004 [283]. Also for future development, sucrose was expected to be at least as competitive (on price) as lignocellulosic biomass for the carbon source [283].
Utilization of sucrose is limited to a few E. coli strains, but can be established in E. coli K-12 by overexpression of cscA and cscB, coding for the invertase and the sucrose transporter [284]. However, as mentioned in the introduction, biofuel synthesis based on sucrose utilization will lead to competition with food production and to the use of arable land. Thus, the use of lignocellulosic biomass would be promising, but it typically includes severe pretreatment of the material, to break down the dense cellulose fibers [285]. Currently, this pretreatment involves acid- or base-catalyzed hydrolysis and the addition of cellulase enzymes, and hence is quite expensive. Furthermore, a sideproduct of this pretreatment is the formation of furfural, which inhibts bacterial growth. This problem has been addressed in a recent publication, and by expression of fucO, ucpA and pntAB and deletion of yqhD, the furfural tolerance of E. coli could be enhanced [286]. Another problem is that lignocellulosic biomass (once degraded to its components) consists of several different sugars that are utilized sequentially, due to the catabolite repression system of E. coli. This sequential degradation leads to many short lag phases, when the bacteria switch from one consumed carbon source to another. To circumvent this problem, regulatory mutant strains can be used [287]. However, degradation of the phenolic compounds from the lignin moiety of lignocellulose is still not possible for E. coli strains and reduces the possible product yield on this substrate. Besides lignocellulose, cheap substrates that can be utilized by E. coli include cheese whey [288, 289] or seaweed hydrolysate [290].
Most studies have addressed either the production of biofuels or the growth of E. coli on alternative carbon sources. However, direct production of FAEE has been reported using hemicellulose sugars [26] and pretreated switchgrass [20, 225].
Conclusions
Of the products that were discussed in this review, free fatty acids and PHAs from genetically engineered E. coli were the subject of most studies and have consequentely yielded the best results. The production of FAEE has only yielded high concentrations if an even higher amount of oleic acid has been supplemented to the medium (in addition to the primary carbon source) [222]. Methyl ketones, fatty alcohols and alkanes are being synthesized only with low yields, but research on their microbial production has just begun. Triacylglycerols can already be produced naturally by many microbes. Engineering of E. coli towards TAG production will only lead to a competetive process if it is possible to combine a high TAG-accumulation with the fast growth of E. coli.
Concerning the productivity of fatty acid synthesis it is difficult to estimate the current state of the art, since many studies have been performed only in batch mode. The theoretical limit of fatty acid production with glucose as the carbon source is roughly 35% (w/w). With some engineered strains, 56 to 85% of the theoretical limit has been reached [201, 291] by metabolic engineering and by modifications in the regulation of fatty acid metabolism. However, the highest-reported product concentrations approached 10 g l-1, with productivities in the range of 0.1 to 0.2 g l-1 h-1[106, 142, 143, 201, 292]. These values indicate that much research still needs to be done to reduce the time for the production of high concentrations of fatty acids. As cell growth and fatty acid production compete for the carbon source, it will be necessary to develop a continuous fermentation or a repeated fed-batch process with high cell-densities that enable high product-concentrations. A problem may then arise, because free fatty acids can impose a considerable stress on the cells if present in high concentrations. Thus, a continuous extraction or the conversion to non-toxic endproducts seems to offer great potential for future processes and strain improvements. PHA or TAG are such end products, but they accumulate in the cells, which makes recycling of living cells impossible. FAEE might be suitable and have been shown not to be growth-inhibitory up to concentrations of 100 g l-1[26]. Finally it is desireable to combine biofuel production with the engineered ability to grow on cheap resources like cellulose or hemicellulose. First attempts have already been made to show the general possibility [26, 225].
Abbreviations
- Acc:
-
Acetyl-CoA carboxylase
- ACP:
-
AcylCarrierProtein
- BC-BCCP:
-
Biotin carboxylase-biotin carboxyl carrier protein
- CRP-cAMP:
-
Cyclic adenosine monophosphate receptor protein-cyclic adenosine monophosphate complex
- FAAE:
-
Fatty acid alkyl ester
- FAEE:
-
Fatty acid ethyl ester
- FFA:
-
Free fatty acid
- (p)ppGpp:
-
(guanosine 5-triphosphate, 3-diphosphate) guanosine 3,5-bispyrophosphate
- PHA:
-
Polyhydroxyalkanoate
- PHB:
-
Polyhydroxybutyrate
- TAG:
-
Triacylglycerol
- tesA:
-
Leaderless version of the gene for the E. coli thioesterase I, that remains in the cytosol
- WS/DGAT:
-
Wax ester synthase/acyl-CoA: Diacylglycerol acetyl transferase.
References
Agency International Energy: Medium-term oil market report 2013. [http://www.iea.org/w/bookshop/add.aspx?id=450] []
Antoni D, Zverlov VV, Schwarz WH: Biofuels from microbes. Appl Microbiol Biotechnol 2007, 77: 23-35. 10.1007/s00253-007-1163-x
Uthoff S, Bröker D, Steinbüchel A: Current state and perspectives of producing biodiesel-like compounds by biotechnology. Microb Biotechnol 2009, 2: 551-565. 10.1111/j.1751-7915.2009.00139.x
Madhavan A, Srivastava A, Kondo A, Bisaria VS: Bioconversion of lignocellulose-derived sugars to ethanol by engineered Saccharomyces cerevisiae . Crit Rev Biotechnol 2012, 32: 22-48. 10.3109/07388551.2010.539551
Amorim HV, Lopes ML, De Castro Oliveira JV, Buckeridge MS, Goldman GH: Scientific challenges of bioethanol production in Brazil. Appl Microbiol Biotechnol 2011, 91: 1267-1275. 10.1007/s00253-011-3437-6
Luque R, Herrero-Davila L, Campelo JM, Clark JH, Hidalgo JM, Luna D, Marinas JM, Romero AA: Biofuels: a technological perspective. Energy Environ Sci 2008, 1: 542-564. 10.1039/b807094f
Atsumi S, Liao JC: Metabolic engineering for advanced biofuels production from Escherichia coli . Curr Opin Biotechnol 2008, 19: 414-419. 10.1016/j.copbio.2008.08.008
Atsumi S, Hanai T, Liao JC: Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 2008, 451: 86-89. 10.1038/nature06450
McDonald RI, Fargione J, Kiesecker J, Miller WM, Powell J: Energy sprawl or energy efficiency: climate policy impacts on natural habitat for the United States of America. PLoS One 2009, 4: e6802. 10.1371/journal.pone.0006802
Tsao C–C, Campbell JE, Spak SN, Carmichael GR, Chen Y: Increased estimates of air-pollution emissions from Brazilian sugar-cane ethanol. Nat Clim Chang 2011, 2: 53-57. 10.1038/nclimate1325
Al-Zuhair S: Production of biodiesel: possibilities and challenges. Biofuels Bioprod Biorefin 2007, 1: 57-66. 10.1002/bbb.2
Michalopoulos A, Landeweerd L, Van der Werf-Kulichova Z, Puylaert PG, Osseweijer P: Contrasts and synergies in different biofuel reports. Interface Focus 2011, 1: 248-254. 10.1098/rsfs.2010.0034
European Biofuels Technology Platform: Biofuel production. [http://www.biofuelstp.eu/fuelproduction.html] []
Delucchi MA: Impacts of biofuels on climate change, water use, and land use. Ann NY Acad Sci 2010, 1195: 28-45. 10.1111/j.1749-6632.2010.05457.x
Fargione J, Hill J, Tilman D, Polasky S, Hawthorne P: Land clearing and the biofuel carbon debt. Science 2008, 319: 1235-1238. 10.1126/science.1152747
Keyzer MA, Merbis M, Voortman R: The biofuel controversy. De Economist 2003, 56: 507-527.
la Grange DC, den Haan R, van Zyl WH: Engineering cellulolytic ability into bioprocessing organisms. Appl Microbiol Biotechnol 2010, 87: 1195-1208. 10.1007/s00253-010-2660-x
Laluce C, Schenberg AC, Gallardo JC, Coradello LF, Pombeiro-Sponchiado SR: Advances and developments in strategies to improve strains of Saccharomyces cerevisiae and processes to obtain the lignocellulosic ethanol–a review. Appl Biochem Biotechnol 2012, 166: 1908-1926. 10.1007/s12010-012-9619-6
Mazzoli R, Lamberti C, Pessione E: Engineering new metabolic capabilities in bacteria: lessons from recombinant cellulolytic strategies. Trends Biotechnol 2012, 30: 111-119. 10.1016/j.tibtech.2011.08.003
Bokinsky G, Peralta-Yahya PP, George A, Holmes BM, Steen EJ, Dietrich J, Lee TS, Tullman-Ercek D, Voigt CA, Simmons BA, Keasling JD: Synthesis of three advanced biofuels from ionic liquid-pretreated switchgrass using engineered Escherichia coli . Proc Natl Acad Sci USA 2011, 108: 19949-19954. 10.1073/pnas.1106958108
Colin VL, Rodríguez A, Cristóbal HA: The role of synthetic biology in the design of microbial cell factories for biofuel production. J Biomed Biotechnol 2011, 2011: 601834.
Kosa M, Ragauskas AJ: Bioconversion of lignin model compounds with oleaginous Rhodococci . Appl Microbiol Biotechnol 2012, 93: 891-900. 10.1007/s00253-011-3743-z
Liu X, Sheng J, Curtiss R III: Fatty acid production in genetically modified Cyanobacteria . Proc Natl Acad Sci USA 2011, 108: 6899-6904. 10.1073/pnas.1103014108
Kalscheuer R, Stölting T, Steinbüchel A: Microdiesel: Escherichia coli engineered for fuel production. Microbiology 2006, 152: 2529-2536. 10.1099/mic.0.29028-0
Nawabi P, Bauer S, Kyrpides N, Lykidis A: Engineering Escherichia coli for biodiesel production utilizing a bacterial fatty acid methyltransferase. Appl Environ Microbiol 2011, 77: 8052-8061. 10.1128/AEM.05046-11
Steen EJ, Kang Y, Bokinsky G, Hu Z, Schirmer A, McClure A, del Cardayre SB, Keasling JD: Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 2010, 463: 559-562. 10.1038/nature08721
Kates M, Wassef MK, Kushner DJ: Radioisotopic studies on the biosynthesis of the glyceryl diether lipids of Halobacterium cutirubrum . Can J Biochem 1968, 46: 971-977. 10.1139/o68-145
Pugh EL, Kates M: Acylation of proteins of the Archaebacteria Halobacterium cutirubrum and Methanobacterium thermoautotrophicum . Biochim Biophys Acta 1994, 1196: 38-44. 10.1016/0005-2736(94)90292-5
Leibundgut M, Maier T, Jenni S, Ban N: The multienzyme architecture of eukaryotic fatty acid synthases. Curr Opin Struct Biol 2008, 18: 714-725. 10.1016/j.sbi.2008.09.008
Schweizer E, Hofmann J: Microbial type I fatty acid synthases (FAS): major players in a network of cellular FAS systems. Microbiol Mol Biol Rev 2004, 68: 501-517. 10.1128/MMBR.68.3.501-517.2004
Harwood JL: Fatty acid metabolism. Annu Rev Plant Physiol Plant Mol Biol 1988, 39: 101-138. 10.1146/annurev.pp.39.060188.000533
Journet EP, Douce R: Enzymic capacities of purified cauliflower bud plastids for lipid synthesis and carbohydrate metabolism. Plant Physiol 1985, 79: 458-467. 10.1104/pp.79.2.458
Kachroo A, Kachroo P: Fatty acid-derived signals in plant defense. Annu Rev Phytopathol 2009, 47: 153-176. 10.1146/annurev-phyto-080508-081820
Liedvogel B, Kleinig H: Fatty acid synthesis in isolated chromoplasts and chromoplast homogenates. ACP stimulation, substrate utilization and cerulenin inhibition. In Biochemistry and Function of Plant Lipids. Edited by: Mazliak P, Benveniste P, Costes C, Douce R. Amsterdam: Elsevier; 1980:107-110.
Bloch K, Vance D: Control mechanisms in the synthesis of saturated fatty acids. Annu Rev Biochem 1977, 46: 263-298. 10.1146/annurev.bi.46.070177.001403
Fernandes ND, Kolattukudy PE: Cloning, sequencing and characterization of a fatty acid synthase-encoding gene from Mycobacterium tuberculosis var. bovis BCG. Gene 1996, 170: 95-99. 10.1016/0378-1119(95)00842-X
Kikuchi S, Rainwater DL, Kolattukudy PE: Purification and characterization of an unusually large fatty acid synthase from Mycobacterium tuberculosis var. bovis BCG. Arch Biochem Biophys 1992, 295: 318-326. 10.1016/0003-9861(92)90524-Z
Holder JW, Ulrich JC, DeBono AC, Godfrey PA, Desjardins CA, Zucker J, Zeng Q, Leach AL, Ghiviriga I, Dancel C, Abeel T, Gevers D, Kodira CD, Desany B, Affourtit JP, Birren BW, Sinskey AJ: Comparative and functional genomics of Rhodococcus opacus PD630 for biofuels development. PLoS Genet 2011, 7: e1002219. 10.1371/journal.pgen.1002219
Kolattukudy PE, Fernandes ND, Azad AK, Fitzmaurice AM, Sirakova TD: Biochemistry and molecular genetics of cell-wall lipid biosynthesis in mycobacteria. Mol Microbiol 1997, 24: 263-270. 10.1046/j.1365-2958.1997.3361705.x
Gattinger A, Schloter M, Munch JC: Phospholipid etherlipid and phospholipid fatty acid fingerprints in selected euryarchaeotal monocultures for taxonomic profiling. FEMS Microbiol Lett 2002, 213: 133-139.
Jones WJ, Holzer GU: The polar and neutral lipid composition of Methanosphaera stadtmanae . Syst Appl Microbiol 1991, 14: 130-134. 10.1016/S0723-2020(11)80290-0
Nishihara M, Nagahama S, Ohga M, Koga Y: Straight-chain fatty alcohols in the hyperthermophilic archaeon Pyrococcus furiosus . Extremophiles 2000, 4: 275-277. 10.1007/s007920070013
Harwood JL, Russell NJ: Lipids in Plants and Microbes. London: George Allen and Unwin; 1984:75-80.
Tornabene TG, Langworthy TA: Diphytanyl and dibiphytanyl glycerol ether lipids of methanogenic archaebacteria. Science 1979, 203: 51-53. 10.1126/science.758677
Pugh EL, Wassef MK, Kates M: Inhibition of fatty acid synthetase in Halobacterium cutirubrum and Escherichia coli by high salt concentrations. Can J Biochem 1971, 49: 953-958. 10.1139/o71-138
Choi-Rhee E, Cronan JE Jr: The biotin carboxylase-biotin carboxyl carrier protein complex of Escherichia coli acetyl-CoA carboxylase. J Biol Chem 2003, 278: 30806-30812. 10.1074/jbc.M302507200
Cronan JE Jr, Waldrop GL: Multi-subunit acetyl-CoA carboxylases. Prog Lipid Res 2002, 41: 407-435. 10.1016/S0163-7827(02)00007-3
Li SJ, Cronan JE Jr: The gene encoding the biotin carboxylase subunit of Escherichia coli acetyl-CoA carboxylase. J Biol Chem 1992, 267: 855-863.
Li SJ, Cronan JE Jr: Growth rate regulation of Escherichia coli acetyl coenzyme A carboxylase, which catalyzes the first committed step of lipid biosynthesis. J Bacteriol 1993, 175: 332-340.
James ES, Cronan JE Jr: Expression of two Escherichia coli acetyl-CoA carboxylase subunits is autoregulated. J Biol Chem 2004, 279: 2520-2527.
Abdel-Hamid AM, Cronan JE Jr: Coordinate expression of the acetyl coenzyme A carboxylase genes, accB and accC , is necessary for normal regulation of biotin synthesis in Escherichia coli . J Bacteriol 2007, 189: 369-376. 10.1128/JB.01373-06
Zhao H, Beckett D: Kinetic partitioning between alternative protein-protein interactions controls a transcriptional switch. J Mol Biol 2008, 380: 223-236. 10.1016/j.jmb.2008.04.068
Karow M, Fayet O, Georgopoulos C: The lethal phenotype caused by null mutations in the Escherichia coli htrB gene is suppressed by mutations in the accBC operon, encoding two subunits of acetyl coenzyme A carboxylase. J Bacteriol 1992, 174: 7407-7418.
Meades G Jr, Benson BK, Grove A, Waldrop GL: A tale of two functions: enzymatic activity and translational repression by carboxyltransferase. Nucleic Acids Res 2010, 38: 1217-1227. 10.1093/nar/gkp1079
Benson BK, Meades G Jr, Grove A, Waldrop GL: DNA inhibits catalysis by the carboxyltransferase subunit of acetyl-CoA carboxylase: implications for active site communication. Protein Sci 2008, 17: 34-42.
Bilder P, Lightle S, Bainbridge G, Ohren J, Finzel B, Sun F, Holley S, Al-Kassim L, Spessard C, Melnick M, Newcomer M, Waldrop GL: The structure of the carboxyltransferase component of acetyl-coA carboxylase reveals a zinc-binding motif unique to the bacterial enzyme. Biochemistry 2006, 45: 1712-1722. 10.1021/bi0520479
Li SJ, Cronan JE Jr: Putative zinc finger protein encoded by a conserved chloroplast gene is very likely a subunit of a biotin-dependent carboxylase. Plant Mol Biol 1992, 20: 759-761. 10.1007/BF00027147
Sasaki Y, Nagano Y, Morioka S, Ishikawa H, Matsuno R: A chloroplast gene encoding a protein with one zinc finger. Nucleic Acids Res 1989, 17: 6217-6227. 10.1093/nar/17.15.6217
Davis MS, Cronan JE Jr: Inhibition of Escherichia coli acetyl coenzyme A carboxylase by acyl-acyl carrier protein. J Bacteriol 2001, 183: 1499-1503. 10.1128/JB.183.4.1499-1503.2001
Davis MS, Solbiati J, Cronan JE Jr: Overproduction of acetyl-CoA carboxylase activity increases the rate of fatty acid biosynthesis in Escherichia coli . J Biol Chem 2000, 275: 28593-28598.
Zha W, Rubin-Pitel SB, Shao Z, Zhao H: Improving cellular malonyl-CoA level in Escherichia coli via metabolic engineering. Metab Eng 2009, 11: 192-198. 10.1016/j.ymben.2009.01.005
Joshi VC, Wakil SJ: Studies on the mechanism of fatty acid synthesis. XXVI. Purification and properties of malonyl-coenzyme A–acyl carrier protein transacylase of Escherichia coli. Arch Biochem Biophys 1971, 143: 493-505. 10.1016/0003-9861(71)90234-7
Verwoert II, Verhagen EF, van der Linden KH, Verbree EC, Nijkamp HJ, Stuitje AR: Molecular characterization of an Escherichia coli mutant with a temperature-sensitive malonyl coenzyme A-acyl carrier protein transacylase. FEBS Lett 1994, 348: 311-316. 10.1016/0014-5793(94)00630-X
Zhang Y, Cronan JE Jr: Transcriptional analysis of essential genes of the Escherichia coli fatty acid biosynthesis gene cluster by functional replacement with the analogous Salmonella typhimurium gene cluster. J Bacteriol 1998, 180: 3295-3303.
Magnuson K, Oh W, Larson TJ, Cronan JE Jr: Cloning and nucleotide sequence of the fabD gene encoding malonyl coenzyme A-acyl carrier protein transacylase of Escherichia coli . FEBS Lett 1992, 299: 262-266. 10.1016/0014-5793(92)80128-4
Garwin JL, Klages AL, Cronan JE Jr: Beta-ketoacyl-acyl carrier protein synthase II of Escherichia coli . Evidence for function in the thermal regulation of fatty acid synthesis. J Biol Chem 1980, 255: 3263-3265.
Zhang X, Agrawal A, San KY: Improving fatty acid production in Escherichia coli through the overexpression of malonyl CoA-acyl carrier protein transacylase. Biotechnol Prog 2012, 28: 60-65. 10.1002/btpr.716
Heath RJ, Rock CO: Inhibition of beta-ketoacyl-acyl carrier protein synthase III (FabH) by acyl-acyl carrier protein in Escherichia coli . J Biol Chem 1996, 271: 10996-11000. 10.1074/jbc.271.18.10996
Choi KH, Heath RJ, Rock CO: Beta-ketoacyl-acyl carrier protein synthase III (FabH) is a determining factor in branched-chain fatty acid biosynthesis. J Bacteriol 2000, 182: 365-370. 10.1128/JB.182.2.365-370.2000
Edwards P, Nelsen JS, Metz JG, Dehesh K: Cloning of the fabF gene in an expression vector and in vitro characterization of recombinant fabF and fabB encoded enzymes from Escherichia coli . FEBS Lett 1997, 402: 62-66. 10.1016/S0014-5793(96)01437-8
D‘Agnolo G, Rosenfeld IS, Vagelos PR: Multiple forms of beta-ketoacyl-acyl carrier protein synthetase in Escherichia coli. J Biol Chem 1975, 250: 5289-5294.
Feng Y, Cronan JE Jr: Escherichia coli unsaturated fatty acid synthesis: complex transcription of the fabA gene and in vivo identification of the essential reaction catalyzed by FabB. J Biol Chem 2009, 284: 29526-29535. 10.1074/jbc.M109.023440
Lai CY, Cronan JE Jr: Beta-ketoacyl-acyl carrier protein synthase III (FabH) is essential for bacterial fatty acid synthesis. J Biol Chem 2003, 278: 51494-51503. 10.1074/jbc.M308638200
Yao Z, Davis RM, Kishony R, Kahne D, Ruiz N: Regulation of cell size in response to nutrient availability by fatty acid biosynthesis in Escherichia coli . Proc Natl Acad Sci USA 2012, 109: E2561-E2568. 10.1073/pnas.1209742109
Tsay JT, Oh W, Larson TJ, Jackowski S, Rock CO: Isolation and characterization of the beta-ketoacyl-acyl carrier protein synthase III gene ( fabH ) from Escherichia coli K-12. J Biol Chem 1992, 267: 6807-6814.
Subrahmanyam S, Cronan JE Jr: Overproduction of a functional fatty acid biosynthetic enzyme blocks fatty acid synthesis in Escherichia coli . J Bacteriol 1998, 180: 4596-4602.
Cronan JE Jr, Birge CH, Vagelos PR: Evidence for two genes specifically involved in unsaturated fatty acid biosynthesis in Escherichia coli . J Bacteriol 1969, 100: 601-604.
Cao Y, Yang J, Xian M, Xu X, Liu W: Increasing unsaturated fatty acid contents in Escherichia coli by coexpression of three different genes. Appl Microbiol Biotechnol 2010, 87: 271-280. 10.1007/s00253-009-2377-x
Heath RJ, Rock CO: Regulation of fatty acid elongation and initiation by acyl-acyl carrier protein in Escherichia coli . J Biol Chem 1996, 271: 1833-1836. 10.1074/jbc.271.4.1833
Heath RJ, Rock CO: Regulation of malonyl-CoA metabolism by acyl-acyl carrier protein and beta-ketoacyl-acyl carrier protein synthases in Escherichia coli . J Biol Chem 1995, 270: 15531-15538. 10.1074/jbc.270.26.15531
Toomey RE, Wakil SJ: Studies on the mechanism of fatty acid synthesis. XV. Preparation and general properties of beta-ketoacyl acyl carrier protein reductase from Escherichia coli . Biochim Biophys Acta 1966, 116: 189-197. 10.1016/0005-2760(66)90001-4
Nomura CT, Taguchi K, Gan Z, Kuwabara K, Tanaka T, Takase K, Doi Y: Expression of 3-ketoacyl-acyl carrier protein reductase ( fabG ) genes enhances production of polyhydroxyalkanoate copolymer from glucose in recombinant Escherichia coli JM109. Appl Environ Microbiol 2005, 71: 4297-4306. 10.1128/AEM.71.8.4297-4306.2005
Nomura CT, Tanaka T, Eguen TE, Appah AS, Matsumoto K, Taguchi S, Ortiz CL, Doi Y: FabG mediates polyhydroxyalkanoate production from both related and nonrelated carbon sources in recombinant Escherichia coli LS5218. Biotechnol Prog 2008, 24: 342-351. 10.1021/bp070303y
Park SJ, Park JP, Lee SY: Metabolic engineering of Escherichia coli for the production of medium-chain-length polyhydroxyalkanoates rich in specific monomers. FEMS Microbiol Lett 2002, 214: 217-222. 10.1111/j.1574-6968.2002.tb11350.x
Lai CY, Cronan JE Jr: Isolation and characterization of beta-ketoacyl-acyl carrier protein reductase ( fabG ) mutants of Escherichia coli and Salmonella enterica serovar Typhimurium. J Bacteriol 2004, 186: 1869-1878. 10.1128/JB.186.6.1869-1878.2004
Jeon E, Lee S, Lee S, Han SO, Yoon YJ, Lee J: Improved production of long-chain fatty acid in Escherichia coli by an engineering elongation cycle during fatty acid synthesis (FAS) through genetic manipulation. J Microbiol Biotechnol 2012, 22: 990-999. 10.4014/jmb.1112.12057
Kass LR, Bloch K: On the enzymatic synthesis of unsaturated fatty acids in Escherichia coli . Proc Natl Acad Sci USA 1967, 58: 1168-1173. 10.1073/pnas.58.3.1168
Cronan JE Jr, Silbert DF, Wulff DL: Mapping of the fabA locus for unsaturated fatty acid biosynthesis in Escherichia coli . J Bacteriol 1972, 112: 206-211.
Silbert DF, Vagelos PR: Fatty acid mutant of E. coli lacking a beta-hydroxydecanoyl thioester dehydrase. Proc Natl Acad Sci USA 1967, 58: 1579-1586. 10.1073/pnas.58.4.1579
Silbert DF: Genetic modification of membrane lipid. Annu Rev Biochem 1975, 44: 315-339. 10.1146/annurev.bi.44.070175.001531
Clark DP, DeMendoza D, Polacco ML, Cronan JE Jr: Beta-hydroxydecanoyl thio ester dehydrase does not catalyze a rate-limiting step in Escherichia coli unsaturated fatty acid synthesis. Biochemistry 1983, 22: 5897-5902. 10.1021/bi00294a032
Heath RJ, Rock CO: Roles of the FabA and FabZ beta-hydroxyacyl-acyl carrier protein dehydratases in Escherichia coli fatty acid biosynthesis. J Biol Chem 1996, 271: 27795-27801. 10.1074/jbc.271.44.27795
Bloch K: The Enzymes. 3rd edition. Edited by: Boyer PD. New York: Academic Press; 1971:441-464.
Mohan S, Kelly TM, Eveland SS, Raetz CR, Anderson MS: An Escherichia coli gene (FabZ) encoding (3 R )-hydroxymyristoyl acyl carrier protein dehydrase. Relation to fabA and suppression of mutations in lipid A biosynthesis. J Biol Chem 1994, 269: 32896-32903.
Bergler H, Fuchsbichler S, Högenauer G, Turnowsky F: The enoyl-[acyl-carrier-protein] reductase (FabI) of Escherichia coli , which catalyzes a key regulatory step in fatty acid biosynthesis, accepts NADH and NADPH as cofactors and is inhibited by palmitoyl-CoA. Eur J Biochem 1996, 242: 689-694. 10.1111/j.1432-1033.1996.0689r.x
Nakashima N, Tamura T, Good L: Paired termini stabilize antisense RNAs and enhance conditional gene silencing in Escherichia coli . Nucleic Acids Res 2006, 34: e138. 10.1093/nar/gkl697
Bergler H, Wallner P, Ebeling A, Leitinger B, Fuchsbichler S, Aschauer H, Kollenz G, Högenauer G, Turnowsky F: Protein EnvM is the NADH-dependent enoyl-ACP reductase (FabI) of Escherichia coli . J Biol Chem 1994, 269: 5493-5496.
Heath RJ, Rock CO: Enoyl-acyl carrier protein reductase ( fabI ) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli . J Biol Chem 1995, 270: 26538-26542. 10.1074/jbc.270.44.26538
Xu HH, Real L, Bailey MW: An array of Escherichia coli clones over-expressing essential proteins: a new strategy of identifying cellular targets of potent antibacterial compounds. Biochem Biophys Res Commun 2006, 349: 1250-1257. 10.1016/j.bbrc.2006.08.166
Thomas J, Rigden DJ, Cronan JE Jr: Acyl carrier protein phosphodiesterase (AcpH) of Escherichia coli is a non-canonical member of the HD phosphatase/phosphodiesterase family. Biochemistry 2007, 46: 129-136. 10.1021/bi061789e
Thomas J, Cronan JE Jr: The enigmatic acyl carrier protein phosphodiesterase of Escherichia coli : genetic and enzymological characterization. J Biol Chem 2005, 280: 34675-34683. 10.1074/jbc.M505736200
Chan DI, Vogel HJ: Current understanding of fatty acid biosynthesis and the acyl carrier protein. Biochem J 2010, 430: 1-19. 10.1042/BJ20100462
Goh S, Boberek JM, Nakashima N, Stach J, Good L: Concurrent growth rate and transcript analyses reveal essential gene stringency in Escherichia coli . PLoS One 2009, 4: e6061. 10.1371/journal.pone.0006061
Keating DH, Carey MR, Cronan JE Jr: The unmodified ( apo ) form of Escherichia coli acyl carrier protein is a potent inhibitor of cell growth. J Biol Chem 1995, 270: 22229-22235. 10.1074/jbc.270.38.22229
Rock CO, Goelz SE, Cronan JE Jr: Phospholipid synthesis in Escherichia coli . Characteristics of fatty acid transfer from acyl-acyl carrier protein to sn-glycerol 3-phosphate. J Biol Chem 1981, 256: 736-742.
Liu T, Vora H, Khosla C: Quantitative analysis and engineering of fatty acid biosynthesis in E. coli . Metab Eng 2010, 12: 378-386. 10.1016/j.ymben.2010.02.003
Jha JK, Sinha S, Maiti MK, Basu A, Mukhopadhyay UK, Sen SK: Functional expression of an acyl carrier protein (ACP) from Azospirillum brasilense alters fatty acid profiles in Escherichia coli and Brassica juncea . Plant Physiol Biochem 2007, 45: 490-500. 10.1016/j.plaphy.2007.03.001
Battesti A, Bouveret E: Acyl carrier protein/SpoT interaction, the switch linking SpoT-dependent stress response to fatty acid metabolism. Mol Microbiol 2006, 62: 1048-1063. 10.1111/j.1365-2958.2006.05442.x
Lu YJ, Zhang YM, Grimes KD, Qi J, Lee RE, Rock CO: Acyl-phosphates initiate membrane phospholipid synthesis in Gram-positive pathogens. Mol Cell 2006, 23: 765-772. 10.1016/j.molcel.2006.06.030
Yao J, Rock CO: Phosphatidic acid synthesis in bacteria. Biochim Biophys Acta 2013, 1831: 495-502. 10.1016/j.bbalip.2012.08.018
Zhang YM, Rock CO: Membrane lipid homeostasis in bacteria. Nat Rev Microbiol 2008, 6: 222-233. 10.1038/nrmicro1839
Ray TK, Cronan JE Jr: Acylation of sn-glycerol 3-phosphate in Escherichia coli . Study of reaction with native palmitoyl-acyl carrier protein. J Biol Chem 1975, 250: 8422-8427.
Heath RJ, Jackowski S, Rock CO: Guanosine tetraphosphate inhibition of fatty acid and phospholipid synthesis in Escherichia coli is relieved by overexpression of glycerol-3-phosphate acyltransferase ( plsB ). J Biol Chem 1994, 269: 26584-26590.
Wahl A, My L, Dumoulin R, Sturgis JN, Bouveret E: Antagonistic regulation of dgkA and plsB genes of phospholipid synthesis by multiple stress responses in Escherichia coli . Mol Microbiol 2011, 80: 1260-1275. 10.1111/j.1365-2958.2011.07641.x
Ogasawara H, Shinohara S, Yamamoto K, Ishihama A: Novel regulation targets of the metal-response BasS-BasR two-component system of Escherichia coli . Microbiology 2012, 158: 1482-1492. 10.1099/mic.0.057745-0
Durfee T, Hansen AM, Zhi H, Blattner FR, Jin DJ: Transcription profiling of the stringent response in Escherichia coli . J Bacteriol 2008, 190: 1084-1096. 10.1128/JB.01092-07
Yoshimura M, Oshima T, Ogasawara N: Involvement of the YneS/YgiH and PlsX proteins in phospholipid biosynthesis in both Bacillus subtilis and Escherichia coli . BMC Microbiol 2007, 7: 69. 10.1186/1471-2180-7-69
Larson TJ, Ludtke DN, Bell RM: sn -Glycerol-3-phosphate auxotrophy of plsB strains of Escherichia coli : evidence that a second mutation, plsX , is required. J Bacteriol 1984, 160: 711-717.
Traxler MF, Summers SM, Nguyen HT, Zacharia VM, Hightower GA, Smith JT, Conway T: The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli . Mol Microbiol 2008, 68: 1128-1148. 10.1111/j.1365-2958.2008.06229.x
Lepore BW, Indic M, Pham H, Hearn EM, Patel DR, van den Berg B: Ligand-gated diffusion across the bacterial outer membrane. Proc Natl Acad Sci U S A 2011, 108: 10121-10126. 10.1073/pnas.1018532108
Campbell JW, Cronan JE Jr: The enigmatic Escherichia coli fadE gene is yafH . J Bacteriol 2002, 184: 3759-3764. 10.1128/JB.184.13.3759-3764.2002
Weimar JD, DiRusso CC, Delio R, Black PN: Functional role of fatty acyl-coenzyme A synthetase in the transmembrane movement and activation of exogenous long-chain fatty acids. Amino acid residues within the ATP/AMP signature motif of Escherichia coli FadD are required for enzyme activity and fatty acid transport. J Biol Chem 2002, 277: 29369-29376. 10.1074/jbc.M107022200
Pech-Canul À, Nogales J, Miranda-Molina A, Àlvarez L, Geiger O, Soto MJ, López-Lara IM: FadD is required for utilization of endogenous fatty acids released from membrane lipids. J Bacteriol 2011, 193: 6295-6304. 10.1128/JB.05450-11
Zhang H, Wang P, Qi Q: Molecular effect of FadD on the regulation and metabolism of fatty acid in Escherichia coli . FEMS Microbiol Lett 2006, 259: 249-253. 10.1111/j.1574-6968.2006.00277.x
Black PN, Zhang Q, Weimar JD, DiRusso CC: Mutational analysis of a fatty acyl-coenzyme A synthetase signature motif identifies seven amino acid residues that modulate fatty acid substrate specificity. J Biol Chem 1997, 272: 4896-4903. 10.1074/jbc.272.8.4896
Yang SY, Li JM, He XY, Cosloy SD, Schulz H: Evidence that the fadB gene of the fadAB operon of Escherichia coli encodes 3-hydroxyacyl-coenzyme A (CoA) epimerase, delta 3-cis-delta 2-trans-enoyl-CoA isomerase, and enoyl-CoA hydratase in addition to 3-hydroxyacyl-CoA dehydrogenase. J Bacteriol 1988, 170: 2543-2548.
Feigenbaum J, Schulz H: Thiolases of Escherichia coli : purification and chain length specificities. J Bacteriol 1975, 122: 407-411.
Duncombe GR, Frerman FE: Molecular and catalytic properties of the acetoacetyl-coenzyme A thiolase of Escherichia coli . Arch Biochem Biophys 1976, 176: 159-170. 10.1016/0003-9861(76)90152-1
Tu X, Hubbard PA, Kim JJ, Schulz H: Two distinct proton donors at the active site of Escherichia coli 2,4-dienoyl-CoA reductase are responsible for the formation of different products. Biochemistry 2008, 47: 1167-1175. 10.1021/bi701235t
Nie L, Ren Y, Schulz H: Identification and characterization of Escherichia coli thioesterase III that functions in fatty acid beta-oxidation. Biochemistry 2008, 47: 7744-7751. 10.1021/bi800595f
Feng Y, Cronan JE Jr: A new member of the Escherichia coli fad regulon: transcriptional regulation of fadM ( ybaW ). J Bacteriol 2009, 191: 6320-6328. 10.1128/JB.00835-09
Campbell JW, Morgan-Kiss RM, Cronan JE Jr: A new Escherichia coli metabolic competency: growth on fatty acids by a novel anaerobic beta-oxidation pathway. Mol Microbiol 2003, 47: 793-805. 10.1046/j.1365-2958.2003.03341.x
Morgan-Kiss RM, Cronan JE Jr: The Escherichia coli fadK ( ydiD ) gene encodes an anerobically regulated short chain acyl-CoA synthetase. J Biol Chem 2004, 279: 37324-37333. 10.1074/jbc.M405233200
Snell KD, Feng F, Zhong L, Martin D, Madison LL: YfcX enables medium-chain-length poly (3-hydroxyalkanoate) formation from fatty acids in recombinant Escherichia coli fadB strains. J Bacteriol 2002, 184: 5696-5705. 10.1128/JB.184.20.5696-5705.2002
Cho BK, Knight EM, Palsson BO: Transcriptional regulation of the fad regulon genes of Escherichia coli by ArcA. Microbiology 2006, 152: 2207-2219. 10.1099/mic.0.28912-0
Fujita Y, Matsuoka H, Hirooka K: Regulation of fatty acid metabolism in bacteria. Mol Microbiol 2007, 66: 829-839. 10.1111/j.1365-2958.2007.05947.x
Kunau W-H, Dommes V, Schulz H: β -oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: a century of continued progress. Prog Lipid Res 1996, 34: 267-342.
Henry MF, Cronan JE Jr: Escherichia coli transcription factor that both activates fatty acid synthesis and represses fatty acid degradation. J Mol Biol 1991, 222: 843-849. 10.1016/0022-2836(91)90574-P
Henry MF, Cronan JE Jr: A new mechanism of transcriptional regulation: release of an activator triggered by small molecule binding. Cell 1992, 70: 671-679. 10.1016/0092-8674(92)90435-F
Feng Y, Cronan JE Jr: Crosstalk of Escherichia coli FadR with global regulators in expression of fatty acid transport genes. PLoS One 2012, 7: e46275. 10.1371/journal.pone.0046275
Dong T, Schellhorn HE: Global effect of RpoS on gene expression in pathogenic Escherichia coli O157:H7 strain EDL933. BMC Genomics 2009, 10: 349. 10.1186/1471-2164-10-349
Liu H, Yu C, Feng D, Cheng T, Meng X, Liu W, Zou H, Xian M: Production of extracellular fatty acid using engineered Escherichia coli . Microb Cell Fact 2012, 11: 41. 10.1186/1475-2859-11-41
Lu X, Vora H, Khosla C: Overproduction of free fatty acids in E. coli : implications for biodiesel production. Metab Eng 2008, 10: 333-339. 10.1016/j.ymben.2008.08.006
Gui L, Sunnarborg A, LaPorte DC: Regulated expression of a repressor protein: FadR activates iclR . J Bacteriol 1996, 178: 4704-4709.
Kumari S, Beatty CM, Browning DF, Busby SJ, Simel EJ, Hovel-Miner G, Wolfe AJ: Regulation of acetyl coenzyme A synthetase in Escherichia coli . J Bacteriol 2000, 182: 4173-4179. 10.1128/JB.182.15.4173-4179.2000
DiRusso CC, Heimert TL, Metzger AK: Characterization of FadR, a global transcriptional regulator of fatty acid metabolism in Escherichia coli . Interaction with the fadB promoter is prevented by long chain fatty acyl coenzyme A. J Biol Chem 1992, 267: 8685-8691.
Pauli G, Ehring R, Overath P: Fatty acid degradation in Escherichia coli : requirement of cyclic adenosine monophosphate and cyclic adenosine monophosphate receptor protein for enzyme synthesis. J Bacteriol 1974, 117: 1178-1183.
Campbell JW, Cronan JE Jr: Bacterial fatty acid biosynthesis: targets for antibacterial drug discovery. Annu Rev Microbiol 2001, 55: 305-332. 10.1146/annurev.micro.55.1.305
My L, Rekoske B, Lemke JJ, Viala JP, Gourse RL, Bouveret E: Transcription of the Escherichia coli fatty acid synthesis operon fabHDG is directly activated by FadR and inhibited by ppGpp. J Bacteriol 2013, 195: 3784-3795. 10.1128/JB.00384-13
Overath P, Pauli G, Schairer HU: Fatty acid degradation in Escherichia coli . An inducible acyl-CoA synthetase, the mapping of old-mutations, and the isolation of regulatory mutants. Eur J Biochem 1969, 7: 559-574.
Nunn WD, Giffin K, Clark D, Cronan JE Jr: Role for fadR in unsaturated fatty acid biosynthesis in Escherichia coli . J Bacteriol 1983, 154: 554-560.
Zhang F, Ouellet M, Batth TS, Adams PD, Petzold CJ, Mukhopadhyay A, Keasling JD: Enhancing fatty acid production by the expression of the regulatory transcription factor FadR. Metab Eng 2012, 14: 653-660. 10.1016/j.ymben.2012.08.009
Farewell A, Diez AA, DiRusso CC, Nystrom T: Role of the Escherichia coli FadR regulator in stasis survival and growth phase-dependent expression of the uspA , fad , and fab genes. J Bacteriol 1996, 178: 6443-6450.
McCue L, Thompson W, Carmack C, Ryan MP, Liu JS, Derbyshire V, Lawrence CE: Phylogenetic footprinting of transcription factor binding sites in proteobacterial genomes. Nucleic Acids Res 2001, 29: 774-782. 10.1093/nar/29.3.774
Zhang YM, Marrakchi H, Rock CO: The FabR (YijC) transcription factor regulates unsaturated fatty acid biosynthesis in Escherichia coli . J Biol Chem 2002, 277: 15558-15565. 10.1074/jbc.M201399200
Zhu K, Zhang YM, Rock CO: Transcriptional regulation of membrane lipid homeostasis in Escherichia coli . J Biol Chem 2009, 284: 34880-34888. 10.1074/jbc.M109.068239
Feng Y, Cronan JE Jr: Complex binding of the FabR repressor of bacterial unsaturated fatty acid biosynthesis to its cognate promoters. Mol Microbiol 2011, 80: 195-218. 10.1111/j.1365-2958.2011.07564.x
Tanaka Y, Tsujimura A, Fujita N, Isono S, Isono K: Cloning and analysis of an Escherichia coli operon containing the rpmF gene for ribosomal protein L32 and the gene for a 30-kilodalton protein. J Bacteriol 1989, 171: 5707-5712.
Podkovyrov S, Larson TJ: Lipid biosynthetic genes and a ribosomal protein gene are cotranscribed. FEBS Lett 1995, 368: 429-431. 10.1016/0014-5793(95)00702-B
Podkovyrov SM, Larson TJ: Identification of promoter and stringent regulation of transcription of the fabH , fabD and fabG genes encoding fatty acid biosynthetic enzymes of Escherichia coli . Nucleic Acids Res 1996, 24: 1747-1752. 10.1093/nar/24.9.1747
Bremer H, Dennis PP: Modulation of chemical composition and other parameters of the cell by growth rate. In Escherichia coli and Salmonella: Cellular and Molecular Biology. 1st edition. Edited by: Neidhardt FC. Washington DC: ASM Press; 1996:1553-1569.
Schneider DA, Gourse RL: Relationship between growth rate and ATP concentration in Escherichia coli : a bioassay for available cellular ATP. J Biol Chem 2004, 279: 8262-8268.
Paul BJ, Barker MM, Ross W, Schneider DA, Webb C, Foster JW, Gourse RL: DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell 2004, 118: 311-322. 10.1016/j.cell.2004.07.009
Paul BJ, Berkmen MB, Gourse RL: DksA potentiates direct activation of amino acid promoters by ppGpp. Proc Natl Acad Sci USA 2005, 102: 7823-7828. 10.1073/pnas.0501170102
Srivatsan A, Wang JD: Control of bacterial transcription, translation and replication by (p) ppGpp. Curr Opin Microbiol 2008, 11: 100-105. 10.1016/j.mib.2008.02.001
Traxler MF, Zacharia VM, Marquardt S, Summers SM, Nguyen HT, Stark SE, Conway T: Discretely calibrated regulatory loops controlled by ppGpp partition gene induction across the ‘feast to famine’ gradient in Escherichia coli . Mol Microbiol 2011, 79: 830-845. 10.1111/j.1365-2958.2010.07498.x
Stent GS, Brenner S: A genetic locus for the regulation of ribonucleic acid synthesis. Proc Natl Acad Sci USA 1961, 47: 2005-2014. 10.1073/pnas.47.12.2005
Eichel J, Chang YY, Riesenberg D, Cronan JE Jr: Effect of ppGpp on Escherichia coli cyclopropane fatty acid synthesis is mediated through the RpoS sigma factor (sigmaS). J Bacteriol 1999, 181: 572-576.
Sammartano LJ, Tuveson RW, Davenport R: Control of sensitivity to inactivation by H2O2 and broad-spectrum near-UV radiation by the Escherichia coli katF locus. J Bacteriol 1986, 168: 13-21.
Small P, Blankenhorn D, Welty D, Zinser E, Slonczewski JL: Acid and base resistance in Escherichia coli and Shigella flexneri : role of rpoS and growth pH. J Bacteriol 1994, 176: 1729-1737.
Hengge-Aronis R, Klein W, Lange R, Rimmele M, Boos W: Trehalose synthesis genes are controlled by the putative sigma factor encoded by rpoS and are involved in stationary-phase thermotolerance in Escherichia coli . J Bacteriol 1991, 173: 7918-7924.
Lange R, Hengge-Aronis R: Growth phase-regulated expression of bolA and morphology of stationary-phase Escherichia coli cells are controlled by the novel sigma factor sigma S. J Bacteriol 1991, 173: 4474-4481.
Battesti A, Majdalani N, Gottesman S: The RpoS-mediated general stress response in Escherichia coli . Annu Rev Microbiol 2011, 65: 189-213. 10.1146/annurev-micro-090110-102946
Wendrich TM, Blaha G, Wilson DN, Marahiel MA, Nierhaus KH: Dissection of the mechanism for the stringent factor RelA. Mol Cell 2002, 10: 779-788. 10.1016/S1097-2765(02)00656-1
Xiao H, Kalman M, Ikehara K, Zemel S, Glaser G, Cashel M: Residual guanosine 3’,5’-bispyrophosphate synthetic activity of relA null mutants can be eliminated by spoT null mutations. J Biol Chem 1991, 266: 5980-5990.
Lazzarini RA, Cashel M, Gallant J: On the regulation of guanosine tetraphosphate levels in stringent and relaxed strains of Escherichia coli . J Biol Chem 1971, 246: 4381-4385.
Spira B, Silberstein N, Yagil E: Guanosine 3’,5’-bispyrophosphate (ppGpp) synthesis in cells of Escherichia coli starved for Pi. J Bacteriol 1995, 177: 4053-4058.
Vinella D, Albrecht C, Cashel M, D‘Ari R: Iron limitation induces SpoT-dependent accumulation of ppGpp in Escherichia coli . Mol Microbiol 2005, 56: 958-970. 10.1111/j.1365-2958.2005.04601.x
Seyfzadeh M, Keener J, Nomura M: spoT -dependent accumulation of guanosine tetraphosphate in response to fatty acid starvation in Escherichia coli . Proc Natl Acad Sci USA 1993, 90: 11004-11008. 10.1073/pnas.90.23.11004
Neidhardt FC: Properties of a bacterial mutant lacking amino acid control of RNA synthesis. Biochim Biophys Acta 1963, 68: 365-379.
Ramagopal S, Davis BD: Localization of the stringent protein of Escherichia coli on the 50S ribosomal subunit. Proc Natl Acad Sci USA 1974, 71: 820-824. 10.1073/pnas.71.3.820
Murray KD, Bremer H: Control of spoT-dependent ppGpp synthesis and degradation in Escherichia coli . J Mol Biol 1996, 259: 41-57. 10.1006/jmbi.1996.0300
Stein JP Jr, Bloch KE: Inhibition of E. coli beta-hydroxydecanoyl thioester dehydrase by ppGpp. Biochem Biophys Res Commun 1976, 73: 881-884. 10.1016/0006-291X(76)90204-7
Hiraoka S, Matsuzaki H, Shibuya I: Active increase in cardiolipin synthesis in the stationary growth phase and its physiological significance in Escherichia coli . FEBS Lett 1993, 336: 221-224. 10.1016/0014-5793(93)80807-7
Taguchi M, Izui K, Katsuki H: Augmentation of cyclopropane fatty acid synthesis under stringent control in Escherichia coli . J Biochem 1980, 88: 1879-1882.
Dennis PP, Nomura M: Stringent control of ribosomal protein gene expression in Escherichia coli . Proc Natl Acad Sci USA 1974, 71: 3819-3823. 10.1073/pnas.71.10.3819
Lazzarini RA, Dahlberg AE: The control of ribonucleic acid synthesis during amino acid deprivation in Escherichia coli . J Biol Chem 1971, 246: 420-429.
Milon P, Tischenko E, Tomsic J, Caserta E, Folkers G, La Teana A, Rodnina MV, Pon CL, Boelens R, Gualerzi CO: The nucleotide-binding site of bacterial translation initiation factor 2 (IF2) as a metabolic sensor. Proc Natl Acad Sci USA 2006, 103: 13962-13967. 10.1073/pnas.0606384103
Edwards AN, Patterson-Fortin LM, Vakulskas CA, Mercante JW, Potrykus K, Vinella D, Camacho MI, Fields JA, Thompson SA, Georgellis D, Cashel M, Babitzke P, Romeo T: Circuitry linking the Csr and stringent response global regulatory systems. Mol Microbiol 2011, 80: 1561-1580. 10.1111/j.1365-2958.2011.07663.x
Cho H, Cronan JE Jr: Defective export of a periplasmic enzyme disrupts regulation of fatty acid synthesis. J Biol Chem 1995, 270: 4216-4219. 10.1074/jbc.270.9.4216
Ruffing AM, Jones HD: Physiological effects of free fatty acid production in genetically engineered Synechococcus elongatus PCC 7942. Biotechnol Bioeng 2012, 109: 2190-2199. 10.1002/bit.24509
Tang X, Feng H, Chen WN: Metabolic engineering for enhanced fatty acids synthesis in Saccharomyces cerevisiae . Metab Eng 2013, 16: 95-102.
Lennen RM, Pfleger BF: Modulating membrane composition alters free fatty acid tolerance in Escherichia coli . PLoS One 2013, 8: e54031. 10.1371/journal.pone.0054031
Zhang X, Li M, Agrawal A, San KY: Efficient free fatty acid production in Escherichia coli using plant acyl-ACP thioesterases. Metab Eng 2011, 13: 713-722. 10.1016/j.ymben.2011.09.007
Zheng Y, Li L, Liu Q, Qin W, Yang J, Cao Y, Jiang X, Zhao G, Xian M: Boosting the free fatty acid synthesis of Escherichia coli by expression of a cytosolic Acinetobacter baylyi thioesterase. Biotechnol Biofuels 2012, 5: 76. 10.1186/1754-6834-5-76
Yu X, Liu T, Zhu F, Khosla C: In vitro reconstitution and steady-state analysis of the fatty acid synthase from Escherichia coli . Proc Natl Acad Sci USA 2011, 108: 18643-18648. 10.1073/pnas.1110852108
Desbois AP, Smith VJ: Antibacterial free fatty acids: activities, mechanisms of action and biotechnological potential. Appl Microbiol Biotechnol 2010, 85: 1629-1642. 10.1007/s00253-009-2355-3
Lennen RM, Kruziki MA, Kumar K, Zinkel RA, Burnum KE, Lipton MS, Hoover SW, Ranatunga DR, Wittkopp TM, Marner WD II, Pfleger BF: Membrane stresses induced by overproduction of free fatty acids in Escherichia coli . Appl Environ Microbiol 2011, 77: 8114-8128. 10.1128/AEM.05421-11
Lennen RM, Politz MG, Kruziki MA, Pfleger BF: Identification of transport proteins involved in free fatty acid efflux in Escherichia coli . J Bacteriol 2013, 195: 135-144. 10.1128/JB.01477-12
Oh HY, Lee JO, Kim OB: Increase of organic solvent tolerance of Escherichia coli by the deletion of two regulator genes, fadR and marR . Appl Microbiol Biotechnol 2012, 96: 1619-1627. 10.1007/s00253-012-4463-8
Dellomonaco C, Clomburg JM, Miller EN, Gonzalez R: Engineered reversal of the beta-oxidation cycle for the synthesis of fuels and chemicals. Nature 2011, 476: 355-359. 10.1038/nature10333
Lennen RM, Braden DJ, West RA, Dumesic JA, Pfleger BF: A process for microbial hydrocarbon synthesis: overproduction of fatty acids in Escherichia coli and catalytic conversion to alkanes. Biotechnol Bioeng 2010, 106: 193-202. 10.1002/bit.22660
Youngquist JT, Rose JP, Pfleger BF: Free fatty acid production in Escherichia coli under phosphate-limited conditions. Appl Microbiol Biotechnol 2013, 97: 5149-5159. 10.1007/s00253-013-4911-0
Ranganathan S, Tee TW, Chowdhury A, Zomorrodi AR, Yoon JM, Fu Y, Shanks JV, Maranas CD: An integrated computational and experimental study for overproducing fatty acids in Escherichia coli . Metab Eng 2012, 14: 687-704. 10.1016/j.ymben.2012.08.008
Li M, Zhang X, Agrawal A, San KY: Effect of acetate formation pathway and long chain fatty acid CoA-ligase on the free fatty acid production in E. coli expressing acy-ACP thioesterase from Ricinus communis . Metab Eng 2012, 14: 380-387. 10.1016/j.ymben.2012.03.007
Bennet BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD: Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli . Nature Chem Biol 2009, 5: 593-599. 10.1038/nchembio.186
Heuser F, Schroer K, Lutz S, Bringer-Meyer S, Sahm H: Enhancement of the NAD (P) (H) pool in Escherichia coli for biotransformation. Eng Life Sci 2007, 7: 343-353. 10.1002/elsc.200720203
Sauer U, Canonaco F, Heri S, Perrenoud A, Fischer E: The soluble and membrane-bound transhydrogenases UdhA and PntAB have divergent functions in NADPH metabolism of Escherichia coli . J Biol Chem 2004, 279: 6613-6619.
Yuan L, Voelker TA, Hawkins DJ: Modification of the substrate specificity of an acyl-acyl carrier protein thioesterase by protein engineering. Proc Natl Acad Sci USA 1995, 92: 10639-10643. 10.1073/pnas.92.23.10639
Babitzke P, Romeo T: CsrB sRNA family: sequestration of RNA-binding regulatory proteins. Curr Opin Microbiol 2007, 10: 156-163. 10.1016/j.mib.2007.03.007
Liu MY, Romeo T: The global regulator CsrA of Escherichia coli is a specific mRNA-binding protein. J Bacteriol 1997, 179: 4639-4642.
Liu MY, Gui G, Wei B, Preston JF 3rd, Oakford L, Yuksel U, Giedroc DP, Romeo T: The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli . J Biol Chem 1997, 272: 17502-17510. 10.1074/jbc.272.28.17502
McKee AE, Rutherford BJ, Chivian DC, Baidoo EK, Juminaga D, Kuo D, Benke PI, Dietrich JA, Ma SM, Arkin AP, Petzold CJ, Adams PD, Keasling JD, Chhabra SR: Manipulation of the carbon storage regulator system for metabolite remodeling and biofuel production in Escherichia coli . Microb Cell Fact 2012, 11: 79. 10.1186/1475-2859-11-79
Dellomonaco C, Rivera C, Campbell P, Gonzalez R: Engineered respiro-fermentative metabolism for the production of biofuels and biochemicals from fatty acid-rich feedstocks. Appl Environ Microbiol 2010, 76: 5067-5078. 10.1128/AEM.00046-10
Eppler T, Boos W: Glycerol-3-phosphate-mediated repression of malT in Escherichia coli does not require metabolism, depends on enzyme IIAGlc and is mediated by cAMP levels. Mol Microbiol 1999, 33: 1221-1231.
Jenkins LS, Nunn WD: Genetic and molecular characterization of the genes involved in short-chain fatty acid degradation in Escherichia coli : the ato system. J Bacteriol 1987, 169: 42-52.
Spratt SK, Ginsburgh CL, Nunn WD: Isolation and genetic characterization of Escherichia coli mutants defective in propionate metabolism. J Bacteriol 1981, 146: 1166-1169.
Lennen RM, Pfleger BF: Engineering Escherichia coli to synthesize free fatty acids. Trends Biotechnol 2012, 30: 659-667. 10.1016/j.tibtech.2012.09.006
Stöveken T, Kalscheuer R, Malkus U, Reichelt R, Steinbüchel A: The wax ester synthase/acyl coenzyme A: diacylglycerol acyltransferase from Acinetobacter sp. strain ADP1: characterization of a novel type of acyltransferase. J Bacteriol 2005, 187: 1369-1376. 10.1128/JB.187.4.1369-1376.2005
Stöveken T, Steinbüchel A: Bacterial acyltransferases as an alternative for lipase-catalyzed acylation for the production of oleochemicals and fuels. Angew Chem Int Ed Engl 2008, 47: 3688-3694. 10.1002/anie.200705265
Shi S, Valle-Rodríguez JO, Khoomrung S, Siewers V, Nielsen J: Functional expression and characterization of five wax ester synthases in Saccharomyces cerevisiae and their utility for biodiesel production. Biotechnol Biofuels 2012, 5: 7. 10.1186/PREACCEPT-1932279820621895
Elbahloul Y, Steinbüchel A: Pilot-scale production of fatty acid ethyl esters by an engineered Escherichia coli strain harboring the p (Microdiesel) plasmid. Appl Environ Microbiol 2010, 76: 4560-4565. 10.1128/AEM.00515-10
Duan Y, Zhu Z, Cai K, Tan X, Lu X: De novo biosynthesis of biodiesel by Escherichia coli in optimized fed-batch cultivation. PLoS One 2011, 6: e20265. 10.1371/journal.pone.0020265
Somerville C: Biofuels. Curr Biol 2007, 17: R115-R119. 10.1016/j.cub.2007.01.010
Park JI, Steen EJ, Burd H, Evans SS, Redding-Johnson AM, Batth T, Benke PI, D‘haeseleer P, Sun N, Sale KL, Keasling JD, Lee TS, Petzold CJ, Mukhopadhyay A, Singer SW, Simmons BA, Gladden JM: A thermophilic ionic liquid-tolerant cellulase cocktail for the production of cellulosic biofuels. PLoS One 2012, 7: e37010. 10.1371/journal.pone.0037010
Kalscheuer R, Stöveken T, Luftmann H, Malkus U, Reichelt R, Steinbüchel A: Neutral lipid biosynthesis in engineered Escherichia coli : jojoba oil-like wax esters and fatty acid butyl esters. Appl Environ Microbiol 2006, 72: 1373-1379. 10.1128/AEM.72.2.1373-1379.2006
Kaiser BK, Carleton M, Hickman JW, Miller C, Lawson D, Budde M, Warrener P, Paredes A, Mullapudi S, Navarro P, Cross F, Roberts JM: Fatty aldehydes in cyanobacteria are a metabolically flexible precursor for a diversity of biofuel products. PLoS One 2013, 8: e58307. 10.1371/journal.pone.0058307
Yu KO, Jung J, Kim SW, Park CH, Han SO: Synthesis of FAEEs from glycerol in engineered Saccharomyces cerevisiae using endogenously produced ethanol by heterologous expression of an unspecific bacterial acyltransferase. Biotechnol Bioeng 2012, 109: 110-115. 10.1002/bit.23311
Saerens SM, Verstrepen KJ, Van Laere SD, Voet AR, Van Dijck P, Delvaux FR, Thevelein JM: The Saccharomyces cerevisiae EHT1 and EEB1 genes encode novel enzymes with medium-chain fatty acid ethyl ester synthesis and hydrolysis capacity. J Biol Chem 2006, 281: 4446-4456. 10.1074/jbc.M512028200
Kalscheuer R, Stöveken T, Malkus U, Reichelt R, Golyshin PN, Sabirova JS, Ferrer M, Timmis KN, Steinbüchel A: Analysis of storage lipid accumulation in Alcanivorax borkumensis : evidence for alternative triacylglycerol biosynthesis routes in bacteria. J Bacteriol 2007, 189: 918-928. 10.1128/JB.01292-06
Alvarez HM, Mayer F, Fabritius D, Steinbüchel A: Formation of intracytoplasmic lipid inclusions by Rhodococcus opacus strain PD630. Arch Microbiol 1996, 165: 377-386. 10.1007/s002030050341
Alvarez AF, Alvarez HM, Kalscheuer R, Wältermann M, Steinbüchel A: Cloning and characterization of a gene involved in triacylglycerol biosynthesis and identification of additional homologous genes in the oleaginous bacterium Rhodococcus opacus PD630. Microbiology 2008, 154: 2327-2335. 10.1099/mic.0.2008/016568-0
Hernández MA, Arabolaza A, Rodriguez E, Gramajo H, Alvarez HM: The atf2 gene is involved in triacylglycerol biosynthesis and accumulation in the oleaginous Rhodococcus opacus PD630. Appl Microbiol Biotechnol 2013, 97: 2119-2130. 10.1007/s00253-012-4360-1
MacEachran DP, Prophete ME, Sinskey AJ: The Rhodococcus opacus PD630 heparin-binding hemagglutinin homolog TadA mediates lipid body formation. Appl Environ Microbiol 2010, 76: 7217-7225. 10.1128/AEM.00985-10
Arabolaza A, Rodriguez E, Altabe S, Alvarez H, Gramajo H: Multiple pathways for triacylglycerol biosynthesis in Streptomyces coelicolor . Appl Environ Microbiol 2008, 74: 2573-2582. 10.1128/AEM.02638-07
Comba S, Menendez-Bravo S, Arabolaza A, Gramajo H: Identification and physiological characterization of phosphatidic acid phosphatase enzymes involved in triacylglycerol biosynthesis in Streptomyces coelicolor . Microb Cell Fact 2013, 12: 9. 10.1186/1475-2859-12-9
Rodriguez E, Navone L, Casati P, Gramajo H: Impact of malic enzymes on antibiotic and triacylglycerol production in Streptomyces coelicolor . Appl Environ Microbiol 2012, 78: 4571-4579. 10.1128/AEM.00838-12
Daniel J, Deb C, Dubey VS, Sirakova TD, Abomoelak B, Morbidoni HR, Kolattukudy PE: Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J Bacteriol 2004, 186: 5017-5030. 10.1128/JB.186.15.5017-5030.2004
Low KL, Shui G, Natter K, Yeo WK, Kohlwein SD, Dick T, Rao SP, Wenk MR: Lipid droplet-associated proteins are involved in the biosynthesis and hydrolysis of triacylglycerol in Mycobacterium bovis bacillus Calmette-Guerin. J Biol Chem 2010, 285: 21662-21670. 10.1074/jbc.M110.135731
Walker RW: Barakat H. Hung JG: The positional distribution of fatty acids in the phospholipids and triglycerides of Mycobacterium smegmatis and M. bovis BCG. Lipids 1970, 5: 684-691.
Voss I, Steinbüchel A: High cell density cultivation of Rhodococcus opacus for lipid production at a pilot-plant scale. Appl Microbiol Biotechnol 2001, 55: 547-555. 10.1007/s002530000576
Kurosawa K, Boccazzi P, de Almeida NM, Sinskey AJ: High-cell-density batch fermentation of Rhodococcus opacus PD630 using a high glucose concentration for triacylglycerol production. J Biotechnol 2010, 147: 212-218. 10.1016/j.jbiotec.2010.04.003
Janßen HJ, Ibrahim MH, Bröker D, Steinbüchel A: Optimization of macroelement concentrations, pH and osmolarity for triacylglycerol accumulation in Rhodococcus opacus strain PD630. AMB Express 2013, 3: 38. 10.1186/2191-0855-3-38
Raetz CR, Newman KF: Neutral lipid accumulation in the membranes of Escherichia coli mutants lacking diglyceride kinase. J Biol Chem 1978, 253: 3882-3887.
Raetz CR, Newman KF: Diglyceride kinase mutants of Escherichia coli : inner membrane association of 1,2-diglyceride and its relation to synthesis of membrane-derived oligosaccharides. J Bacteriol 1979, 137: 860-868.
Rucker J, Paul J, Pfeifer BA, Lee K: Engineering E. coli for triglyceride accumulation through native and heterologous metabolic reactions. Appl Microbiol Biotechnol 2013, 97: 2753-2759. 10.1007/s00253-013-4714-3
Kaczmarzyk D, Fulda M: Fatty acid activation in cyanobacteria mediated by acyl-acyl carrier protein synthetase enables fatty acid recycling. Plant Physiol 2010, 152: 1598-1610. 10.1104/pp.109.148007
Hsieh HJ, Su CH, Chien LJ: Accumulation of lipid production in Chlorella minutissima by triacylglycerol biosynthesis-related genes cloned from Saccharomyces cerevisiae and Yarrowia lipolytica . J Microbiol 2012, 50: 526-534. 10.1007/s12275-012-2041-5
Merchant SS, Kropat J, Liu B, Shaw J, Warakanont J: TAG, you’re it! Chlamydomonas as a reference organism for understanding algal triacylglycerol accumulation. Curr Opin Biotechnol 2012, 23: 352-363. 10.1016/j.copbio.2011.12.001
Williams CG: On the constitution of the essential oil of rue. Philos Trans R Soc Lond 1858, 148: 199-204. 10.1098/rstl.1858.0011
Forney FW, Markovetz AJ: The biology of methyl ketones. J Lipid Res 1971, 12: 383-395.
Yu G, Nguyen TT, Guo Y, Schauvinhold I, Auldridge ME, Bhuiyan N, Ben-Israel I, Iijima Y, Fridman E, Noel JP, Pichersky E: Enzymatic functions of wild tomato methylketone synthases 1 and 2. Plant Physiol 2010, 154: 67-77. 10.1104/pp.110.157073
Park J, Rodriguez-Moya M, Li M, Pichersky E, San KY, Gonzalez R: Synthesis of methyl ketones by metabolically engineered Escherichia coli . J Ind Microbiol Biotechnol 2012, 39: 1703-1712. 10.1007/s10295-012-1178-x
Goh EB, Baidoo EE, Keasling JD, Beller HR: Engineering of bacterial methyl ketone synthesis for biofuels. Appl Environ Microbiol 2012, 78: 70-80. 10.1128/AEM.06785-11
Müller J, MacEachran D, Burd H, Sathitsuksanoh N, Bi C, Yeh YC, Lee TS, Hillson NJ, Chhabra SR, Singer SW, Beller HR: Engineering of Ralstonia eutropha H16 for autotrophic and heterotrophic production of methyl ketones. Appl Environ Microbiol 2013, 79: 4433-4439. 10.1128/AEM.00973-13
Keasling JD, Hu Z, Somerville C, Church G, Berry D, Friedman L, Schirmer A, Brubaker S, Del Cardayre SB: Production of fatty acids and derivatives thereof. 2007.
Doan TT, Carlsson AS, Hamberg M, Bulow L, Stymne S, Olsson P: Functional expression of five Arabidopsis fatty acyl-CoA reductase genes in Escherichia coli . J Plant Physiol 2009, 166: 787-796. 10.1016/j.jplph.2008.10.003
Akhtar MK, Turner NJ, Jones PR: Carboxylic acid reductase is a versatile enzyme for the conversion of fatty acids into fuels and chemical commodities. Proc Natl Acad Sci USA 2013, 110: 87-92. 10.1073/pnas.1216516110
Schirmer A, Rude MA, Li X, Popova E, del Cardayre SB: Microbial biosynthesis of alkanes. Science 2010, 329: 559-562. 10.1126/science.1187936
Harger M, Zheng L, Moon A, Ager C, An JH, Choe C, Lai YL, Mo B, Zong D, Smith MD, Egbert RG, Mills JH, Baker D, Pultz IS, Siegel JB: Expanding the product profile of a microbial alkane biosynthetic pathway. ACS Synth Biol 2013, 2: 59-62. 10.1021/sb300061x
Howard TP, Middelhaufe S, Moore K, Edner C, Kolak DM, Taylor GN, Parker DA, Lee R, Smirnoff N, Aves SJ, Love J: Synthesis of customized petroleum-replica fuel molecules by targeted modification of free fatty acid pools in Escherichia coli . Proc Natl Acad Sci USA 2013, 110: 7636-7641. 10.1073/pnas.1215966110
Rude MA, Baron TS, Brubaker S, Alibhai M, del Cardayre SB, Schirmer A: Terminal olefin (1-alkene) biosynthesis by a novel p450 fatty acid decarboxylase from Jeotgalicoccus species. Appl Environ Microbiol 2011, 77: 1718-1727. 10.1128/AEM.02580-10
Matsumoto K, Taguchi S: Enzyme and metabolic engineering for the production of novel biopolymers: crossover of biological and chemical processes. Curr Opin Biotechnol 2013, 24: 1054-1060. 10.1016/j.copbio.2013.02.021
Steinbüchel A, Hustede E, Liebergesell M, Pieper U, Timm A, Valentin H: Molecular basis for biosynthesis and accumulation of polyhydroxyalkanoic acids in bacteria. FEMS Microbiol Rev 1992, 9: 217-230.
Rehm BH, Krüger N, Steinbüchel A: A new metabolic link between fatty acid de novo synthesis and polyhydroxyalkanoic acid synthesis. The phaG gene from Pseudomonas putida KT2440 encodes a 3-hydroxyacyl-acyl carrier protein-coenzyme a transferase. J Biol Chem 1998, 273: 24044-24051. 10.1074/jbc.273.37.24044
Lageveen RG, Huisman GW, Preusting H, Ketelaar P, Eggink G, Witholt B: Formation of polyesters by Pseudomonas oleovorans : effect of substrates on formation and composition of poly-( R )-3-hydroxyalkanoates and poly-( R )-3-hydroxyalkenoates. Appl Environ Microbiol 1988, 54: 2924-2932.
Kim Do Y, Kim HW, Chung MG, Rhee YH: Biosynthesis, modification, and biodegradation of bacterial medium-chain-length polyhydroxyalkanoates. J Microbiol 2007, 45: 87-97.
Fiedler S, Steinbüchel A, Rehm BH: PhaG-mediated synthesis of Poly (3-hydroxyalkanoates) consisting of medium-chain-length constituents from nonrelated carbon sources in recombinant Pseudomonas fragi . Appl Environ Microbiol 2000, 66: 2117-2124. 10.1128/AEM.66.5.2117-2124.2000
Olivera ER, Carnicero D, Jodra R, Minambres B, García B, Abraham GA, Gallardo A, Román JS, García JL, Naharro G, Luengo JM: Genetically engineered Pseudomonas: a factory of new bioplastics with broad applications. Environ Microbiol 2001, 3: 612-618. 10.1046/j.1462-2920.2001.00224.x
Sun Z, Ramsay JA, Guay M, Ramsay BA: Carbon-limited fed-batch production of medium-chain-length polyhydroxyalkanoates from nonanoic acid by Pseudomonas putida KT2440. Appl Microbiol Biotechnol 2007, 74: 69-77. 10.1007/s00253-006-0655-4
Langenbach S, Rehm BH, Steinbüchel A: Functional expression of the PHA synthase gene phaC1 from Pseudomonas aeruginosa in Escherichia coli results in poly (3-hydroxyalkanoate) synthesis. FEMS Microbiol Lett 1997, 150: 303-309. 10.1016/S0378-1097(97)00142-0
Qi Q, Rehm BH, Steinbüchel A: Synthesis of poly (3-hydroxyalkanoates) in Escherichia coli expressing the PHA synthase gene phaC2 from Pseudomonas aeruginosa : comparison of PhaC1 and PhaC2. FEMS Microbiol Lett 1997, 157: 155-162. 10.1111/j.1574-6968.1997.tb12767.x
Klinke S, Ren Q, Witholt B, Kessler B: Production of medium-chain-length poly (3-hydroxyalkanoates) from gluconate by recombinant Escherichia coli . Appl Environ Microbiol 1999, 65: 540-548.
Rehm BH, Steinbüchel A: Heterologous expression of the acyl-acyl carrier protein thioesterase gene from the plant Umbellularia californica mediates polyhydroxyalkanoate biosynthesis in recombinant Escherichia coli . Appl Microbiol Biotechnol 2001, 55: 205-209. 10.1007/s002530000541
Park SJ, Lee SY: Biosynthesis of poly (3-hydroxybutyrate- co-3-hydroxyalkanoates) by metabolically engineered Escherichia coli strains. Appl Biochem Biotechnol 2004, 113–116: 335-346.
Zheng Z, Gong Q, Liu T, Deng Y, Chen JC, Chen GQ: Thioesterase II of Escherichia coli plays an important role in 3-hydroxydecanoic acid production. Appl Environ Microbiol 2004, 70: 3807-3813. 10.1128/AEM.70.7.3807-3813.2004
Nomura CT, Tanaka T, Gan Z, Kuwabara K, Abe H, Takase K, Taguchi K, Doi Y: Effective enhancement of short-chain-length-medium-chain-length polyhydroxyalkanoate copolymer production by coexpression of genetically engineered 3-ketoacyl-acyl-carrier-protein synthase III ( fabH ) and polyhydroxyalkanoate synthesis genes. Biomacromolecules 2004, 5: 1457-1464. 10.1021/bm049959v
Agnew DE, Stevermer AK, Youngquist JT, Pfleger BF: Engineering Escherichia coli for production of C (1) (2)-C (1) (4) polyhydroxyalkanoate from glucose. Metab Eng 2012, 14: 705-713. 10.1016/j.ymben.2012.08.003
Vickers CE, Klein-Marcuschamer D, Krömer JO: Examining the feasibility of bulk commodity production in Escherichia coli . Biotechnol Lett 2012, 34: 585-596. 10.1007/s10529-011-0821-3
J-il C, Lee SY: Process analysis and economic evaluation for Poly (3-hydroxybutyrate) production by fermentation. Bioprocess Eng 1997, 17: 335-342. 10.1007/s004490050394
Willke T, Vorlop K: Biotransformation of glycerol into 1,3-propanediol. Eur J Lipid Sci Tech 2008, 110: 831-840. 10.1002/ejlt.200800057
Tao L, Aden A: The economics of current and future biofuels. In Vitro Cell Dev Biol 2009, 45: 199-217.
International Energy Agency: Biofuels for transport: an international perspective. Paris: International Energy Agency; 2004.
Sabri S, Nielsen LK, Vickers CE: Molecular control of sucrose utilization in Escherichia coli W, an efficient sucrose-utilizing strain. Appl Environ Microbiol 2013, 79: 478-487. 10.1128/AEM.02544-12
Rumbold K, van Buijsen HJ, Gray VM, van Groenestijn JW, Overkamp KM, Slomp RS, van der Werf MJ, Punt PJ: Microbial renewable feedstock utilization: a substrate-oriented approach. Bioeng Bugs 2010, 1: 359-366. 10.4161/bbug.1.5.12389
Wang X, Yomano LP, Lee JY, York SW, Zheng H, Mullinnix MT, Shanmugam KT, Ingram LO: Engineering furfural tolerance in Escherichia coli improves the fermentation of lignocellulosic sugars into renewable chemicals. Proc Natl Acad Sci USA 2013, 110: 4021-4026. 10.1073/pnas.1217958110
Kim JH, Block DE, Mills DA: Simultaneous consumption of pentose and hexose sugars: an optimal microbial phenotype for efficient fermentation of lignocellulosic biomass. Appl Microbiol Biotechnol 2010, 88: 1077-1085. 10.1007/s00253-010-2839-1
Viitanen MI, Vasala A, Neubauer P, Alatossava T: Cheese whey-induced high-cell-density production of recombinant proteins in Escherichia coli . Microb Cell Fact 2003, 2: 2. 10.1186/1475-2859-2-2
Rosales-Colunga LM, Alvarado-Cuevas ZD, Razo-Flores E, Rodríguez Ade L: Maximizing hydrogen production and substrate consumption by Escherichia coli WDHL in cheese whey fermentation. Appl Biochem Biotechnol 2013, 171: 704-715. 10.1007/s12010-013-0394-9
Mazumdar S, Bang J, Oh MK: L-Lactate production from seaweed hydrolysate of Laminaria japonica using metabolically engineered Escherichia coli . Appl Biochem Biotechnol 2013, 171: 704-715. 10.1007/s12010-013-0394-9
Zhang F, Carothers JM, Keasling JD: Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat Biotechnol 2012, 30: 354-359. 10.1038/nbt.2149
Xu P, Gu Q, Wang W, Wong L, Bower AG, Collins CH, Koffas MA: Modular optimization of multi-gene pathways for fatty acids production in E. coli. Nat Commun 2013, 4: 1409.
Acknowledgements
We acknowledge support by Deutsche Forschungsgemeinschaft and the Open Access Publication Fund of the University of Muenster.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HJ conceptualized, researched and wrote the manuscript. AS conceptualized, helped to draft the manuscript and critically revised it. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Janßen, H.J., Steinbüchel, A. Fatty acid synthesis in Escherichia coli and its applications towards the production of fatty acid based biofuels. Biotechnol Biofuels 7, 7 (2014). https://doi.org/10.1186/1754-6834-7-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1754-6834-7-7