
Abstract
Background
Attention-deficit/hyperactivity disorder (ADHD) is a common neurobehavioral psychiatric disorder that afflicts children, with a reported prevalence of 2.4% to 19.8% worldwide. Stimulants (methylphenidate [MPH] and amphetamine) are considered first-line ADHD pharmacotherapy. MPH is a catecholamine reuptake inhibitor, whereas amphetamines have additional presynaptic activity. Although MPH and amphetamine can effectively manage ADHD symptoms in most pediatric patients, many still fail to respond optimally to either. After administration, the prodrug stimulant lisdexamfetamine dimesylate (LDX) is converted to l-lysine and therapeutically active d-amphetamine in the blood. The objective of this study was to evaluate the clinical efficacy of LDX in children with ADHD who remained symptomatic (ie, nonremitters; ADHD Rating Scale IV [ADHD-RS-IV] total score > 18) on MPH therapy prior to enrollment in a 4-week placebo-controlled LDX trial, compared with the overall population.
Methods
In this post hoc analysis of data from a multicenter, randomized, double-blind, forced-dose titration study, we evaluated the clinical efficacy of LDX in children aged 6-12 years with and without prior MPH treatment at screening. ADHD symptoms were assessed using the ADHD-RS-IV scale, Conners' Parent Rating Scale-Revised short form (CPRS-R), and Clinical Global Impressions-Improvement scale, at screening, baseline, and endpoint. ADHD-RS-IV total and CPRS-R ADHD Index scores were summarized as mean (SD). Clinical response for the subgroup analysis was defined as a ≥ 30% reduction from baseline in ADHD-RS-IV score and a CGI-I score of 1 or 2. Dunnett test was used to compare change from baseline in all groups. Number needed to treat to achieve one clinical responder or one symptomatic remitter was calculated as the reciprocal of the difference in their proportions on active treatment and placebo at endpoint.
Results
Of 290 randomized participants enrolled, 28 received MPH therapy at screening, of which 26 remained symptomatic (ADHD-RS-IV > 18). ADHD-RS-IV total scores, changes from baseline, clinical responsiveness, and rates of symptomatic remission in this subgroup were comparable to the overall population. The safety and tolerability profiles for LDX were comparable to other stimulants currently available.
Conclusion
In this analysis, children with significant clinical ADHD symptoms despite MPH treatment improved during treatment with LDX and experienced similar improvements in their symptoms as the overall study population.
Trial Registration
ClinicalTrials.gov: NCT00556296
Similar content being viewed by others

Background
Attention-deficit/hyperactivity disorder (ADHD) is one of the most common neurobehavioral psychiatric disorders that afflicts children [1], with a reported prevalence of 2.4% to 19.8% worldwide [2] using the criteria from the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) from the American Psychiatric Association [3]. Two Canadian studies of children and adolescents, using earlier diagnostic criteria to examine ADHD prevalence, estimated a prevalence of 6.3% in an Ontario study of participants (aged 4 to 16 years) [4], and 3.3% to 8.9% in a comparable population (aged 6 to 14 years) in Quebec [5].
Stimulants have long been used to treat ADHD symptoms. The Texas Consensus Conference Panel on Pharmacotherapy of Childhood ADHD algorithm [6] considered psychostimulants as first-line pharmacotherapy treatments for ADHD; However, the Canadian ADHD Resource Alliance (CADDRA) guidelines consider long-acting stimulants and atomoxetine as first-line agents in the management of ADHD [7]. The stimulant types most commonly used in ADHD treatment are methylphenidate (MPH) and amphetamine. These have similar subjective effects [8] yet differ in their mechanisms of action--MPH is a dopamine and norepinephrine reuptake inhibitor, while amphetamines have additional presynaptic activity--stimulating the release of dopamine, norepinephrine, and serotonin [9]. Although both are considered efficacious, a meta-analysis of 23 studies comparing the efficacy of immediate-release (IR) formulations of MPH and amphetamine in the treatment of children with ADHD revealed small but statistically significant differences in favor of amphetamine [10]. A comparative review of controlled crossover studies [11] found that clinical response rates for IR formulations of MPH and amphetamine ranged from 57% to 68% and 69% to 77%, respectively. The review also estimated that 87% to 92% participants respond to at least one of these stimulants. However, although MPH and amphetamine can effectively manage ADHD symptoms in most pediatric patients, many patients still fail to respond optimally to either.
Lisdexamfetamine dimesylate (LDX; Vyvanse®) is a prodrug stimulant with a novel delivery mechanism, approved in Canada [12] and the United States [13] for the treatment of ADHD in children 6 to 12 years of age, adolescents aged 13 to 17 years, and adults. LDX is a therapeutically inactive molecule. LDX is converted, primarily in the blood, to l-lysine and therapeutically active d-amphetamine [14]. In Canada, the approved dosages range from 20 to 60 mg capsules for once daily oral administration and in the United States from 20 to 70 mg also once daily [12, 13].
LDX has been shown to be effective from 1.5 to 13 hours postdose in children [15], and from 2 to 14 hours postdose in adults [16]. In a randomized controlled trial (RCT), LDX was associated with improvements in clinical symptoms of ADHD in children while maintaining a safety profile similar to other stimulant medications [17].
In this post hoc analysis from the RCT, the efficacy of LDX in a subset of children, who had significant ADHD symptoms at study enrollment despite receiving MPH treatment, was evaluated to determine clinical response to LDX therapy in these study participants. Based on previous findings that some patients fail to achieve optimal response to either MPH or amphetamine, children who were previously treated with MPH and continue to have ADHD symptoms may be responsive to amphetamine-based ADHD treatment.
Methods
The methods used in this study for the overall study population have been described previously [17]. This was a multicenter, randomized, double-blind, forced-dose titration, parallel-group study, conducted in accordance with the Guideline for Good Clinical Practice from the World Health Organization and the Declaration of Helsinki and its amendments.
Participants
Biederman et al previously described full inclusion/exclusion criteria [17]. Briefly, children aged 6 to 12 years who met DSM-IV-TR criteria for a primary diagnosis of ADHD [18] and had a ADHD Rating Scale IV (ADHD-RS-IV) [19, 20] score of ≥ 28 at baseline after washout were eligible for inclusion, regardless of medication used for ADHD at screening.
Study Design
The study comprised a 1-week screening period; a 1-week washout period of prior psychoactive medications; and 4-weeks of double-blind treatment. During screening, participants received an initial ADHD-RS-IV evaluation. Participants receiving medication for ADHD at enrollment were allowed to continue their medication during the screening evaluation. After screening, the parents/caregivers of eligible participants were instructed to discontinue their prior ADHD medications, if they had not already done so.
Baseline assessments were made after the 1-week washout. Participants were randomized in a 1:1:1:1 ratio (using a block-randomization schedule) to receive double-blind, oral administration of LDX 30 mg/day for 4 weeks, 50 mg/day (30 mg/day for week 1, 50 mg/day for weeks 2 to 4), 70 mg/day (30 mg/day for week 1, 50 mg/day for week 2, 70 mg/day for weeks 3 and 4), or placebo for 4 weeks.
Efficacy Outcome Measures
The primary efficacy outcome was the change in mean ADHD-RS-IV total score from baseline to treatment endpoint, defined as the last postrandomization week for which a score was obtained. ADHD-RS-IV total score assessments were based on investigator interviews with the caregiver and child regarding symptom severity during the preceding week.
Secondary efficacy measures included ADHD-RS-IV total scores at screening, baseline, and endpoint; percent change in ADHD-RS-IV total score; the Conners' Parent Rating Scale-Revised (CPRS-R: Short Form) [21]; and the investigator-rated Clinical Global Impressions (CGI) scale [22]. The CGI-Severity (CGI-S) assessment was conducted at the baseline visit and the CGI-Improvement (CGI-I) assessment was conducted at subsequent visits.
Efficacy was assessed in the overall efficacy population, all participants who had ADHD-RS-IV scores recorded at baseline and at least one other postrandomization time point.
Post Hoc Efficacy Analyses
This post hoc efficacy analysis assessed treatment effects of LDX and placebo in participants receiving MPH prior to entering the present study, who had available screening data and significant ADHD symptoms prior to discontinuing their MPH regimen. Efficacy was further evaluated according to mean daily MPH dose received (≥ 1 mg/kg vs < 1 mg/kg) during prior treatment.
Rates of symptomatic remission and clinical response were evaluated throughout the study in participants receiving prior MPH therapy and the efficacy population.
Steele et al [23] suggested that treatment response be considered as an improvement in symptom scores from baseline of 25% to 30%. However, reductions from baseline do not take into account potential differences in baseline severity of disease. Participants with severe symptoms at baseline may be considered responders but still exhibit symptoms. Hence, a clinical response definition that includes a percent reduction in symptoms and a measure of global clinical improvement, such as the CGI-I, may be a better measure of clinical response to treatment. Moreover, other studies have shown that a 1-level change on the CGI-I was consistent with an estimated 10- to 15-point or 25% to 30% change from baseline in ADHD-RS-IV total score [24].
In the primary analysis [17], Biederman et al reported on the ADHD-RS-IV (primary outcome measure) and CGI-I (secondary outcome measure) as continuous measures. In this present analysis, clinical response to LDX treatment was defined as a dual criteria of ≥ 30% reduction in ADHD-RS-IV total score from baseline and a CGI-I score of 1 or 2 at endpoint based on data from previous reports defining response [23, 25]; symptomatic remission was defined as ADHD-RS-IV total score of ≤ 18 [26]. Conversely, nonremitters on prior MPH were defined as participants with an ADHD-RS-IV total score > 18 while receiving MPH prior to entering the study. Number-needed-to-treat (NNT) for 1 participant to achieve a therapeutic clinical response or symptomatic remission at treatment endpoint was calculated to translate the efficacy data into more clinically meaningful terms.
Safety Assessments
Safety assessments, in enrolled participants who received at least 1 dose of study medication, have been reported previously [17]. Briefly, these included adverse events (AEs), electrocardiograms (ECGs), blood pressure (BP), heart rate, and laboratory assessments. Treatment-emergent AEs (TEAEs) were coded using the Medical Dictionary for Regulatory Activities version 7.1 [27]. TEAEs referred to events with onset after the first date of treatment and no later than 3 days following termination of treatment. No separate assessments were performed in nonremitters on prior MPH due to low sample numbers and no reason to expect differences in safety/tolerability in these participants.
Statistical Analyses
ADHD-RS-IV total and CPRS-R ADHD Index scores were summarized as mean (standard deviation [SD]). Mean change in ADHD-RS-IV total score for the overall population was assessed using 2-way analysis of covariance. Dunnett test for multiple mean comparisons with least-squares adjustment was used to compare change from baseline in the 3 active treatment groups versus placebo. NNT to achieve 1 clinical responder or 1 symptomatic remitter was calculated as the reciprocal of the difference in proportions of clinical responders or symptomatic remitters on active treatment and placebo at treatment endpoint.
Results
Participant Demographics and Disposition
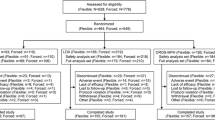
In total, 297 children were enrolled at 40 study sites in the United States, of which 7 children discontinued prior to randomization, and 290 were randomized to receive LDX (n = 218) or placebo (n = 72). Of these, 285 had a postrandomization symptom assessment and were included in the efficacy population. Full demographic data for this population have been previously reported [17].
Of the 290 randomized participants, 28 were receiving MPH treatment at screening and 26 of these were classified as nonremitters on prior MPH at the screening visit, prior to randomization (Table 1). Median age was 9 years and 11/26 (42.3%) female and 15/26 (57.7%) male participants were included. Prior treatment for 19 (73.1%) participants was osmotic, controlled-release MPH (OROS MPH), alone or in combination with another ADHD medication (1 participant in combination with IR dex-MPH [d-MPH], 1 with IR mixed amphetamine salts); 2 (7.7%) participants received prior treatment with extended release (ER) MPH; 3 (7.7%) participants received prior treatment with IR MPH; 1 (3.8%) participant was previously treated with sustained release MPH (SR MPH); 1 (3.8%) participant was prior treated with MPH controlled delivery (MPH CD) (Table 1). Sixteen participants (61.5%) received an average daily dose of ≥ 1 mg/kg MPH, and 10 (38.5%) an average daily dose of < 1 mg/kg MPH.
Changes in ADHD-RS-IV Total Scores
Mean (SD) screening, baseline, and endpoint ADHD-RS-IV total scores for nonremitters during prior MPH treatment, nonremitters stratified according to prior MPH dosage received, and overall efficacy population are shown in Figure 1.

ADHD-RS-IV total scores in (A) nonremitters during prior MPH treatment; and (B) the overall efficacy population.
The mean (SD) change in ADHD-RS-IV total score from baseline with LDX treatment was -24.0 (12.56) (Figure 2), corresponding to a mean (SD) percentage reduction of 57 (29.9%) in the 19 nonremitters on prior MPH treatment. The mean (95% confidence interval [CI]) placebo-adjusted ADHD-RS-IV total score reduction for this group was -17.6 (-29.65, -5.49; P = .0063).

ADHD-RS-IV total scores in prior MPH participants receiving LDX and classified as nonremitters.
Changes in ADHD-RS-IV and CGI-I Scores: Remitters and Responders
Of the 26 nonremitters on prior MPH at screening, 12 (63.2%) participants receiving LDX and 1 (14.3%) receiving placebo were classified as remitters during the study (Figure 3). Similar patterns of symptomatic remission with LDX treatment were observed in the overall efficacy population. As well, patterns of symptomatic remission were similar with placebo treatment in the overall efficacy population and in nonremitters on prior MPH. The NNT (95% CI) to achieve symptomatic remission with LDX at treatment endpoint was 2.0 (1.21, 6.63) in nonremitters and 2.1 (1.74, 2.72) in the overall study population.

Percentage of symptomatic remitters* during the study. *Symptomatic remitters = participants who achieved ADHD-RS-IV total scores ≤ 18.
Of nonremitters on prior MPH, clinical response was achieved in 15 (78.9%) treated with LDX and 3 (42.9%) treated with placebo, respectively. In the overall efficacy population, 169 (79.3%) treated with LDX and 21 (29.2%) treated with placebo achieved clinical response (Figure 4). Of the 169 LDX clinical responders, 54 (32.0%) received 30 mg/d LDX, 55 (32.5%) received 50 mg/d LDX, and 60 (35.5%) received 70 mg/d LDX. NNT (95% CI) to achieve clinical response with LDX at treatment endpoint was 2.0 (1.21, 6.63) in nonremitters on prior MPH, versus 1.8 (1.51, 2.22) in the overall population.

Percentage of clinical responders* during the study. *Clinical responders = participants who achieved ≥ 30% reduction in ADHD-RS-IV total scores from baseline and CGI-I scores of 1 or 2.
Changes in CPRS-R ADHD Index Scores
Mean (SD) morning, afternoon, and evening CPRS-R ADHD index scores at baseline and endpoint in nonremitters on prior MPH are shown in Figure 5. The mean changes from baseline morning, afternoon, and evening CPRS-R ADHD index scores were -14.7 (10.90), -12.2 (12.89), and -13.4 (11.69) for the LDX groups, respectively, and -1.3 (14.92), -0.1 (9.01), and 0.4 (11.25) for the placebo group, respectively. These data were similar to the CPRS-R ADHD index scores observed in the overall population [17].

CPRS-R ADHD index scores in prior MPH participants with ADHD-RS-IV total scores > 18 at screening. *Data available for 18 participants receiving LDX at baseline, afternoon, and evening time points.
Safety and Tolerability
Full safety analyses have been reported previously [17]. In the safety population, 196/290 (68%) participants reported one or more TEAEs; 21/290 (7.2%) discontinued due to TEAE. TEAEs with an incidence ≥ 5% in the combined LDX group were decreased appetite, insomnia, headache, upper abdominal pain, irritability, weight loss, vomiting, nausea, dizziness, and nasopharyngitis and, in the placebo group, were headache, cough, nasal congestion, nasopharyngitis, and upper abdominal pain. No serious AEs were observed during the study. More than 95% of TEAEs were mild or moderate in intensity and most began during the first week of treatment and abated over time [17]. Mean (SE) change from baseline at endpoint for pulse (bpm) ranged from 0.3 (1.20) to 4.1 (1.17) in all LDX groups and was -0.7 (1.17) in the placebo group. The systolic BP change for all LDX groups ranged from 0.4 (1.08) to 2.6 (1.05) mm Hg and for placebo was 1.3 (1.05) mm Hg. For diastolic BP the change ranged from 0.6 (0.93) to 2.3 (0.91) mm Hg for all LDX groups and was 0.6 (0.91) mm Hg for the placebo group. LDX treatment was not associated with any significant changes in mean BP, ECG parameters, and laboratory values.
Discussion
In this post hoc analysis, LDX showed efficacy when given to children with significant clinical ADHD symptoms despite prior MPH treatment. Efficacy outcomes were similar to the results of the overall population assessed in the clinical trial.
Among participants previously treated with MPH, more than half were receiving doses (average daily dose ≥ 1 mg/kg) considered generally effective according to the regimens administered in randomized, controlled trials [28, 29]. Conversely, just under half may have received suboptimal doses. Moreover, none of these measures differed from those observed in the overall study population. Although this study was not powered to detect differences between the treatment groups, the percentage of clinical responders in the overall study group was comparable regardless of LDX dose received. Similarly, no apparent differences occurred between the NNTs to achieve clinical response or symptomatic remission for the overall efficacy population and nonremitters on prior MPH. The NNT values calculated are comparable or superior to those reported elsewhere in the literature for symptomatic remission and clinical response to MPH and atomoxetine, which range from approximately 1.9 to 5.3 depending on formulation and types of raters [30].
Differential responses to MPH and amphetamine may explain a successful clinical response to LDX in participants who had significant ADHD symptoms despite prior MPH therapy. In 2 separate crossover studies [31, 32] comparing the efficacy of MPH and dextroamphetamine, most children with ADHD who did not respond to 1 stimulant responded to the other. A bimodal pattern of clinical response to atomoxetine has been described, with no obvious demographic or clinical predictors of clinical response [33].
Clinical trial design may have contributed to the observed clinical response to LDX treatment in nonremitters on prior MPH. LDX treatment was administered in a forced-dose titration, while prior MPH therapy was provided according to community standards and included potential suboptimal dosing. Use of different definitions of therapeutic response may have altered the rates observed.
This post hoc analysis has limitations. The classification of participants as nonremitters on prior MPH considers only the ADHD-RS-IV total score at screening and may not reflect the participants' overall clinical response to MPH. It should be noted that switching from MPH formulations to LDX was done as part of the study protocol and not purely as a clinical practice decision.
The 4-week study duration limits the ability to extrapolate the findings to the long-term treatment generally required in managing ADHD. This study was not prospectively designed or powered to detect differences between the treatment groups. A prospective study would be required to confirm these preliminary findings.
Conclusions
In this post hoc analysis of children who had significant clinical ADHD symptoms despite previous MPH treatment, LDX demonstrated efficacy and clinical response in the subpopulation assessed. Efficacy outcomes in this population were similar to those in the overall study population.
Abbreviations
- ADHD:
-
attention-deficit/hyperactivity disorder
- ADHD-RS-IV:
-
ADHD Rating Scale IV
- AE:
-
adverse event
- BP:
-
blood pressure
- CADDRA:
-
Canadian ADHD Resource Alliance
- CD:
-
controlled delivery
- CGI-S:
-
Clinical Global Impressions-Severity
- CGI-I:
-
Clinical Global Impressions-Improvement
- CI:
-
confidence interval
- CPRS-R:
-
Short Form: Conners' Parent Rating Scale-Revised
- dMPH:
-
dexmethylphenidate
- DSM-IV-TR Diagnostic and Statistical Manual of Mental Disorders:
-
, Fourth Edition, Text Revision
- ECG:
-
electrocardiogram
- ER:
-
extended-release
- IR:
-
immediate-release
- LDX:
-
lisdexamfetamine dimesylate
- MAS:
-
mixed amphetamine salts
- MPH:
-
methylphenidate
- NNT:
-
number-needed-to-treat
- OROS:
-
osmotic release oral system
- RCT:
-
randomized controlled trial
- SR:
-
sustained-release
- TEAE:
-
treatment-emergent AE
References
Pliszka S, the AACAP Work Group on Quality Issues: Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007, 46: 894-921.
Faraone SV, Sergeant J, Gillberg C, Biederman J: The worldwide prevalence of ADHD: is it an American condition?. World Psychiatry. 2003, 2: 104-13.
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders DSM-IV. 1994, Washington, DC: American Psychiatric Association
Szatmari P, Offord DR, Boyle MH: Ontario Child Health Study: prevalence of attention deficit disorder with hyperactivity. J Child Psychol Psychiatry. 1989, 30: 219-30. 10.1111/j.1469-7610.1989.tb00236.x.
Breton JJ, Bergeron L, Valla JP, Berthiaume C, Gaudet N, Lambert J, St-Georges M, Houde L, Lepine S: Quebec child mental health survey: prevalence of DSM-III-R mental health disorders. J Child Psychol Psychiatry. 1999, 40: 375-84. 10.1111/1469-7610.00455.
Pliszka SR, Crismon ML, Hughes CW, Conners CK, Emslie GJ, Jensen PS, McCracken JT, Swanson JM, Lopez M, The Texas Consensus Conference Panel on Pharmacotherapy of Childhood Attention-Deficit/Hyperactivity Disorder: The Texas Children's Medication Algorithm Project: revision of the algorithm for pharmacotherapy of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2006, 45: 642-57. 10.1097/01.chi.0000215326.51175.eb.
Canadian ADHD Resource Alliance: Canadian ADHD Practice Guidelines. CAP-Guidelines. Accessed October 4, 2011, [http://www.caddra.ca/cms4/pdfs/caddraGuidelines2011.pdf]3
Heishman SJ, Henningfield JE: Discriminative stimulus effects of d-amphetamine, methylphenidate, and diazepam in humans. Psychopharmacology (Berl). 1991, 103: 436-42. 10.1007/BF02244241.
Heal DJ, Cheetham SC, Smith SL: The neuropharmacology of ADHD drugs in vivo: insights on efficacy and safety. Neuropharmacology. 2009, 57: 608-18. 10.1016/j.neuropharm.2009.08.020.
Faraone SV, Buitelaar J: Comparing the efficacy of stimulants for ADHD in children and adolescents using meta-analysis. Eur Child Adolesc Psychiatry. 2009, 19: 353-64.
Arnold LE: Methylphenidate vs. amphetamine: comparative review. J Atten Disord. 2000, 3: 200-211. 10.1177/108705470000300403.
Vyvanse Product Monograph: 2011, Saint-Laurent, Québec: Shire Canada Inc
Vyvanse [package insert]. 2011, Wayne, PA: Shire US Inc
Pennick M: Absorption of lisdexamfetamine dimesylate and its enzymatic conversion to d-amphetamine. Neuropsychiatr Dis Treat. 2010, 6: 317-27.
Wigal SB, Kollins SH, Childress AC, Squires L, for the 311 Study Group: A 13-hour laboratory school study of lisdexamfetamine dimesylate in school-aged children with attention-deficit/hyperactivity disorder. Child Adolesc Psychiatry Ment Health. 2009, 3: 17.
Wigal T, Brams M, Gasior M, Gao J, Squires L, Giblin J, on behalf of the 316 Study Group: Randomized, double-blind, placebo-controlled, crossover study of the efficacy and safety of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder: novel findings using the adult workplace environment design. Behav Brain Funct. 2010, 6: 34-10.1186/1744-9081-6-34.
Biederman J, Krishnan S, Zhang Y, McGough JJ, Findling RL: Efficacy and tolerability of lisdexamfetamine dimesylate (NRP-104) in children with attention-deficit/hyperactivity disorder: a phase III, multicenter, randomized, double-blind, forced-dose, parallel-group study. Clin Ther. 2007, 29: 450-463. 10.1016/S0149-2918(07)80083-X.
American Psychiatric Association: Attention-deficit and disruptive behavior disorders. Diagnostic and Statistical Manual of Mental Disorders DSM-IV-TR. 2000, Washington, DC: American Psychiatric Association, 85-93. Text Revision, Fourth
DuPaul GJ, Power TJ, Anastopoulos AD, Reid R: ADHD Rating Scale-IV: Checklists, Norms, and Clinical Interpretation. 1998, New York, NY: Guilford Press
Faries DE, Yalcin I, Harder D, Heiligenstein JH: Validation of the ADHD Rating Scale as a clinician administered and scored instrument. J Atten Disord. 2001, 5: 107-15. 10.1177/108705470100500204.
Conners CK, Sitarenios G, Parker JDA, Epstein JN: The Revised Conners' Parent Rating Scale (CPRS-R): factor structure, reliability, and criterion validity. J Abnorm Child Psychol. 1998, 26: 257-68. 10.1023/A:1022602400621.
Guy W: Clinical global impressions. ECDEU Assessment Manual for Psychopharmacology. 1976, Rockville, MD: US Department of Health, Education, and Welfare; Public Health Service, Alcohol, Drug Abuse and Mental Health Administration, NIMH Psychopharmacology Research Branch, 218-222.
Steele M, Jensen PS, Quinn DMP: Remission versus response as the goal of therapy in ADHD: a new standard for the field?. Clin Ther. 2006, 28: 1892-908. 10.1016/j.clinthera.2006.11.006.
Goodman D, Faraone SV, Adler LA, Dirks B, Hamdani M, Weisler R: Interpreting ADHD rating scale scores: linking ADHD rating scale scores and CGI levels in two randomized controlled trials of lisdexamfetamine dimesylate in ADHD. Primary Psychiatry. 2010, 17: 44-52.
Swanson JM, Kraemer HC, Hinshaw SP, Arnold LE, Conners CK, Abikoff HB, Clevenger W, Davies M, Elliott GR, Greenhill LL, Hechtman L, Hoza B, Jensen PS, March JS, Newcorn JH, Owens EB, Pelham WE, Schiller E, Severe JB, Simpson S, Vitiello B, Wells K, Wigal T, Wu M: Clinical relevance of the primary findings of the MTA: success rates based on severity of ADHD and ODD symptoms at the end of treatment. J Am Acad Child Adolesc Psychiatry. 2001, 40: 168-79. 10.1097/00004583-200102000-00011.
Spencer T, Biederman J, Wilens T, Steingard R, Geist D: Nortriptyline treatment of children with attention-deficit hyperactivity disorder and tic disorder or Tourette's syndrome. J Am Acad Child Adolesc Psychiatry. 1993, 32: 205-10. 10.1097/00004583-199301000-00029.
Medical Dictionary for Regulatory Activities (MedDRA), Version 7.1. 2009, Northrop Gruman Corporation
Pelham WE, Gnagy EM, Burrows-Maclean L, Williams A, Fabiano GA, Morrisey SM, Chronis AM, Forehand GL, Nguyen CA, Hoffman MT, Lock TM, Fielbelkorn K, Coles EK, Panahon CJ, Steiner RL, Meichenbaum DL, Onyango AN, Morse GD: Once-a-day Concerta® methylphenidate versus three-times-daily methylphenidate in laboratory and natural settings. Pediatrics. 2001, 107: E105-10.1542/peds.107.6.e105.
Steele M, Weiss M, Swanson J, Wang J, Prinzo RS, Binder CE: A randomized, controlled effectiveness trial of OROS-methylphenidate compared to usual care with immediate-release methylphenidate in attention deficit-hyperactivity disorder. Can J Clin Pharmacol. 2006, 13: e50-e62.
Banaschewski T, Coghill D, Santosh P, Zuddas A, Asherson P, Buitelaar J, Danckaerts M, Dopfner M, Faraone SV, Rothenberger A, Sergeant J, Steinhausen HC, Sonuga-Barke EJ, Taylor E: Long-acting medications for the hyperkinetic disorders. A systematic review and European treatment guideline. Eur Child Adolesc Psychiatry. 2006, 15: 476-95. 10.1007/s00787-006-0549-0.
Arnold LE, Christopher J, Huestis R, Smeltzer DJ: Methylphenidate vs dextroamphetamine vs caffeine in minimal brain dysfunction: controlled comparison by placebo washout design with Bayes' analysis. Arch Gen Psychiatry. 1978, 35: 463-73. 10.1001/archpsyc.1978.01770280073008.
Elia J, Borcherding BG, Rapoport JL, Keysor CS: Methylphenidate and dextroamphetamine treatments of hyperactivity: are there true nonresponders?. Psychiatry Res. 1991, 36: 141-55. 10.1016/0165-1781(91)90126-A.
Newcorn JH, Sutton VK, Weiss MD, Sumner CR: Clinical responses to atomoxetine in attention-deficit/hyperactivity disorder: the Integrated Data Exploratory Analysis (IDEA) study. J Am Acad Child Adolesc Psychiatry. 2009, 48: 511-18. 10.1097/CHI.0b013e31819c55b2.
Acknowledgements
Clinical research was funded by the sponsor, Shire Canada Inc. Under the direction of the authors, Kira Belkin and William Perlman, employees of Excerpta Medica, and Huda Ismail Abdullah, PhD, and Michael Pucci, PhD, employees of SCI Scientific Communications & Information (SCI), provided writing assistance for this publication. Editorial assistance in formatting, proofreading, copy editing, and fact checking was also provided by Excerpta Medica and SCI. Robert Morgan from Shire Canada Inc. also reviewed and edited the manuscript for scientific accuracy. Shire Canada Inc. provided funding to Excerpta Medica and SCI for support in writing and editing this manuscript. Although the sponsor was involved in the design, collection, analysis, interpretation, and fact checking of information, the content of this manuscript, the ultimate interpretation, and the decision to submit it for publication in Child and Adolescent Psychiatry and Mental Health was made by the authors independently.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Dr Jain or Saundra Jain receives or has received grant research support from Abbott, Addrenex, Aspect, Forest, Lilly, and Pfizer; served as a consultant for Addrenex, Impax, Lilly, and Shire; served on a speaker's bureau for Cyberonics, GlaxoSmithKline, Jazz, Pfizer, Shire, and Takeda; received honorarium from Cyberonics, Forest, Jazz, Lilly, Pfizer, Roche, Shire, and Takeda. Dr Babcock is an employee of Shire and holds stock and/or stock options in Shire. Dr Burtea is formerly an employee of Shire Canada Inc. and holds stock and/or stock options in Shire Canada Inc. Dr Dirks is an employee of Shire and holds stock and/or stock options in Johnson & Johnson and Shire. Mr Adeyi is an employee of Shire and holds stock and/or stock options in Shire. Dr Scheckner is an employee of Shire and holds stock and/or stock options in Shire. Dr Lasser is an employee of Shire and holds stock and/or stock options in Shire.
Authors' contributions
RJ was an investigator on the parent study and participated in data acquisition, analysis, interpretation, and presentation. RJ was fully involved in drafting the manuscript and revising the intellectual content of this manuscript. He has given final approval of this version. TBabcock was the associate director, Scientific Publications, Clinical Development, and Medical Affairs for this study, and made substantial contributions to the analysis and interpretation of the data. He was deeply involved in drafting the manuscript and revising the intellectual content. He has given final approval of this version. TBurtea was the medical director, Global Clinical Development and Medical Affairs for this study and made substantial contributions to the analysis, and interpretation of the data. He was deeply involved in drafting the manuscript and revising the intellectual content. He has given final approval of this version. BD was the director, Clinical Development and Medical Affairs for this study, and made substantial contributions to the analysis and interpretation of the data. He was deeply involved in drafting the manuscript and revising the intellectual content. He has given final approval of this version. BA was a statistician involved in all post hoc data analysis, interpretation, and presentation. Statistician BA was fully involved in drafting and revising the intellectual content of this manuscript. Statistician BA has given final approval to this version. BS was the associate director, Scientific Publications, Clinical Development, and Medical Affairs for this study, and made substantial contributions to the analysis and interpretation of the data. He was deeply involved in drafting the manuscript and revising the intellectual content. He has given final approval of this version. RL was the senior director, Clinical Development and Medical Affairs for this study, and made substantial contributions to the analysis and interpretation of the data. He was deeply involved in drafting the manuscript and revising the intellectual content. He has given final approval of this version.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Jain, R., Babcock, T., Burtea, T. et al. Efficacy of lisdexamfetamine dimesylate in children with attention-deficit/hyperactivity disorder previously treated with methylphenidate: a post hoc analysis. Child Adolesc Psychiatry Ment Health 5, 35 (2011). https://doi.org/10.1186/1753-2000-5-35
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1753-2000-5-35