
Abstract
Attention-deficit/hyperactivity disorder (ADHD) is a neurodevelopmental disorder characterized by symptoms of inattention, hyperactivity, and impulsivity associated with clinically significant impairment in functioning. ADHD has an early onset, but frequently persists, with a prevalence estimate of 4% in adults. Dasotraline is a novel compound that is a potent inhibitor of dopamine and norepinephrine transporters that achieves stable plasma concentrations with once-daily dosing. In this study, adult outpatients meeting DSM-IV-TR criteria for ADHD were randomized to 4 weeks of double-blind, once-daily treatment with dasotraline 4 and 8 mg/day or placebo. The primary efficacy end point was change from baseline at week 4 in the ADHD Rating Scale, Version IV (ADHD RS-IV) total score. Secondary efficacy end points included the Clinical Global Impression, Severity (CGI-S) scale, modified for ADHD symptoms. Least squares (LS) mean improvements at week 4 in ADHD RS-IV total score were significantly greater for dasotraline 8 mg/day vs placebo (−13.9 vs −9.7; P=0.019), and nonsignificantly greater for 4 mg/day (−12.4; P=0.076). The LS mean improvements in modified CGI-S were significantly greater at week 4 for dasotraline 8 mg/day vs placebo (−1.1 vs −0.7; P=0.013), and for 4 mg/day vs placebo (−1.1 vs −0.7; P=0.021). The most frequent adverse events reported were insomnia, decreased appetite, nausea, and dry mouth. Discontinuations due to treatment-emergent adverse events were 10.3% and 27.8% of patients in 4 and 8 mg/day treatment groups, respectively. This study provides preliminary evidence that once-daily dosing with dasotraline, a long-acting, dual monoamine reuptake inhibitor, may be a safe and efficacious treatment for adult ADHD.
Similar content being viewed by others

INTRODUCTION
Attention-deficit/hyperactivity disorder (ADHD) is a neurodevelopmental disorder characterized by persistent symptoms of hyperactivity (eg, restlessness, impulsiveness) and inattention (eg, distractibility, forgetfulness) associated with clinically significant impairment in social, academic, and occupational functioning (American Psychiatric Association, 2013). ADHD affects 5–7% of children (based on DSM-IV-TR criteria; Polanczyk et al, 2014), and frequently persists into adulthood, with prevalence estimates of 4% in adults (Faraone and Biederman, 2005; Kessler et al, 2006).
Dopamine (DA) and norepinephrine (NE) have been implicated in the pathophysiology of ADHD, and drugs that increase DA and NE neurotransmission are clinically useful in ADHD symptom management (Faraone et al, 2005).
Several classes of drugs have demonstrated efficacy in the treatment of ADHD, including short- and long-acting stimulant medications (eg, methylphenidate, amphetamine), and nonstimulant medications (eg, atomoxetine, clonidine, guanfacine; Faraone and Glatt, 2010; McDonagh et al, 2011). Currently available treatments for ADHD tend to produce high rates of adverse CNS and cardiovascular and gastrointestinal effects, with abuse liability a risk for the stimulant class of medications. Therefore, there is a need for additional treatment options that may offer reduced abuse liability and a different tolerability profile.
Dasotraline [(1R,4S)-4-(3,4-Dichlorophenyl)-1,2,3,4-tetrahydronaphthalen-1-amine] is a novel compound with DNRI pharmacology. Dasotraline acts as a potent inhibitor of human DA transporters (DAT; dopamine uptake IC50 3 nM) and NE transporters (NET; norepinephrine uptake IC50 4 nM), and a weaker inhibitor of human serotonin transporters (SERT; serotonin uptake IC50 15 nM) (Data on file, Sunovion, 2014).
In a behavioral paradigm of ADHD in rats (delay discounting test), administration of dasotraline significantly increased percent choice of delayed reward, a measure of impulsive choice (Data on file, Sunovion, 2014). The effects were similar to those observed with a methylphenidate control.
In humans, dasotraline has a tmax of 10–12 h, a  of 47–77 h, achieves steady-state plasma concentration by 2 weeks of daily dosing, and is metabolized by oxidative pathways (Data on file, Sunovion, 2014).
of 47–77 h, achieves steady-state plasma concentration by 2 weeks of daily dosing, and is metabolized by oxidative pathways (Data on file, Sunovion, 2014).
The pharmacokinetic and pharmacodynamic characteristics of dasotraline suggest that it may have a favorable pharmacologic profile for the treatment of ADHD, including sustained treatment effects throughout a once-per-day 24 h dosing interval, and a reduced risk for abuse and diversion, thus providing an enhanced treatment approach for patients with ADHD (Minzenberg (2012)).
The primary objective of this study was to evaluate the efficacy of dasotraline for the treatment of adult patients with ADHD. Secondary objectives were to evaluate the safety and tolerability of dasotraline, and to assess the relationship between the plasma concentrations of dasotraline, improvement in efficacy measures, and plasma concentrations of 3,4-dihydroxyphenylglycol (DHPG).
MATERIALS AND METHODS
Patients
The trial enrolled adult outpatients, ages 18–55 years, inclusive, with a primary diagnosis of ADHD based on DSM-IV-TR criteria, and confirmed by Conners’ Adult ADHD Diagnostic Interview (CAADID; Epstein et al, 2001) Part 2. In addition, patients were required to have been prescribed at least one previous trial of medication (stimulant or nonstimulant). Patients were also required to have a minimum ADHD RS-IV (with adult prompts) total score of ≥26 at Screen (if receiving no current treatment) and Baseline, or ≥22 (if currently being treated for ADHD); an ADHD RS-IV total score of ≥26 at Baseline for all patients; and a Clinical Global Impression, Severity (CGI-S; modified to evaluate overall severity of ADHD symptoms) score of ≥4 at Screen and Baseline (moderate-or-greater symptom severity). Reasons for exclusion included the following: any active medical condition or any clinically significant abnormality on physical examination, laboratory testing, or ECG that could interfere with appropriate participation in the study; any history (based on DSM-IV-TR criteria using the Mini International Neuropsychiatric Interview; Sheehan et al, 1998), of bipolar disorder, schizophrenia, or other psychotic disorder; current Axis II personality disorder; history in the past 12 months of substance abuse or dependence, or positive breath alcohol test or urine drug screen at the Screen visit. Potential patients were also excluded if they had received treatment in the previous 6 months with lithium, or anticonvulsant or antipsychotic medication; or were currently being treated with any antidepressant, stimulant, sedating antihistamine medication, or an α-2 adrenergic receptor agonist (including clonidine and guanfacine).
The study was approved by an institutional review board at each investigational site and was conducted in accordance with the International Conference on Harmonisation Good Clinical Practices guidelines and with the ethical principles of the Declaration of Helsinki. Before study entry, all patients reviewed and signed an informed consent document explaining study procedures and potential risks. An independent data and safety monitoring board reviewed and monitored patient data throughout the study.
Study Design
This multicenter, randomized, double-blind, placebo-controlled, parallel-group study of dasotraline in adults with ADHD was conducted between December 2012 and November 2013 at 30 sites in the United States. The study consisted of 3 periods, a screening period of no more than 27 days, a double-blind 4-week treatment period, and a 2 week washout period. During the screening period, psychiatric, medical, and laboratory screening tests were completed, and any current medication for ADHD was discontinued (for a minimum of 5 half-lives, or 3 days, before randomization, whichever was longer). Patients who continued to meet all study inclusion and exclusion criteria at baseline were randomized in a 1 : 1 : 1 ratio via an interactive response system (IXRS) to receive 4 weeks of double-blind treatment with dasotraline in a fixed daily dose of either 4 or 8 mg or placebo. Study medication was provided in blister packs of either dasotraline 2 mg, or identically matched placebo capsules, and was taken once daily at approximately the same time in the evening, without regard to meals. At the end of 4 weeks of double-blind treatment, study medication was abruptly discontinued, and patients returned for weekly assessment visits during a 2-week washout period.
Dose Selection
Dasotraline at 4 and 8 mg/day were selected to target steady-state plasma concentrations of 8 and 18 ng/ml, and were estimated to correspond with DAT receptor occupancies of 56% and 71%, respectively, based on the results of a previous PET study with dasotraline (DeLorenzo et al, 2011). DAT receptor occupancies above 50% have been associated with clinically therapeutic effects for methylphenidate (Hannestad et al, 2010) and atomoxetine (Ding et al, 2014).
Concomitant Medications
Concomitant use of zolpidem, zaleplon, and eszopiclone were permitted on ≤3 nights per week during the study for the management of treatment-emergent insomnia, but not prophylactically, and not within 8 h of any psychiatric assessment. Patients treated for hypertension with stable doses of β-adrenergic receptor antagonists were permitted to continued treatment.
Efficacy Assessments
The primary efficacy measure was the ADHD Rating Scale, Version IV (ADHD RS-IV, with adult prompts; Adler et al, 2008), an 18-item scale based on DSM-IV-TR criteria that consists of a total score, and Hyperactivity/Impulsivity and Inattentiveness subscale scores. Additional efficacy measures included the Clinical Global Impression-Severity of Illness that was modified to evaluate the overall severity of ADHD symptoms (National Institute of Mental Health (NIMH), 1985), the Wender-Reimherr Adult Attention Deficit Disorder Scale (WRAADDS; Marchant et al, 2013) that measures the severity of ADHD symptoms in adults, and the 24-item Brief Psychiatric Rating Scale that assesses the presence and severity of psychotic symptoms (Guy, 1976). Efficacy assessments were obtained at baseline and weekly intervals.
Cognitive function was evaluated at baseline, week 2, and week 4 using a computerized cognitive assessment battery (Cognitive Drug Research (CDR); Wesnes, 2005) that measured attention, working memory, and episodic memory.
Safety Assessments
Safety and tolerability were assessed by physical examinations, 12-lead ECGs, vital signs, hematology, chemistry, urinalysis, and monitoring of adverse events. The severity of insomnia and insomnia-related interference with daytime functioning was assessed using the 7-item Insomnia Severity Index (ISI; Bastien et al, 2001); risk of drug withdrawal was evaluated using the 20-item Physician Withdrawal Checklist (PWC; Rickels et al, 2008); an Abuse Potential Monitoring Plan was implemented that included monitoring of sentinel adverse events and medication handling irregularities. Potential treatment-emergent suicidal ideation was monitored using the Columbia Suicide Severity Rating Scale (C-SSRS; Posner et al, 2011). Acute subjective response to study drug (drug liking) was monitored weekly using the 3-item Drug Effects Questionnaire (DEQ; Morean et al, 2013).
Blood draws for dasotraline plasma concentrations and DHPG/NE plasma levels were collected weekly. Plasma concentrations for dasotraline, DHPG, and NE were determined using a validated bioanalytical method via HPLC with MS/MS detection. Interassay coefficient of variation ranged from 2.0 to 2.7%; lower limit of quantitation (LLOQ) was 10.0 pg/ml.
Statistical Analysis
The primary efficacy variable was the change from baseline in ADHD RS-IV total score at week 4 in the dasotraline 4 and 8 mg groups compared with placebo. A mixed model for repeated measures (MMRM) was employed using the modified intent-to-treat (ITT) population. The MMRM model included treatment, visit (as a categorical variable), pooled center, baseline ADHD RS-IV total score, and treatment-by-visit interaction. An unstructured covariance matrix was used for the within-subject correlation. The Kenward–Roger approximation was used to estimate denominator degrees of freedom. The primary efficacy comparisons of only dasotraline 4 and 8 mg vs placebo at end point (week 4) were adjusted using the Hochberg procedure. Missing values were not imputed for this MMRM analysis. The MMRM assumed that data were missing at random (MAR). A random effects pattern mixture model with two patterns (completers and dropouts) demonstrated that MAR was a reasonable assumption for the primary efficacy end point. Secondary efficacy variables (change from baseline at each study week in the CGI-S, WRAADDS, ADHD RS-IV Inattentiveness, and Hyperactivity/Impulsivity subscale scores) were analyzed using the same MMRM method.
Analysis of responder rates (defined as ≥30% reduction in ADHD RS-IV-total score) was performed using a logistic regression model that included baseline ADHD RS-IV total score, pooled center, and treatment, and was done for both observed case (OC) and last observation carried forward (LOCF) values at week 4.
Based on a clinically relevant improvement in the ADHD RS-IV total score reported in a meta-analysis of acute clinical trials (McGough and Faraone, 2009), it was estimated that a sample size of 85 patients per treatment group would be needed to provide 83% power for significant comparisons of both dasotraline dosages (vs placebo), or 94% power for at least one significant comparison. A prespecified upward sample-size adjustment was based on an estimated study attrition of 15%; the sample size was further increased to 112 per treatment group based on discontinuations observed during the course of the study.
Pharmacokinetic analyses
Observed values of plasma concentrations for dasotraline and DHPG were summarized descriptively at each visit by treatment group in both the PK and PD populations. The relationship between selected efficacy variables and dasotraline plasma concentrations and DHPG concentrations was analyzed using population PK/PD methods and will be reported separately.
RESULTS
Patients and Disposition
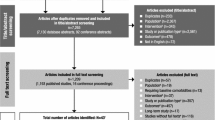
A total of 341 patients were randomized to 4-week double-blind treatment (Figure 1); 337 received at least one dose of study medication (safety population), and 331 had both baseline and at least one postbaseline efficacy assessment (modified ITT population). Baseline demographic and clinical characteristics were similar across the three treatment groups (Table 1). The majority of patients were male (59%), with a mean age of 34 years.

Patient disposition.
The 4-week treatment completion rates were 90.9% for the placebo group, 82.8% for dasotraline 4 mg/day group, and 51.3% for dasotraline 8 mg/day group (Figure 1). The discontinuation rates due to adverse events were 10.3% for the dasotraline 4 mg/day group, 27.8% for the 8 mg/day group, and 1.8% for the placebo group.
Efficacy
The least squares (LS) mean improvement from baseline to week 4 in the ADHD RS-IV total score (the primary end point) was significantly greater for the dasotraline 8 mg/day group vs the placebo group (−13.9 vs −9.7; P=0.019; effect size=0.41), and nonsignificantly greater for the 4 mg/day group (−12.4; P=0.076, effect size=0.25; Table 2). Both the 4 and 8 mg/day doses showed numerical advantages compared with placebo beginning at week 2 (Figure 2).

LS mean change in ADHD RS-IV total scores (MMRM).
Improvement from baseline in both the ADHD RS-IV Hyperactivity/Impulsivity and Inattentiveness subscale scores were significantly greater for the dasotraline 8 mg/day group, and nonsignificantly greater for the 4 mg/day group (Table 2).
Treatment with 8 mg/day dasotraline was associated with significantly greater week 4 responder rates (≥30% reduction in ADHD RS-IV total score; LOCF) vs the placebo group (52.3% vs 38.2%; P=0.029; NNT=8). Treatment with 4 mg/day dasotraline was associated with nonsignificantly higher week 4 responder rates vs the placebo group (49.1% vs 38.2%; P=0.109; NNT=10).
Improvement from baseline in the modified CGI-S scale was significantly greater at week 4 for both the dasotraline 4 and 8 mg/day dosage groups (Table 2). Improvement from baseline in the WRAADDS total score was nonsignificantly greater at week 4 for both the dasotraline 4 and 8 mg/day dosage groups (Table 2). On the WRAADDS attention difficulties subscore, LS mean improvement from baseline was significantly greater at week 4 for the dasotraline 8 mg/day group vs the placebo group (−1.3 vs −0.8; P=0.008), and for the dasotraline 4 mg/day group vs the placebo group (−1.1 vs −0.8; P=0.025). On the WRAADDS disorganization subscore, LS mean improvement from baseline was significantly greater at week 4 for the dasotraline 8 mg/day group vs the placebo group (−1.0 vs −0.8; P=0.037); and was nonsignificantly greater for the dasotraline 4 mg/day group vs the placebo group (−1.0 vs −0.8; P=0.056).
On the computerized cognitive assessment battery, no significant main effects for dasotraline were observed for measures of attention, working memory, or episodic memory.
Mean Brief Psychiatric Rating Scale (BPRS) scores were relatively low at baseline for all three study treatment groups (31.3–31.7), and did not worsen at week 4 in the dasotraline 4 mg/day (−1.3), 8 mg/day (−1.6), and placebo (−1.8) groups.
Safety
Treatment-emergent adverse events (TEAEs) are summarized in Table 3. The percentages of patients with TEAEs were similar between the 4 and 8 mg dasotraline groups (77.6% and 81.1%, respectively) and were higher than the percentage of TEAEs in the placebo group (59.1%). Insomnia, decreased appetite, and dry mouth were the most frequent adverse events. The majority of adverse events were rated as mild or moderate; the incidence of events rated as ‘severe’ was 13.5% in the dasotraline 8 mg/day group, 6.0% in the dasotraline 4 mg/day group, and 2.7% in the placebo group. The most common adverse events leading to discontinuation (and occurring in ≥2 patients) in the dasotraline 4 and 8 mg/day and placebo groups, respectively, were insomnia (2.6, 10.8, and 0%), anxiety (2.6, 1.8, and 0%), and panic attack (0, 2.7, and 0%). Overall, discontinuations due to adverse events were 10.3%, 27.8%, and 1.8% in the dasotraline 4 and 8 mg/day and placebo groups respectively.
There were 5 serious TEAEs that occurred in 4 patients: 2 patients in the dasotraline 4 mg/day group (ectopic pregnancy and psychotic disorder) and 2 patients in the dasotraline 8 mg/day group (mediastinitis and bacteremia in one patient and adjustment disorder with depressed mood). The SAEs were reported as unrelated to study drug, except for psychotic disorder that was possibly related.
Insomnia severity index
Small LS mean changes were observed in the ISI total score for each dasotraline treatment group from week 1 (4 mg, +0.89; 8 mg, +2.17; placebo, −1.53) to week 4 (4 mg, +0.34; 8 mg, +0.99; placebo, −2.48).
Drug liking and withdrawal
No evidence of drug liking was observed on the DEQ, with mean item scores remaining within 5 mm of the 0 point at all assessment weeks. No evidence of drug misuse or diversion was detected through the Abuse Potential Monitoring Plan. No signs or symptoms of withdrawal were observed upon discontinuation of study drug, with a mean maximum change on the PWC of −1.3 in the dasotraline 4 mg/day group, −0.5 in the dasotraline 8 mg/day group, and −0.3 in the placebo group from week 4 to week 6.
Suicidal ideation
On the C-SSRS, three patients (one in each treatment group) reported mild, treatment-emergent suicidal ideation, but with no plan.
Weight
The mean change from baseline to week 4 in weight was −1.07 kg for the 4 mg/day dasotraline group, −2.90 kg for the 8 mg/day dasotraline group, and +0.24 kg for the placebo group.
Vital signs and ECG
Mean change from baseline to week 4 in supine heart rate was +5.7, +6.2, and −1.2 b.p.m., respectively, for the dasotraline 4 and 8 mg/day groups and the placebo group. Mean change from baseline to week 4 in supine systolic/diastolic blood pressure was −0.2/+0.3, +3.3/+2.5 mm Hg, and +0.4/−0.5 mm Hg, respectively, for the dasotraline 4 and 8 mg/day groups and the placebo group. On the ECG, minimal mean changes were observed at week 4 in QTcF of +1.1, +1.6, and −0.2 ms, respectively, for the dasotraline 4 and 8 mg/day groups and the placebo group. No patients treated with dasotraline had clinically significant increases in QTcF duration (>60 ms).
Laboratory
Laboratory measures did not identify any safety concerns for dasotraline.
Plasma Concentrations of Dasotraline and DHPG
For the 4 and 8 mg/day doses, mean dasotraline plasma levels increased during the first 2 weeks of treatment, and then plateaued at weeks 3 and 4 (Figure 3a). Plasma concentrations of DHPG decreased in the first week of treatment for both the 4 and 8 mg/day doses of dasotraline, and then plateaued at subsequent weeks (Figure 3b).

Mean (95% CI) plasma concentrations for dasotraline (a) and DHPG (b).
DISCUSSION
We report here the first placebo-controlled efficacy trial of dasotraline in adult patients with ADHD. In this 4-week, fixed-dose study, 8 mg/day dasotraline significantly improved symptoms of ADHD assessed by the ADHD RS-IV total score (the primary efficacy end point), and the ADHD RS-IV hyperactivity/impulsivity and inattentiveness subscale scores. Dasotraline at 4 mg/day was associated with trend-significant improvement on the ADHD RS-IV total and subscale scores. Dasotraline significantly improved global ADHD symptom severity, assessed by the CGI-S scale (modified for ADHD), at both doses studied. Treatment response (at least 30% improvement on the ADHD RS-IV total score) favored dasotraline vs placebo with NNT values of 8–10.
For the 8 mg/day dose, a trend separation from placebo on the ADHD RS-IV total score was apparent by week 2; this became significant at weeks 3 and 4. The dasotraline 4 mg/day dose showed a similar time course of response on the ADHD RS-IV total score, with numeric separation observed by week 2 and at all subsequent study visits. The effect sizes for the dasotraline 4 mg/day (0.25) and 8 mg/day (0.41) doses observed in this study at week 4 overlapped with the range and 95% confidence intervals of those reported in meta-analyses of the DAT/NET inhibitor methylphenidate (Faraone et al, 2004; Koesters et al, 2009), but were smaller than the effect sizes reported for amphetamine-based medications (Faraone and Glatt, 2010). Given the prolonged elimination half-life of dasotraline, requiring ∼2 weeks to achieve steady state, it is possible that a longer duration of study treatment (beyond 4 weeks) could have provided additional therapeutic benefit.
The pharmacokinetics and pharmacology of dasotraline differentiate it from other agents that have demonstrated efficacy for the treatment of ADHD. In contrast to the pharmacokinetics of amphetamine, methylphenidate, and atomoxetine, dasotraline has slow absorption and a long-elimination half-life resulting in stable plasma concentrations of over 24 h with once-daily dosing. Pharmacologically, dasotraline is a dual dopamine and norepinephrine reuptake inhibitor (DNRI), unlike psychostimulants such as amphetamine that facilitate the direct release of dopamine and norepinephrine (Rothman et al, 2001), and unlike atomoxetine, a nonstimulant that inhibits only norepinephrine transporters (Wong et al, 1982). In this study, dasotraline at 4 mg/day resulted in mean concentrations at steady state of 6 ng/ml. A prior human PET study demonstrated that dasotraline plasma concentrations of 4.5 ng/ml were associated with 50% DAT occupancy, with no significant serotonin transporter occupancy (DeLorenzo et al, 2011). Dasotraline at 4 and 8 mg/day also decreased circulating DHPG levels, indicative of central inhibition of norepinephrine transporters (Vincent et al, 2004).
The results of this study support the concept that symptoms of ADHD may be improved by providing constant, steady-state inhibition of DA and NE reuptake. This pharmacologic profile suggests the potential for sustained treatment effects throughout the 24 h dosing interval, and contrasts with the peak and trough pattern of effects within the dosing interval induced by current ADHD medications, attributable to either their faster pharmacodynamics (methylphenidate medications) or their facilitated release of dopamine (amphetamine-based medications).
The most frequent adverse events reported for dasotraline in this study were insomnia, decreased appetite, and dry mouth, consistent with DNRI pharmacology. The majority of adverse events were transient, and mild to moderate in severity. Insomnia occurred in 35% of patients at the 4 mg/day dose, and in 45% of patients at the 8 mg/day dose. Insomnia led to study discontinuation in a smaller proportion of patients, 2.6% for the 4 mg/day dose and 10.8% for the 8 mg/day dose. The rates of insomnia at the 4 mg/day dose were comparable to rates observed for stimulant drugs such as methylphenidate (Peterson et al, 2008), whereas higher rates were observed for the 8 mg/day dose. Rates of insomnia were also higher for dasotraline compared with atomoxetine (Stattera US Prescribing Information (2015)), whereas rates of nausea were lower in this study on dasotraline (10% at 8 mg/day vs 30%; Stattera US Prescribing Information (2015)). The pharmacokinetics of dasotraline, with slow onset to peak effect and prolonged elimination half-life, may have contributed to the reduced incidence of nausea, while increasing the incidence of insomnia. Future studies will investigate whether gradual titration to higher doses, and use of flexible dosing, will be associated with reduced rates of insomnia or other DNRI-related adverse events.
In this study, no evidence of drug liking was observed on the DEQ for either dose of dasotraline, nor was any drug misuse or diversion detected through the study Abuse Potential Monitoring Plan. In addition, no signs or symptoms of withdrawal were observed on the PWC for either dose of dasotraline. Delayed absorption and/or maintenance of steady plasma concentrations is known to reduce the abuse potential of stimulant drugs (Volkow et al, 1995; Kollins et al, 1998; Spencer et al, 2012). For example, tesofensine, a drug with slow absorption and elimination rates in humans, and substantial DAT occupancy (Appel et al, 2014), demonstrated low abuse potential comparable to atomoxetine and bupropion in a study of recreational stimulant users (Schoedel et al, 2010). Dasotraline has been evaluated at single doses of up to 36 mg in a randomized, double-blind, crossover study of recreational stimulant users, and was found to have abuse potential no greater than placebo and significantly less than that of methylphenidate (data on file). These findings are consistent with the delayed time to peak serum concentration and prolonged elimination half-life of dasotraline.
Monitoring of vital signs and laboratory parameters did not identify any clinically significant safety concerns for dasotraline. Small increases were noted in mean heart rate and, for the 8 mg/day dose only, in mean systolic and diastolic blood pressure. Dasotraline had no notable effect on QTc or other ECG parameters. No increased risk of suicidal ideation was noted on the C-SSRS for either dasotraline dose group compared with placebo.
The limitations of this study include, as noted above, the short (4 weeks) treatment period, the lack of titration in the higher dose treatment arm, and the requirement that all patients in the study have a history of medication treatment for ADHD. The latter requirement may have biased the treatment population by increasing recruitment of ADHD patients with higher levels of chronicity and/or current stimulant use. In fact, plasma samples collected during study treatment identified several patients with plasma concentrations of amphetamine or methylphenidate in the placebo treatment group (n=14; 13.5%) and in the dasotraline treatment group (4 mg/day, n=7, 6.1%; 8 mg/day, n=9, 8.4%). All efficacy and safety data from these patients were included in the analyses and results reported here; exclusion of these subjects from the primary efficacy analysis did not change the results.
In conclusion, the results of this fixed-dose, placebo-controlled, proof-of-concept trial found once-daily dosing with dasotraline, a novel, long-acting dopamine and norepinephrine reuptake inhibitor, to be an efficacious treatment of ADHD in adults. Dasotraline was generally well tolerated. Higher rates of insomnia were observed at 8 mg/day compared with 4 mg/day in this fixed-dose study where patients were initiated at these doses without titration. Further evaluation of the clinical utility of dasotraline in ADHD is warranted.
FUNDING AND DISCLOSURE
Drs Koblan, Hopkins, Sarma, Jin, Goldman, and Loebel are employees of Sunovion Pharmaceuticals. Dr Kollins, in the past 3 years, has received research support and/or consulting fees from the following sources: Akili Interactive, Alcobra Pharmaceuticals, Atentiv, Ironshore Pharmaceuticals, Arbor Pharmaceuticals, NEOS Pharmaceuticals, Purdue Canada, Rhodes Pharmaceuticals, Shire Pharmaceuticals, Sunovion Pharmaceuticals, Tris Pharma, the National Institutes of Health (NIDA, NIEHS), the Center for Scientific Review, and the Environmental Protection Agency.
References
Adler LA, Faraone SV, Spencer TJ, Michelson D, Reimherr FW, Glatt SJ et al (2008). The reliability and validity of self- and investigator ratings of ADHD in adults. J Atten Disord 11: 711–719.
American Psychiatric Association (2013) Diagnostic and Statistical Manual of Mental Disorders. 5th edn. Washington, DC.
Appel L, Bergström M, Buus Lassen J, Långström B (2014). Tesofensine, a novel triple monoamine re-uptake inhibitor with anti-obesity effects: dopamine transporter occupancy as measured by PET. Eur Neuropsychopharmacol 24: 251–261.
Bastien CH, Vallières A, Morin CM (2001). Validation of the Insomnia Severity Index as an outcome measure for insomnia research. Sleep Med 2: 297–307.
DeLorenzo C, Lichenstein S, Schaefer K, Dunn J, Marshall R, Organisak L et al (2011). SEP-225289 serotonin and dopamine transporter occupancy: a PET study. J Nucl Med 52: 1150–1155.
Ding YS, Naganawa M, Gallezot JD, Nabulsi N, Lin SF, Ropchan J et al (2014). Clinical doses of atomoxetine significantly occupy both norepinephrine and serotonin transports: Implications on treatment of depression and ADHD. Neuroimage 86: 164–171.
Epstein JN, Johnson DE, Conners CK (2001) Conners’ Adult ADHD Diagnostic Interview for DSM-IV. Multi-Health Systems: North Tonawanda, NY.
Faraone SV, Biederman J (2005). What is the prevalence of adult ADHD? Results of a population screen of 966 adults. J Attention Disord 9: 384–391.
Faraone SV, Glatt SJ (2010). A comparison of the efficacy of medications for adult attention-deficit/ hyperactivity disorder using meta-analysis of effect sizes. J Clin Psychiatry 71: 754–763.
Faraone SV, Perlis RH, Doyle AE, Smoller JW, Goralnick JJ, Holmgren MA et al (2005). Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry 57: 1313–1323.
Faraone SV, Spencer T, Aleardi M, Pagano C, Biederman J (2004). Meta-analysis of the efficacy of methylphenidate for treating adult attention-deficit/hyperactivity disorder. J Clin Psychopharmacol 24: 24–29.
Guy W (1976) ECDEU Assessment Manual for Psychopharmacology, Revised. US Department of Health, Education, and Welfare Pub. No. (ADM). National Institute of Mental Health: Rockville, MD, pp 76–338.
Hannestad J, Gallezot JD, Planeta-Wilson B, Lin SF, Williams WA, van Dyck CH et al (2010). Clinically relevant doses of methylphenidate significantly occupy norepinephrine transporters in humans in vivo. Biol Psychiatry 68: 854–860.
Kessler RC, Adler L, Barkley R, Biederman J, Conners CK, Demler O et al (2006). The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry 163: 716–723.
Koesters M, Becker T, Kilian R, Fegert JM, Weinmann S (2009). Limits of meta-analysis: methylphenidate in the treatment of adult attention-deficit hyperactivity disorder. J Psychopharmacol 23: 733–744.
Kollins SH, Rush CR, Pazzaglia PJ, Ali JA (1998). Comparison of acute behavioral effects of sustained-release and immediate-release methylphenidate. Exp Clin Psychopharmacol 6: 367–374.
Marchant BK, Reimherr FW, Robison D, Robison RJ, Wender PH (2013). Psychometric properties of the Wender-Reimherr Adult Attention Deficit Disorder Scale. Psychol Assess 25: 942–950.
McDonagh MS, Peterson K, Thakurta S, Low A (2011) Drug class review: Pharmacologic treatments for attention deficit hyperactivity disorder. Update 4 final report. Prepared by the Oregon Evidence-based Practice Center for the Drug Effectiveness Review Project. Oregon Health & Science University: Portland, OR.
McGough JJ, Faraone SV (2009). Estimating the size of treatment effects: moving beyond p values. Psychiatry (Edgmont) 6: 21–29.
Minzenberg MJ (2012). Pharmacotherapy for attention-deficit/hyperactivity disorder: from cells to circuits. Neurotherapeutics 9: 610–621.
Morean ME, de Wit H, King AC, Sofuoglu M, Rueger SY, O'Malley SS (2013). The drug effects questionnaire: psychometric support across three drug types. Psychopharmacology (Berl) 227: 177–192.
National Institute of Mental Health (NIMH) (1985). CGI (Clinical Global Impression Scale)–NIMH. Psychopharmacol Bull 21: 839–843.
Peterson K, McDonagh MS, Fu R (2008). Comparative benefits and harms of competing medications for adults with attention-deficit hyperactivity disorder: a systematic review and indirect comparison meta-analysis. Psychopharmacology (Berl) 197: 1–11.
Polanczyk GV, Willcutt EG, Salum GA, Kieling C, Rohde LA (2014). ADHD prevalence estimates across three decades: an updated systematic review and meta-regression analysis. Int J Epidemiol 43: 434–442.
Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA et al (2011). The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 168: 1266–1277.
Rickels K, Garcia-Espana F, Mandos LA, Case GW (2008). Physician Withdrawal Checklist (PWC-20). J Clin Psychopharmacol 28: 447–451.
Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI et al (2001). Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 39: 32–41.
Schoedel KA, Meier D, Chakraborty B, Manniche PM, Sellers EM (2010). Subjective and objective effects of the novel triple reuptake inhibitor tesofensine in recreational stimulant users. Clin Pharmacol Ther 88: 69–78.
Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E et al (1998). The Mini-International Neuropsychiatric Interview (MINI): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry 59: 22–33.
Spencer TJ, Biederman J, Martin JM, Moorehead TM, Mirto T, Clarke A et al (2012). Importance of pharmacokinetic profile and timing of coadministration of short- and long-acting formulations of methylphenidate on patterns of subjective responses and abuse potential. Postgrad Med 124: 166–173.
Stattera US Prescribing Information (2015). Accessed on 23 January 2015 at http://pi.lilly.com/us/strattera-pi.pdf.
Vincent S, Bieck PR, Garland EM, Loghin C, Bymaster FP, Black BK et al (2004). Clinical assessment of norepinephrine transporter blockade through biochemical and pharmacological profiles. Circulation 109: 3202–3207.
Volkow ND, Ding YS, Fowler JS, Wang GJ, Logan J, Gatley JS et al (1995). Is methylphenidate like cocaine? Studies on their pharmacokinetics and distribution in the human brain. Arch Gen Psychiatry 52: 456–463.
Wender PH (1995) Attention-Deficit Hyperactivity Disorder in Adults. Oxford University Press: New York, NY.
Wesnes K (2005). The Cognitive Drug Research computerised assessment system: application to clinical trials. Memory: Basic Concepts, Disorders and Treatment. In: De Deyn P, Thiery E, D’Hooge R (eds). ACCO, pp 453–472.
Wong DT, Threlkeld PG, Best KL, Bymaster FP (1982). A new inhibitor of norepinephrine uptake devoid of affinity for receptors in rat brain. J Pharmacol Exp Ther 222: 61–65.
Acknowledgements
We thank the participants of this study and the study investigators involved in the conduct of this trial. Editorial assistance with manuscript preparation was provided by Dr Edward Schweizer of Paladin Consulting Group, and paid for by Sunovion Pharmaceuticals. The authors are entirely responsible for the scientific content of the paper. This study was sponsored by Sunovion Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Koblan, K., Hopkins, S., Sarma, K. et al. Dasotraline for the Treatment of Attention-Deficit/Hyperactivity Disorder: A Randomized, Double-Blind, Placebo-Controlled, Proof-of-Concept Trial in Adults. Neuropsychopharmacol 40, 2745–2752 (2015). https://doi.org/10.1038/npp.2015.124
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.124
- Springer Nature Switzerland AG
This article is cited by
-
What pharmacological interventions are effective in binge-eating disorder? Insights from a critical evaluation of the evidence from clinical trials
International Journal of Obesity (2022)
-
Lifetime evolution of ADHD treatment
Journal of Neural Transmission (2021)
-
Genome mining and characterisation of a novel transaminase with remote stereoselectivity
Scientific Reports (2019)
-
Treatment strategies for ADHD: an evidence-based guide to select optimal treatment
Molecular Psychiatry (2019)
-
Progress in Developing Pharmacologic Agents to Treat Bulimia Nervosa
CNS Drugs (2019)