
Abstract
Background
Hantzsch 1,4-dihydropyridines (Hantzsch1,4-DHP) have been extensively utilized as the analogs of nicotinamide adenine dinucleotide (NADH) coenzyme to study the mechanism and various redox processes. During the redox processes 1,4-DHP systems undergo transformation into the corresponding pyridine derivatives through oxidation. Consequently, the interest in this aromatization reaction, investigation of a wide range of 1, 4-DHPs continues to attract the attention of researchers. Herein, we report the preparation of pyridine derivatives and the crystal structures determined by X-ray crystallographic methods.
Results
The crystal structures and conformational studies of two organic compounds, namely ethyl 2-methyl-4-phenyl-5-oxo-5H-indeno [1,2-b] pyridine-3-carboxylate (I) and ethyl 2-methyl-4-(4 chlorophenyl)-5-oxo-5H-indeno [1,2-b] pyridine-3-carboxylate (II) are reported. The terminal ethyl group of the compound I is disordered over two positions with the refined occupancies of 0.645 & 0.355 and C8 one dimensional zig-zag chain running along 101 direction through C-H…O type of intermolecular interactions. In the compound II, C-H…O interactions connect the molecules to form an R22 (16) dimer running along 011 direction.
Conclusion
The crystal structures ethyl 2-methyl-4-phenyl-5-oxo-5H-indeno [1,2-b] pyridine-3-carboxylate and ethyl 2-methyl-4-(4 chlorophenyl)-5-oxo-5H-indeno [1,2-b] pyridine-3-carboxylate have been investigated in detail. The terminal ethyl group of compound I is disordered. In compound II, the substitution of Cl atom in the phenyl ring alters the configuration of carboxylate group with respect to the pyridine indane ring.

Similar content being viewed by others


Background
Hantzsch 1,4-dihydropyridines (Hantzsch1,4-DHP) have been extensively utilized as the analogs of nicotinamide adenine dinucleotide (NADH) coenzyme to study the mechanism and the synthetic potential of various redox processes [1, 2]. Hantzsch 1,4-DHP based drugs such as nifedipine and niguldipine are widely used as calcium channel blockers for the treatment of cardiovascular disorders including angina, hypertension and cardiac arrhythmias [3]. During the redox processes and in the course of drug metabolism [4], 1,4-DHP systems are oxidatively transformed into the corresponding pyridine derivatives. Consequently, this aromatization reaction continues to attract the attention of researchers to establish a general protocol applicable to a wide range of 1,4-dihydropyridines. A number of methods and reagents have been reported recently in the literature for this purpose [5–14].
Some of these methods suffer from disadvantages such as the use of strong or toxic oxidants, the requirement of severe conditions or need excess of the oxidants. Other drawbacks are the long reaction times, production of by-products, the lower yields of products and/or the requirement of tedious work-up procedures.
N-Bromosuccinimide (NBS) is a versatile reagent for the oxidation of primary and secondary alcohols, α-hydroxycarboxylic acids [15], α-hydroxycarboxylic esters [16], hydrazines and hydrazones [15]. In addition, NBS is preferred for allylic bromination. While hydroxy acids like malic acid, tartaric acid, citric acid etc. are converted to aldehydes and ketones, polyhydric alcohols (glycol, glycerol and hexitols) are quantitatively decomposed to carbon dioxide and water [17] with NBS. NBS also promotes reactions of sterically hindered cresols via p-benzoquinone methide [18].
Having synthesized a number of 1, 4-dihydropyridines derived from indane-1,3-dione, we have dehydrogenated them to the corresponding pyridines. The reagent of the choice for effecting dehydrogenation is NBS in methanol (Schemes 1 and 2). This reagent was earlier employed to effect dehydrogenation of simple dihydropyridines [19].

Synthesis scheme of the dihydropyridines.

Synthesis scheme of the compounds I and II.
Experimental
The title compounds reported in the present work were prepared by the following procedure [19, 20].
Preparation of 4a-b
To an alcoholic solution (50 mL) of indane-1,3-dione 2 (0.01 mol), appropriate aromatic aldehydes 1a-b (0.01 mol), ethyl acetoacetate 3 (0.01 mol), ammonium acetate (0.02 mol) and a drop of piperidine were added and the mixture was refluxed for 1 hr. The reaction mixture was concentrated to half of its original volume and allowed to cool in an ice-chest. The solid 4a-b thus separated was filtered, washed with ice cold aqueous ethanol and crystallized from petroleum ether (60–80°C)-chloroform (1: 1) (Scheme 1).
Preparation of 5a-b
To a solution of ethyl 2-methyl-4-aryl-5-oxo-1H,4H-indeno [1,2-b] dihydropyridine-3-carboxylate 4a-b (0.5 g, 1.87 mmol) in methanol (10.0 mL), N-bromosuccinimide (0.33 g, 1.87 mmol) was added and the reaction mixture was stirred at room temperature. The colour of the solution changes immediately and the reaction proceeds instantaneously within five minutes. The course of the reaction was monitored by TLC. The reaction mixture was diluted with water (50 mL) and extracted with chloroform (3 × 20 mL). The organic layer was separated, dried over anhydrous sodium sulfate and filtered (Scheme 2). Evaporation of the solvent afforded the products ethyl 2-methyl-4-phenyl-5-oxo-5H-indeno [1,2-b] pyridine-3-carboxylate (Scheme 3) or ethyl 2-methyl-4-(4-chlorophenyl)-5-oxo-5H-indeno [1,2-b] pyridine-3-carboxylate respectively in excellent yields (Scheme 4). For compound (5a): Yield 96%; M.p. 212˚C. For compound (5b): Yield 89%; M.p. 198˚C.

Scheme showing the structural formula of compound I.

Scheme showing the structural formula of compound II.
Results and Discussion
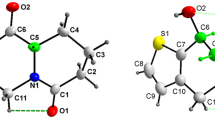
In both the compounds, the indenopyridine ring is almost planar, with r.m.s deviation of 0.035(2) Å [C3] and 0.087(2) Å [C11] for compounds I and II, respectively. The keto atom O substituted in the indenopyridine in both the molecules are slightly out of plane [0.048(2) & 0.217(1) Å for I & II]. The substitution of the Cl atom in the phenyl ring plays a vital role while packing the molecules in the unit cell and promotes the change of conformation of the carboxylate group. This is evidenced from the torsion angle values of [C10-C11-15-O2] and [C12-C11-C15-O2] 114.5(2)° & -63.5(2)° for (I) and −74.9(2)° & 114.0(1)° for (II), respectively. The terminal ethyl group in compound I is disordered over two positions with refined occupancies of 0.645 & 0.355. The phenyl ring and indenopyridine rings are oriented by an angle of 67.8(1)˚ in compound (I) which is almost similar in compound (II) amounting the value of 55.2(1)˚. The overall conformations in both the molecules are similar as can be seen from the superimposed rmsd value 0.154 Å (Figure 1). Both the structures are stabilized by C-H…O type of intra and intermolecular interactions. In compound I, molecules at (x, y, z) and (x + 1/2, −y − 1/2, z + 1/2) are linked through intermolecular C20-H20…O1 hydrogen bond to form a C8 zig-zag chain (Figure 2) running along 101 direction [21]. The combination of C5-H5…O3 and C22-H22…O1 intermolecular hydrogen bonds, lead to the formation of a R22 (16) ring motif chain running along [0 1 1] direction (Figure 3), observed in compound II.

The conformation of both the molecules, as seen from the superimposition of the planar indenopyridine rings.

Figure showing the intermolecular hydrogen bonds resulting in C8 zig-zag motif in compound (I).

Figure showing the intermolecular hydrogen bonds resulting in R 2 2 (16) ring motifs chain running along 0 1 1 direction in compound (II).
X-ray Crystallography
Single crystal X-ray intensity data for the compounds (I) and (II) were collected using a Bruker Kappa APEX II area-detector diffractometer with MoKα (0.71073 Å) radiation at room temperature (293 K). The data reduction was carried out using the program SAINT [22]. The absorption corrections were applied using the Multi-scan method using SADABS program [23]. The structures of both the compounds were solved by direct methods using SHELXS97 [24] and all the non-hydrogen atoms were refined anisotropically by full-matrix least-squares on F2 taking all the unique reflections using SHELXL97 [24]. The hydrogen attached with carbon atoms were placed in their calculated positions and included in the isotropic refinement using the riding model with C–H = 0.93 Å (−CH) or 0.97 Å (−CH2) Å or 0.96 Å (−CH3) Å with Uiso (H) = 1.2Ueq (parent C atom). The crystal data, experimental conditions and structure refinement parameters for the compounds (I) and (II) are presented in Table 1. Table 2 gives the geometry of the intra and intermolecular interactions. The molecular structure of compounds (I) and (II) with the atom numbering scheme using ORTEP3 [25] are given in Figure 4 and Figure 5, respectively. The least-squares plane, geometrical and puckering parameters of both the compounds were calculated using PLATON software package [26–28].

ORTEP plot of compound (I) showing with atoms ellipsoids are drawn at 40% probability level.

ORTEP plot of compound (II) showing with atoms ellipsoids drawn at 40% probability level.
Conclusions
The title compounds were synthesized, crystallized and the crystal structures have been determined by single-crystal X-ray diffraction methods. The terminal ethyl group of the compound I is disordered over two positions with the refined occupancies of 0.645 & 0.355. C-H…O intermolecular hydrogen bond builds up a one dimensional zig-zag chain running along 101 directions. In compound II, C-H…O hydrogen bonds connect the molecules to form a R22 (16) dimer chain running along 011 direction.
References
Stout DM, Meyers AI: Recent advances in the chemistry of dihydropyridines. Chem Rev. 1982, 82: 223-243. 10.1021/cr00048a004.
Kill RJ, Widdowson DA: Bioorganic Chemistry. Edited by: Van Tamelen EE. 1978, New York: Academic, 239-
Triggle DJ: Comprehensive Medicinal Chemistry. Edited by: Emmett JC. 1990, Oxford: Pergamon, Vol. 3, Ch. 14.1
Janis RA, Triggle DJ: New developments in Ca2+ channel antagonists. J Med Chem. 1983, 26: 775-785. 10.1021/jm00360a001.
Nasr-Esfahani M, Moghadam M, Tangestaninejad S, Mirkhani V: Biomimetic oxidation of Hantzsch 1,4-dihydropyridines with tetra-n-butylammonuimperiodate catalyzed by tetraphenylporphyrinatomanganese (III) chloride [Mn (TPP) Cl]. Bioorg Med Chem Lett. 2005, 13: 3276-3278.
Moghadam M, Nasr-Esfahani M, Tangestaninejad S, Mirkhani V: Mild and efficient oxidation of Hantzsch 1,4-dihydropyridines with sodium periodate catalyzed by a new polystyrene-bound Mn (TPP) Cl. Bioorg Med Chem Lett. 2006, 16: 2026-2030. 10.1016/j.bmcl.2005.12.072.
Balogh M, Hermecz I, Meszaros Z, Laszlo P: Aromatization of 1, 4-Dihydropyridines by Clay-Supported Metal Nitrates. Helv Chim Acta. 1984, 67: 2270-2272. 10.1002/hlca.19840670834.
Eynde JJV, Orazio RD, Haverabeke YV: Potassium permanganate, a versatile reagent for the aromatization of Hantzsch 1,4-dihydropyridines. Tetrahedron. 1994, 50: 2479-2484. 10.1016/S0040-4020(01)86964-7.
Eynde JJV, Mayence A, Maquestiau A: A novel application of the oxidizing properties of pyridiniumchlorochromate: aromatization of Hantzsch 1,4-dihydropyridines. Tetrahedron. 1992, 48: 463-468. 10.1016/S0040-4020(01)89008-6.
Delgado F, Alvarez C, Garcia O, Penieres G, Marques C: Unusual oxidative dealkylation of certain 4-alkyl-1, 4-dihydropyridines with MnO2/bentonite using microwave irradiation in the absence of solvent (II). Synth Commun. 1991, 21: 2137-2141. 10.1080/00397919108055446.
Mashraqui SH, Karnik MA: Bismuth nitrate pentahydrate. A convenient reagent for the oxidation of Hantzsch 1, 4-dihydropyridines. Synthesis. 1998, 5: 713-714.
Wang B, Hu Y: The Aromatization of Hantzsch 1,4-Dihydropyridines by Tetrakis-Pyridine Cobalt (II) Dichromate (TPCD). Synth Commun. 1999, 29: 4193-4199. 10.1080/00397919908085893.
Maquestiau A, Mayence A, Eynde JJV: Ultrasound-promoted aromatization of hantzsch 1,4-Dihydropyridines by clay-supported cupric nitrate. Tetrahedron Lett. 1991, 32: 3839-3840. 10.1016/S0040-4039(00)79390-7.
Khadikar B, Borkat S: Silica Gel Supported Ferric Nitrate: A Convenient Oxidizing Reagent. Synth Commun. 1998, 28: 207-212. 10.1080/00397919808005712.
Barakat MZ, Wahap MFA, Sadr MM: Some reactions with N-Bromosuccinimide. J Am Chem Soc. 1955, 77: 1670-1672. 10.1021/ja01611a077.
Kurse PF, Geurkink JN, Grist KL: Studies with N-Halo Reagents. II. New Syntheses of β-Bromo-α-keto Esters, Ethyl Phenylglyoxylate and Phenacyl Bromide Using N-Bromosuccinimide. J Am Chem Soc. 1954, 76: 5796-5797. 10.1021/ja01651a061.
Abdel-wahab MF, Barakat MZ: Die Verwendung von N-Bromosuccinimid bei Bromierungs-und Abbaureaktionen. Monatsh Chem. 1957, 88: 692-701. 10.1007/BF00901354.
Duar S, Turk J, Speigle J, Cobin J, Masnovi J, Baker JR: NBS-Promoted Reactions of Symmetrically Hindered Methylphenols via p-Benzoquinone Methide. J Org Chem. 2000, 65: 108-115. 10.1021/jo9911185.
Nagarajan R, Anthonyraj JCA, Muralidharan D, Saikumar C, Perumal PT: A convenient method for the oxidation of Hantzch 1,4-dihydropyridines with N-bromosuccinimide. Indian J Chem. 2006, 45B: 826-828.
Edayadulla N, Ramesh P: Synthesis of 2-methyl-4-aryl-5oxo-1H, 4H-indeno [1,2-b] dihydropyridine-3-carboxylates. Indian J Heterocyclic Chem. 2007, 16: 371-374.
Bernstein J, Davis RE, Shimoni L, Chang NL: Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew Chem Int Ed Engl. 1995, 34: 1555-1573. 10.1002/anie.199515551.
Bruker : APEX2 and SAINT. 2004, Madison, Wisconsin, USA: Bruker AXS Inc
Sheldrick GM: SADABS. Version 2.03. 2001, Germany: University of Göttingen
Sheldrick GM: A short history of SHELX. Acta Crystallogr A. 2008, A64: 112-122.
Farrugia LJ: ORTEP-3 for Windows - a version of ORTEP-III with a Graphical User Interface (GUI). J Appl Crytallogr. 1997, 30: 565-
Cremer D, Pople JA: A General Definition of Ring Puckering Coordinates. J Am Chem Soc. 1975, 97: 1354-1358. 10.1021/ja00839a011.
Nardelli M: Ring asymmetry parameters from out-of-plane atomic displacements. Acta Appl Crystallogr. 1983, C39: 1141-1142.
Spek AL: PLATON. J Appl Crytallogr A. 2003, 36: 7-13. 10.1107/S0021889802022112.
Acknowledgements
One of the authors NE is grateful to Mother Teresa Women’s University, Kodaikanal, Tamilnadu-India and DST-CURIE programme for their encouragement and providing facilities for doing this research work. NE also put forth his heartfelt thanks to his guide Dr. P. Ramesh (Late) for the encouragement and motivation of the research work. PR thanks Prof. Kyeong Kyu Kim, Laboratory of Structural Biology, Department of Molecular Cell Biology, Samsung Biomedical Research Institute, Sungkyunkwan University School of Medicine, Suwon 440–746, South Korea and the National Research Foundation of Korea for the financial support in the form of postdoctoral fellowship (2012K2A4A1034867 and 2011–0030915).
Additional material
Crystallographic data (excluding structure factors) for the structures of compounds (I) and (II) reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers, CCDC 996464 and CCDC 996465, respectively. Copies of the data can be obtained free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1 EZ, UK. (fax: +44-(0)1223-336033 or email: deposit@ccdc.cam.ac.uk).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PR collected the X-ray data and solved the crystal structures under the guidance of MNP. NE synthesized the title compounds under the guidance of Prof. PR (Late). PR and NE contributed equally to this work. All authors have read and approved the final manuscript.
Ramesh Pandian, Edayadulla Naushad contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pandian, R., Naushad, E., Vijayakumar, V. et al. Synthesis and crystal structures of 2-methyl-4-aryl-5-oxo-5H-indeno [1,2-b] pyridine carboxylate derivatives. Chemistry Central Journal 8, 34 (2014). https://doi.org/10.1186/1752-153X-8-34
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1752-153X-8-34