
Abstract
Background
Sertraline is a well known antidepressant drug which belongs to a class called selective serotonin reuptake inhibitor. Most published methods do not enable studying the stability of this drug in different stress conditions.
Results
Two new methods were developed for the determination of sertraline (SER). Both methods are based on coupling with 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl) in borate buffer of pH 7.8 and measuring the reaction product spectrophotometrically at 395 nm (Method I) or spectrofluorimetrically at 530 nm upon excitation at 480 nm (Method II). The response-concentration plots were rectilinear over the range 2-24 μg/mL and 0.25-5 μg/mL for methods I and II respectively with LOD of 0.18 μg/mL and 0.07 μg/mL, and LOQ of 0.56 μg/mL and 0.21 μg/mL for methods I and II, respectively.
Conclusion
Both methods were applied to the analysis of commercial tablets and the results were in good agreement with those obtained using a reference method. The fluorimetric method was further applied to the in vivo determination of SER in human plasma. A proposal of the reaction pathway was presented. The spectrophotometric method was extended to stability study of SER. The drug was exposed to alkaline, acidic, oxidative and photolytic degradation according to ICH guidelines. Moreover, the method was utilized to investigate the kinetics of oxidative degradation of the drug. The apparent first order rate constant and t1/2 of the degradation reaction were determined.

Similar content being viewed by others


Background
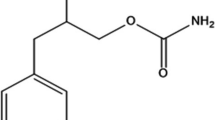
Sertraline is selective serotonin reuptake inhibitor which is clinically effective for the treatment of depression, obsessive-compulsive disorder, depression relapse and social phobia [1]. Chemically it is (1S,4S)-4 [3,4-dichlorophenyl]-1,2,3,4 tetrahydro-N-methyl-1-naphthylamine [2] (Figure 1). Various methods have been reported for the determination of sertraline (SER) including spectrophotometry through it's reaction with some acidic dyes [3] and some haloquinones [4].

Structural formula of SER.
A good guide to the work published is found in the comprehensive monographs in its analytical profile series [5] It was also determined in biological fluids and dosage forms by GC [6–8] MS,GC [9] and HPLC methods [10–13].
Most of these methods are either tedious or require highly sophisticated instrumentation. Therefore, our target was to develop simple and sensitive spectrofluorimetric and spectrophotometric methods for the determination of SER in pharmaceutical preparations and biological fluids.
4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl) is a useful derivatizing agent for primary amines, secondary amines, thiols, etc.. Several pharmaceutical compounds have been determined through this approach [14–20].
During preparation of this manuscript, a paper on the spectrofluorimetric determination of SER appeared [21]. However, the paper did not deal neither with the determination of drug in plasma nor with the stability study of the drug. Since the purpose of stability studies is to monitor possible changes to a product or a material over a time at different storage conditions, it is expected that, only those methods that are stability-indicating should be used.
The parent drug stability guidelines issued by the international conference of harmonization (ICH) [22] requires that analytical test procedure should indicate stability. Therefore, the present study was extended to establish the inherent stability of SER under different stress conditions such as, alkaline, acidic, oxidative and photolytic conditions.
Experimental
Apparatus
Spectrophotometric analysis was carried out on a Shimadzu (Kyoto, Japan) UV-1601 PC, UV-Visible double-beam spectrophotometer with matched 1 cm path-length quartz cells. Absorption spectra were recorded on a fast scan speed, setting slit width to be 1 nm and sampling interval to be auto.
The spectrofluorimetric measurements were made using Perkin Elmer LS 45 Luminescence Spectrometer equipped with 150 Watt Xenon arc lamp and quartz cell (1 cm).
Materials and Reagents
All reagents and solvents were of Analytical Reagent grade.
The pure sample of sertraline (SER) was kindly supplied by the Egyptian Company for Chemicals and Pharmaceuticals (ADWIA) (10th of Ramadan City, Egypt), with a purity of 99.78% as determined by the reference method [23]. The following preparations containing the drug were purchased from local pharmacies:
-
▪ Seserine® tablets, batch # 080371, labeled to contain 56.0 mg SER HCl equivalent to 50.0 mg SER/tablet, product of ADWIA Co. S.A.E, 10th of Ramadan City, Egypt.
-
▪ Serlift® tablets, batch # 810102, labeled to contain SER HCl equivalent to 100.0 mg SER/tablet, product of Global Napi Pharmaceuticals for REXCEL EGYPT.
-
▪ Lustral® tablets, batch # 9201, labeled to contain 50.0 mg of SER/tablet, product of Pfizer Egypt S.A.E. Cairo.
- Plasma was kindly provided by Mansoura University Hospital, and kept frozen until use after gentle thawing.
- 4-Chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl ) purchased from Sigma ( St.Louis USA). A stock solution containing 0.2% (w/v) of the reagent was freshly prepared in methanol.
- Methanol, hydrochloric acid and diethyl ether (Merck, Darmstadt, Germany).
- Borate buffer (0.2 M, pH 7.8), phosphate buffer (0.2M, pH 12).
- 1.0 M aqueous solution of sodium hydroxide (BDH, UK).Hydrogen peroxide (BDH, UK); 0.1% (v/v) aqueous solution
Standard solution
A stock solution was prepared by dissolving 20.0 mg of SER in 100 mL of methanol and was further diluted with the same solvent as appropriate. The standard solutions were stable for seven days when kept in the refrigerator.
General Procedure
Construction of the Calibration Curve
a) Spectrophotometric method (Method I)
To a set of 10 mL volumetric flasks, aliquot volumes containing the drug were quantitatively transferred to obtain final concentrations of 2-24 μg/mL of SER. 2.0 mL of borate buffer (pH 7.8) followed by 1 mL of NBD-Cl solution (0.2% w/v) and were mixed well. The solutions were heated in a thermostatically controlled water bath at 60°C for 25 min. The reaction was stopped by cooling under tap water, and then 0.2 mL of concentrated HCl was added. The solutions were quantitatively transferred to 10 mL volumetric flasks and each flask was made up to volume with methanol. The absorbance was measured at 395 nm against a reagent blank. The calibration graph was constructed by plotting the absorbance versus the final concentration of the drug (μg/mL). Alternatively, the corresponding regression equation was derived.
b) Spectrofluorimetric method (Method II)
The procedure adopted for spectrophotometric method was performed after dilution of the standard solution to obtain the working concentration range of 0.25-5 μg/mL. The fluorescence intensity of the resulting solutions was measured at 530 nm after excitation at 480 nm. The corrected fluorescence intensity (ΔF) was plotted vs the final concentrations of the drug (μg/mL) to get the calibration curve. Alternatively, the regression equation was derived.
Applications
Procedure for commercial tablets
Ten tablets were finely powdered and pulverized well. A weighed quantity of the powdered tablets equivalent to 20 mg of SER was transferred into a 100 mL volumetric flask, about 80 mL of methanol was added and the flask was sonicated for 15 min. The volume was completed with methanol, mixed well and filtered. Aliquots containing suitable concentrations of SER were analyzed as described under Construction of the Calibration Graph. The nominal content of the tablets was determined either from the calibration curve or using the corresponding regression equation.
Procedure for spiked human plasma
Control samples of plasma (1 mL) were spiked with increasing quantities of SER to give a final drug concentration of 0.2-0.4 μg/mL. 2 mL of acetone was added to 1.0 mL of the spiked plasma and was shaked gently. Then the mixture was centrifuged at 6000 rpm for 10 min. The clear supernatant was separated and then 1 mL phosphate buffer (pH 12) was added. After which 3 × 5 mL diethyl ether was used for extraction and the mixture was centrifuged. The ethereal layer was evaporated to dryness. The residue was reconstituted in 2.0 mL of methanol, and then method II was performed as described under Construction of the Calibration Graph. The nominal content of the drug was determined using the corresponding regression equation.
Stability Indication of the Method
Forced Degradation Studies
1- Alkaline Degradation
Solutions for alkaline degradation were prepared by dissolving 100 mg of SER in the least amount of methanol, and the volume was completed to 100 mL in a volumetric flask with 5.0 M sodium hydroxide, then the solution was heated in a boiling water bath for three hours. Then aliquot volumes of these solutions were transferred to 10 mL volumetric flasks and treated as described under method I under Construction of the Calibration Graph.
2- Acidic Degradation
The same method for alkaline degradation was followed using 5.0 M hydrochloric acid instead of 5.0 M sodium hydroxide. Then aliquot volumes of these solutions were transferred to 10 mL volumetric flasks and treated also as described under method I under Construction of the Calibration Graph.
3- Photolytic Degradation
Solutions for photolytic degradations were prepared by dissolving 100 mg of SER in the least amount of methanol, and the volume was completed to 25 mL in a volumetric flask with the same solvent. The solution was exposed to UV-Lamp at a wavelength of 245 nm at a distance of 15 cm placed in a wooden cabinet for 24 and 48 hours. Then aliquot volumes of these solutions were transferred to 10 mL volumetric flasks and treated as described for method I under Construction of the Calibration Graph.
Oxidative Degradation Studies
For the kinetic study of the oxidative degradation, aliquots of SER stock solution were quantitatively transferred to 50 mL volumetric flasks so that the final concentration is 100 μg/mL, then 5 mL of 1% (v/v) H2O2 was added so that its final concentration is 0.1% (v/v) and the solution was heated at different temperature settings using a thermostatically controlled water bath then allowed to cool. Each flask was completed to volume with methanol and aliquots of these solutions were transferred to 10 mL volumetric flasks so that the final concentration is 20 μg/mL. The procedure described for method I under Construction of the Calibration Graph was performed.
Results and Discussion
In the present study, SER was found to react with NBD-Cl in presence of borate buffer (pH 7.8) producing a yellow color peaking at 395 nm. (Figure 2) and resulting in a strong fluorescence at 530 nm after excitation at 480 nm (Figure 3).

Absorption Spectra. (a) SER (20 μg/mL) in borate buffer of pH 7.8. (b) The reaction product of SER (20 μg/mL) with NBD-Cl in borate buffer of pH 7.8. (c) Blank solution with NBD-Cl in borate buffer of pH 7.8.

Fluorescence spectra of the reaction product. (A, B) SER ( 2.5 μg/mL) with 0.2% NBD-Cl at pH 7.8. (A', B') blank with 0.2% NBD-Cl at pH 7.8.
Optimization of Experimental parameters
The spectrofluorimetric and spectrophotometric properties of the colored product as well as the different experimental parameters affecting the color development and its stability were carefully studied and optimized. Such factors were changed individually while others were kept constant. The factors include pH, type of buffer, temperature, time of heating, effect of surfactants and effect of diluting solvent.
Effect of pH
The influence of pH on the absorbance of the reaction product was evaluated. Maximum absorbance intensity was obtained at pH 7.7 and remained constant up to 7.9 after which the absorbance of the reaction product began to decrease gradually until pH 9. Therefore, pH of 7.8 ± 0.1 was chosen as the optimum one (Figure 4). Other buffers having the same pH value such as phosphate and hexamine were tried and compared with 0.2 M borate buffer. Borate buffer was found to be superior to other buffers having the same pH value since the net fluorescence intensity was highest in case of borate buffer. This is probably, because the rate of hydrolysis of NBD-Cl to NBD-OH was much slower. This result is in agreement with that of Miyano et al [24].

Effect of pH of borate buffer 0.2 M on the absorbance of the reaction product of SER (20 μg/mL) with NBD-Cl.
Effect of concentration of NBD-Cl
The influence of the concentration of NBD-Cl was studied using different volumes of 0.2% w/v solution of the reagent. It was found that, the reaction of NBD-Cl with SER started upon using 0.2 mL of the reagent in the presence of borate buffer (pH 7.8). Increasing the volume of the reagent, produces a proportional increase in the absorbance of the reaction product up to 0.8 mL and remains constant up to 1.2, after which further increase produces a gradual decrease in the absorbance value. Therefore, 1 mL of 0.2% w/v of NBD-Cl solution was chosen as the optimal volume of the reagent (Figure 5).

Effect of volume of NBD-Cl 0.2% (w/v) on the absorbance of the reaction product of SER (20 μg/mL) with NBD-Cl.
Effect of heating temperature
Different temperature settings were used with constant heating time. Increasing the temperature of the water bath was found to produce a proportional increase in the absorbance of the reaction product up to 55°C and remained constant until 65°C after which further increase in the temperature produces a gradual decrease in the absorbance value, so the optimum temperature for study was 60°C ± 5°C (Figure 6).

Effect of heating temperature on the absorbance of the reaction product of SER (20 μg/mL) with NBD-Cl.
Effect of time
The time of heating is an essential part of the experiment. Different time intervals were tested to ascertain the time after which the solution attains its highest absorbance. It was found that after 25 min, the reaction product reaches the highest absorbance (Figure 7). It was observed that heating time for 25 min is adequate and the absorbance of the reaction product is stable for about 40 min. at room temperature.

Effect of heating time on the absorbance of the reaction product of SER (20 μg/mL) with NBD-Cl.
The fluorescence intensity or absorbance value of the hydrolysis product of NBD-Cl, namely,4-hydroxy-7-nitrobenzo-2-oxa-1,3-diazole (NBD-OH) is quenched by decreasing the pH of the reaction medium to less than 1. Therefore, acidification of the reaction mixture prior to measurement of the fluorescence intensity or absorbance value remarkably decreased the background fluorescence or absorbance due to the formation of NBD-OH without affecting the drug reagent adduct, hence the sensitivity was increased.
Effect of diluting solvent
Dilution with different solvents such as methanol, water, acetone, dimethylsulfoxide and dimethylformamide was studied. The highest absorbance value was achieved upon diluting with methanol.
Effect of surfactant
The influence of concentration of different surfactants on the absorbance of the reaction product was studied hopefully they may enhance the absorbance. It was found that by using different concentrations of sodium dodecyl sulphate or cetrimide, there was a negligible increase in the absorbance, while using gelatine decreases the absorbance; therefore the study was performed omitting any surfactant.
Analytical parameters for Sertraline
Validation of the proposed methods
The validity of the methods was tested regarding linearity, specificity, accuracy, repeatability and precision according to ICH Q2B recommendations [22].
Linearity
The absorbance-concentration plot was linear over the range of 2-24 μg/mL with minimum detection limit (LOD) of 0.18 μg/mL. While, the fluorescence-concentration plot was found to be linear over the range of 0.25-5 μg/mL with LOD of 0.07 μg/mL. Linear regression analysis of the data gave the following equations:
Where A is the absorbance in 1-cm cell, F is the corrected fluorescence intensity, and C is the concentration of the drug in μg/mL.
The limits of quantification (LOQ) and the limits of detection (LOD) were calculated according to ICH Q2B [22] using the following equations:
Where Sa = The standard deviation of the intercept of regression line
b = Slope of the calibration curve.
The results are shown in Table 1.
LOQ were 0.56 and 0.21 μg/mL for methods I and II respectively, while LOD were 0.18 and 0.07 μg/mL for methods I and II respectively.
These small values allowed the determination of SER in spiked human plasma using method II.
The proposed methods were evaluated by studying the accuracy as percent relative error (% Er) (table 1) and precision as percent relative standard deviation (% RSD) and the results are shown in table 1. The small values of % Er and % RSD indicates high accuracy and high precision of the proposed methods.
Accuracy
To test the validity of the proposed methods, they were applied to the determination of pure sample of SER over the working concentration ranges. The results obtained were in good agreement with those obtained using reference method [23]. Using Student's t-test and variance ratio F-test [25], revealed no significant difference between the performance of both methods and reference method regarding the accuracy and precision, respectively. The reference method recommended an HPLC determination of SER in pure and dosage forms using a mixture of 35% acetonitrile: methanol (92:8, v/v) and 65% of sodium acetate buffer at pH 4.5 as mobile phase with UV detection at 230 nm.
The validity of the two methods were evaluated by statistical analysis of the regression lines regarding the standard deviation of the residuals (Sy/x), the standard deviation of the intercept (Sa) and standard deviation of the slope (Sb). The results are given in Table 1. The small values of the figures point out to the low scattering of the calibration graph and the precision of the methods.
Precision
Repeatability
The repeatability was performed by applying the proposed methods for the determination of two concentrations of SER in pure form on three successive times, and the results are listed in Table 2.
Intermediate precision
It was performed through repeated analysis of SER in pure form, using the concentrations shown in Table 2 for a period of 3 successive days. The results are also summarized in Table 2.
Robustness of the method
The robustness of the methods adopted is demonstrated by the constancy of the fluorescence intensity/absorbance value with the deliberated minor changes in the experimental parameters such as, change in pH 7.8 ±0.1, change in the volume of NBD-Cl (0.2% w/v), 1.0 ± 0.2 mL, the change in reaction time 25 ± 5 min. These minor changes that may take place during the experimental operation didn't affect the fluorescence intensity or the absorbance value of the reaction products.
Pharmaceutical Applications
The proposed methods were then applied to the determination of SER in its tablets. The methods were tested for linearity, selectivity and accuracy according to ICH Q2B recommendations [22].
Selectivity
The selectivity of the method was investigated by observing any interference encountered from the common tablet excepients, such as talc, lactose, starch, avisil, gelatine, and magnesium stearate. These excepients did not interfere with the proposed methods. As revealed by a blank experiment using tablets additives but omitting sertraline.
Accuracy
The results of the pharmaceutical applications of the proposed methods were statistically compared with those obtained using the reference method [23]. Statistical analysis [25], of the results, using Student's t-test and variance ratio F-test revealed no significant difference between the performance of the proposed and reference methods regarding the accuracy and precision, respectively (Table 3).
Analysis of Biological fluids
The high sensitivity of the proposed fluorimetric method allowed the determination of SER in spiked human plasma (Figure 8). Sertraline is given orally in a dose of 100 mg three times daily; this leads to a final blood level concentration of about 0.25 μg/mL i.e can be determined by the proposed method. The results are shown in Table 4. The extraction procedure described by Kim et.al [26] was adopted here. The results are satisfactorily accurate and precise.

Fluorescence emission spectra of plasma spiked with SER. (a) Blank, (b) 0.25 μg/mL, (c) 0.3 μg/mL, and (d) 0.35 μg/mL.
Precision
The within-day precision was evaluated through replicate analysis of plasma samples spiked with increasing concentrations of the drug. The percentage recoveries based on the average of five separate determinations were 101.05 ± 1.65, thus indicating the high precision of the method (Table 4).
The inter-day precision was also evaluated through replicate analysis of plasma samples spiked with 0.4 μg/mL of drug on three successive days. The percentage recoveries based on the average of four separate determinations were 101.94 ± 1.194. The results are also shown in Table 4.
Mechanism of the reaction
The stoichiometry of the reaction was studied adopting the limiting logarithmic method [27]. The absorbance of the reaction product was alternatively measured in the presence of excess of either NBD Cl or SER. A plot of log absorbance versus log [NBD-Cl] and log [SER] gave straight lines, the values of the slopes were 0.79 and 0.83 respectively (Figure 9). Hence, it is concluded that, the molar reactivity of the reaction is 0.79/0.83, i.e. the reaction proceeds in the ratio of 1: 1. By analogy to previous study [16], it is confirmed that one molecule of the drug reacts with one molecule of NBD-Cl. Sertraline in alkaline medium reacts with NBD-Cl through its secondary amino group to give the following final reaction product. A schematic proposal of the reaction pathway is given in Scheme 1.

Limiting logarithmic plots for the molar reactivity of SER with NBD-Cl. (A) log A vs log [NBD-Cl]. (B) log A vs log [SER].

Proposed reaction pathway between NBD-Cl and sertraline.
Stability indication of the method
Forced degradation studies of SER
In order to establish whether the analytical method was stability-indicating or not, SER was exposed to different forced degradation studies. The stability of SER has been studied under extreme challenge conditions as well as the more traditional pharmaceutical challenge conditions. Methanol was used as a diluting solvent in all forced degradation studies. The drug was found to be highly stable under forced alkaline, acidic or photolytic degradation conditions as revealed from the constant absorbance values of samples treated with concentrated solutions of NaOH or HCl and UV-light.
Oxidative degradation studies
Degradation kinetics study
For the kinetic study, 0.1% (v/v) of hydrogen peroxide was used for the oxidative degradation of the drug. The degradation was found to follow first order kinetics and to be temperature dependent (Figure 10). The apparent first order degradation rate constant (Kobs) and the half life time (t1/2) at each temperature were calculated (Table 5). Plotting log Kobs values versus 1/T, the Arrhenius plot was obtained (Figure 11). The activation energy was calculated and found to be 6.38 k.Cal.Mol -1.

Semilogarithmic plot of SER (20.0 μg/mL) versus different heating times with 0.1% (v/v) H 2 O 2 then with 0.2% NBD-Cl using the above conditions. (A = 80°C, B = 70°C, C = 60°C, D = 50°C ).

Arrhenius plot for the degradation of SER (20.0 μg/mL) in 0.1%(v/v) H 2 O 2 .
The tolerance limit (concentration of interfering substance causing 3.0% relative error) of the degradation product was measured after mixing increasing concentrations of the degradation product with 10 μg/mL pure SER. The tolerance limit was found to be 20%, i.e. up to 20% from the degradation product will not cause interference in the proposed spectrophotometric method (Table 6).
Pathway of degradation
By analogy to previous report [28] discussing the degradation of isradipine (containing secondary amino group) by hydrogen peroxide, it is suggested that the degradation pathway is via the formation of N- oxide derivative (Scheme 2).

Proposed oxidative degradation pathway of sertraline with hydrogen peroxide.
Conclusion
Two sensitive, simple and time saving methods were developed for the determination of SER either per se or in pharmaceutical preparations. By virtue of high sensitivity of the proposed fluorimetric method, it could be applied to the determination of SER in spiked human plasma with good accuracy and precision. The spectrophotometric method is also considered as stability indicating assay.
References
Nickell PV, Ward HE, Booth RG: Antianxiety agents. Burger's Medicinal Chemistry and Drug Discovery. Edited by: Wolff ME. 1997, John Wiley and Sons, New York, 5: 153-194.
The British Pharmacopoeia 2009. 2009, The stationary office: London, Electronic version
Onal A, Kepekci SE, Cetin SM, Ertürk S: Spectrophotometric determination of certain antidepressants in pharmaceutical preparations. J AOAC Int. 2006, 89: 966-971.
Darwish IA: Development and validation of spectrophotometric methods for determination of fluoxetine, sertraline and paroxetine in pharmaceutical dosage forms. J AOAC Int. 2005, 88: 38-45.
Parasrampuria J: Sertraline hydrochloride. Analytical Profiles of Drug Substances and Excipients. Edited by: Brittain HG. 1996, Academic Press: New York, 24: 443-486.
Rogowsky D, Marr M, Long G, Moore C: Determination of sertraline and desmethylsertraline in human serum using copolymeric bonded-phase extraction, liquid chromatography and gas chromatography-mass spectrometry. J Chromatogr Biomed Appl. 1994, 655: 138-141. 10.1016/0378-4347(94)00112-X.
Logan BK, Friel PN, Case GA: Analysis of sertraline (Zoloft) and its major metabolite in post-mortem specimens by gas and liquid chromatography. J Anal Toxicol. 1994, 18: 139-142.
Fouda HG, Ronfeld RA, Weidler DJ: Gas-chromatographic - mass-spectrometric analysis and preliminary human pharmacokinetics of sertraline, a new antidepressant drug. J Chromatogr Biomed Appl. 1987, 61: 197-202.
Tremaine LM, Joerg EA: Automated gas-chromatographic - electron-capture assay for the selective serotonin uptake-blocker, sertraline. J Chromatogr Biomed Appl. 1989, 88: 423-429.
Gupta RN, Dziurdzy SA: Therapeutic monitoring of sertraline. Clin Chem. 1994, 40: 498-499.
Wiener HL, Kramer HK, Reith MEA: Separation and determination of sertraline and its metabolite, demethylsertraline, in mouse cerebral cortex by reversed-phase high-performance liquid chromatography. J Chromatogr Biomed Appl. 1990, 92: 467-472.
Patel J, Spencer EP, Flangan RJ: HPLC of sertraline and norsertraline in plasma or serum. Biomed Chromtogr. 1990, 10: 351-354.
McIntyre IM, King CV, Staikos V, Gall J, Drummer OH: A fatality involving moclobemide, sertraline, and pimozide. J Forensic Sci. 1997, 42: 951-953.
El-Emam AA, Hansen SH, Moustafa MA, El-Ashry SM, El-Sherbiny DT: Determination of lisinopril in dosage forms and spiked human plasma through derivatization with 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (NBD-C1) followed by spectrophotometry or HPLC with fluorimetric detection. J Pharm Biomed Anal. 2004, 34: 35-44. 10.1016/j.japna.2003.08.021.
El-Enany N: Spectrofluorimetric and spectrophotometric determination of gliclazide in pharmaceuticals by derivatization with 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole. J AOAC Int. 2003, 86: 209-214.
El-Enany N, Belal F, Rizk M: Kinetic spectrophotometric determination of isoxsuprine in dosage forms through derivatization with 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl). Sci Pharm. 2006, 74: 99-119. 10.3797/scipharm.2006.74.99.
El-Enany N, El-Sherbiny D, Belal F: Spectrophotometric, spectrofluorometric and HPLC determination of desloratadine in dosage forms and human plasma. Chem Pharm Bull. 2007, 55: 1662-70. 10.1248/cpb.55.1662.
Ulu ST: Determination of tianeptine in human plasma using high-performance liquid chromatography with fluorescence detection. J Chromatogr B. 2006, 834: 62-67. 10.1016/j.jchromb.2006.02.027.
Erturk S, Cetin SM, Atmaca S, Ersoy L, Baktr G: A sensitive HPLC method for the determination of Fluoxetine and Norfluoxetine in human plasma with fluorescence detection. Ther Drug Monit. 2005, 27: 38-43. 10.1097/00007691-200502000-00009.
Farshchia A, Ghiasi G, Bahrami G: High performance liquid chromatography determination of sertraline in human. Iranian Journal of Pharmaceutical Sciences. 2009, 5: 171-178.
Mahmoud AM, Darwish IA, Khalil NY: Fluorimetric study for the reaction between sertraline and 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole: kinetics, mechanism and application for the determination of sertraline in tablets. J Fluorescence. 2010, 20: 607-13. 10.1007/s10895-009-0595-3.
ICH Harmonized Tripartite Guideline: Validation of Analytical Procedures. 2005, Text and Methodology, Q2(R1), Current Step 4 Version, Parent Guidelines on Methodology , [http://www.bioforum.org.il/Uploads/Editor/karen/q2_r1_step4.pdf]
Malfará WR, Bertucci C, Costa Queiroz ME, Dreossi Carvalho SA, Pires Bianchi Mde L, Cesarino EJ, Crippa JA, Costa Queiroz RH: Reliable HPLC method for therapeutic drug monitoring of frequently prescribed tricyclic and nontricyclic antidepressants. J Pharm Biomed Anal. 2007, 44: 955-962. 10.1016/j.jpba.2007.04.005.
Miyano H, Toyo'oka T, Miyano H: Further studies on the reaction of amines and proteins with 4-fluoro-7-nitrobenzofurazan. Anal Chim Acta. 1985, 170: 81-87.
Miller JN, Miller JC: Statistics and chemometrics for analytical chemistry. 2005, Prentice Hall: England, 39-73. 5
Kim KM, Jung BH, Choi MH, Woo JS, Paeng KJ, Chung BC: Rapid and sensitive determination of sertraline in human plasma using gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2002, 769: 333-339. 10.1016/S1570-0232(02)00027-2.
Rose J: Advanced physico-chemical experiments. 1964, Pitman: London, England
Bartlett MG, Spell JC, Mathis PS, Elgany MF, El Zeany BE, Elkawy MA, Stewart JT: Determination of degradation products from the calcium-channel blocker isradipine. J Pharm Biomed Anal. 1998, 18: 335-345. 10.1016/S0731-7085(98)00047-8.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MIW designed the proposed method and analyzed the data statistically. FFB proposed, planned and supervised the whole work. NME coordinated the study and modified the text. HE carried out the experimental work. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Walash, M.I., Belal, F.F., El-Enany, N.M. et al. Development and validation of stability indicating method for determination of sertraline following ICH guidlines and its determination in pharmaceuticals and biological fluids. Chemistry Central Journal 5, 61 (2011). https://doi.org/10.1186/1752-153X-5-61
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1752-153X-5-61