
Abstract
Background
A novel rapid, accurate, and stability-indicating reversed-phase high performance liquid chromatographic (RP-HPLC) and first derivative spectrophotometric determination were explained for the assay of vortioxetine (VRT) in bulk and pharmaceutical formulations. For RP-HPLC method, optimal separation and determination of VRT were achieved with a Waters Symmetry C18, (100 × 4.6 mm, 3.5 μm) analytical column using a mobile phase consisting of methanol:0.05 M potassium dihydrogen phosphate (pH:3.0 ± 0.05) (30:70, v/v) in isocratic mode with flow rate of 1.3 mL min−1. Injection volume was 20 μL. The maximum absorption wavelength of VRT is 225.0 nm; hence, 225.0 nm was studied as the detection wavelength and column at 50 °C temperature. The caffeine was used as the internal standard (IS). On the other hand, the first derivative spectrophotometric method for the analysis of vortioxetine was performed by measuring the amplitude at 251.7 and 272.6 nm.
Result
The HPLC method was found to be linear in the concentration ranges of 10.0-70.0 μg mL−1 with the coefficient value R2 of 0.9998, and the mean recovery value was 100.7%. Further stability studies were done through exposure of the analyte solutions to various stress conditions: acid, alkali hydrolysis, chemical oxidation, and exposure to UV radiation. For the first derivative spectrophotometric method, linearity was observed in the concentration range 6.0-30.0 μg mL−1 (for 237.7 nm R2 = 0.9999 and for 257.2 nm R2 = 0.9997).
Conclusion
The methods were validated in accordance with ICH guidelines with respect to linearity, accuracy, specificity, limit of detection, precision, and limit of quantification.
Similar content being viewed by others

Background
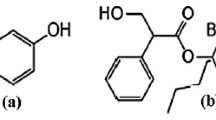
Vortioxetine (VRT) (Fig. 1) is the first in a novel group of drugs known as a serotonergic (5-HT) antidepressant, chemically known as 1-[2-(2,4-dimethylphenylsulfanyl)-phenyl]-piperazine. Vortioxetine (VRT) binds with high affinity to the serotonin transporter and its antidepressant effects are considering to be secondary to enhancing serotonin in the central nervous system by way of inhibition of reuptake [1]. Providers may consider 5 mg per day for patients who cannot tolerate higher doses. VRT can be stopped abruptly, but a reduction to 10 mg per day is recommended for patients taking 15 mg or more per day for a week before complete [2,3,4,5].

Structure of vortioxetine
A new film-coated tablet of VRT has been newly developed by Lundbeck Pharm. Pharmacopeias do not provide any monographs for VRT and pharmaceutical dosage form. Until now, no references describing of VRT in degradation studies have been found in the literature. UPLC-MS/MS method was reported for the determination of VRT in rat and human plasma [6, 7]. Additionally, a few methods were presented for the quantitative determination of vortioxetine and its major human metabolite in biological fluids besides analyzing tablets and impurities using LC-MS [8,9,10,11,12], LC-MS/MS [13,14,15,16], HPLC [17,18,19], and HILIC-MS [20]. Spectrophotometric UV-metric and potentiometric pH-metric methods were used for the determination of VRT [21].
In the current study, the RP-HPLC assay of VRT in the film-coated tablets was optimized and validated. The presented method was carried out for the determination of VRT under different stressed conditions in addition to the analysis of coated tablet forms. A stability-indicating method has not been described in any of the cited literature, so the optimization of a rapid, accurate, precise, and specific stability-indicating RP-HPLC method for the quantitation of VRT in coated tablet dosage forms is required.
Methods
Chemicals
Vortioxetine reference substance was supplied from Lundbeck Pharm. Ind. The purity of the drug is 98.87%. A commercial preparation, Brintellix® film-coated tablets (produced by Lundbeck Pharm. Ind., Turkey, containing 10.0 mg of VRT per coated tablet) was assayed. Methanol, potassium dihydrogen phosphate, ammonium acetate, and acetonitrile of HPLC grade were received from Merck Chem. Ind. All reagents employed in this study were of analytical-reagent grade.
Apparatus
Instrument used in the study was Dionex Ultimate focused 3000-9 equipped with auto sampler and DAD. The wavelength of the UV detector was set at 225.0 nm. Peak areas were integrated by using the computer Chromeleon Client software program. Absorbance measurements were measured on a double beam UV-VIS spectrophotometer model UV-1601 by using quartz cells with 10.0 mm path length.
Chromatographic conditions
An isocratic mobile phase consisted of potassium dihydrogen phosphate buffer (pH:3.0 ± 0.05):methanol in the ratio (30:70 volumetric ratio) filtered and degassed through a membrane filter of 0.45 μm porosity before use. The analytical column was a Waters Symmetry RP-18 (3.5 μm, 100 cm × 4.6 mm) column. The flow rate of 1.3 mL min−1 and the detector was set at 225.0 nm. All determinations were made at 50 °C and the injection volume was 20.0 μL. The working solutions and mobile phases were prepared daily.
Construction of the calibration graph and linearity
For the HPLC method, the standard stock solution was prepared separately by dissolving VRT and caffeine (CAF) (internal standard) in the degassed mobile phase, respectively. Based on these stock solutions, the working standard solution, VRT, was prepared in a linear working range of 10.0-70.0 μg mL−1 and caffeine as internal standard at the amount of 20.0 μg mL−1 into 10.0 mL volumetric flasks using the mobile phase. Five replicate measurements were made for all concentrations. The injection volume of each sample is 20 μL. To calculate the concentration of VRT, the ratio of VRT to IS peak area was calculated. The linear working graph was constructed and the calibration equal was then stored. In the first derivative spectrophotometric method, Beer’s law range, linear regression equations, and correlation coefficients determined.
Validation parameters
Accuracy as recovery
The accuracy of the suggested method has quantified the percentage of analytical recovery of VRT by using the standard addition method. The recovery test was performed at three levels: 30.0, 40.0, and 50.0 μg mL−1 of the pure sample. Five replicate experiments were performed and recoveries (%), RSD (%) were found.
Precision
So as to control the precision experiment, intraday and inter-day experiments were evaluated. The method was found to be in precision at three different independent (low, medium, and high) amounts during the same day, and 5 different days. The relative standard deviations were found.
Sensitivity
The LOD and LOQ parameters were estimated as described at ICH guidelines by the formulas given below; where Sb is the standard deviation of the intercept of the linear line and a is the slope of the regression equation [22].
Robustness
The robustness of the proposed RP-HPLC method was carried out by altering the experimental conditions such as analytical column, column temperature, flow rate, changing pH, and mobile phase. The robustness of the derivative spectrophotometric method was established by introducing small changes in experimental conditions like wavelength ± 1 nm. Making a deliberate change in wavelength was taken place and RSD of absorbance found to be less than 2, specify that the method was robust.
Degradation studies
Degradation studies were conducted to determine specificity and stability-indicating properties of the suggested method. That these stress conditions were performed under the following acidic, alkaline, oxidative, and photodegradation conditions. VRT (20.0 mg) and CAF (20.0 mg) were exactly weighed. The weighed active ingredients were taken into 100.0 mL volumetric flasks and diluted with the mobile phase and made up to its volume. The prepared stock solutions were used of forced degradation experiments.
For acidic or alkaline degradation studies, 1.0 mL of standard stock VRT solution was transferred to a 10.0-mL volumetric flask in the presence of IS. A total of 2.0 mL of 0.1 M HCl or 0.1 M NaOH solution was added and stirred. The volumetric flask was heated in the water bath at 80 °C for 1 h, cooled to room temperature, and neutralized with 0.1 M NaOH or 0.1 M HCl to pH 7.0. The above solution has diluted the volume with the mobile phase.
For the oxidation degradation study, 1.0 mL of standard stock solution of VRT in the presence of IS was transferred into a 10.0-mL volumetric flask. A total of 2.0 mL of 3.0 H2O2 solution was added and mixed well. The volumetric flask was heated in the water bath at 80 °C for 1 h, cooled to room temperature. The above solution has diluted the volume with the mobile phase.
For photodegradation study, analytically pure sample of VRT and CAF were transferred into a petri dish and exposed into the sunlight for 48 h. The solids were dissolved with the mobile phase.
Filter the degradation solutions with 0.45-μm syringe filters and place in vials of the RP-HPLC system for all degradation studies.
Application to pharmaceutical formulation
At first, ten tablets of Brintellix were weighed and finely powdered in a mortar. The contents were diluted with the mobile phase. Quantity of one tablet weigh was weighed and diluted with the mobile phase in a 100.0-mL volumetric flask. The volumetric flask was shaken for 20.0 min by an ultrasonic shaker. The excipient was separated by filtration. After the filtration process, the appropriate volume of filtered solution and IS was transferred into a 100.0-mL volumetric flask and completed with the mobile phase. All solutions were filtered through 0.45 μm Millipore membrane filters before injections into the HPLC system. The concentration of VRT was calculated by calibration graph prepared with stock solution.
Results
The proposed RP-HPLC method was applied for the amount of VRT in pharmaceutical formulation. To optimize the RP-HPLC assay parameters, various variables effect on peak shape, retention, and resolution as well as suitable internal standard was investigated. As stationary phase, Waters®–Symmetry C18 (150 mm × 4.6 mm, 3.5 μm), Luna® C18 (3.0 μm, 4.6 × 50 mm) and Kromasil® C18, (3.5 μm, 4.6 × 100 mm) column were tested so as to obtain a sharp peak. The results obtained in the different stationary phases were presented below in Table 1. The best results were achieved by using Waters Symmetry C18.
A series of aqueous mobile phases containing buffer solutions and methanol were tested in the initial stage as mobile phase, different combinations of organic modifiers, buffer solutions in different pH values and flow rate effects were examined. DAD detector was set at 220.0, 223.0, 225.0, 230.0, and 235.0 nm for all conditions that the optimum wavelength was found to be 225.0 nm. The pH of the mobile phase was changed over the range of 3.0-6.0 that the optimum pH was found to be 3.0. The best separation was attained when using a mobile phase combination of 0.05 M KH2PO4:methanol (30:70, by volume ratio). Different internal standards were investigated to achieve the best resolution between VRT and the internal standard, and CAF was selected as an internal standard.
The column temperature effect on retention and resolution of VRT and CAF (IS) was examined, and no significant effect was detected from the range between 20-60 °C. Therefore, all investigations were performed at 50 °C. The UV detection was carried out at 225.0 nm because it showed the greatest response that was preferred for studies. Under the previously optimized conditions, the retention times of VRT and CAF (IS) were 0.88 and 1.78 min; in this way, chromatographic separation was finished within 2.0 min (Fig. 2). Both VRT and CAF separated as a clear symmetrical peaks (symmetry factor of 0.91 and 1.11 for VRT and CAF, respectively) which were well separated with minimal tailing and good resolution.

Chromatograms of (a) 20.0 μg mL−1, (b) 30.0 μg mL−1, (c) 40.0 μg mL−1, (d) 50.0 μg mL −1, (e) 60.0 μg mL−1, and (f) 70.0 μg mL−1, VRT, and 20.0 μg mL−1 internal standard CAF in mobile phase
Subsequently, the degradation of sample solutions under stress conditions was analyzed. The thermal, acid, base, oxidative, and photolytic degraded solutions of VRT and CAF pass the purity test (Table 2). The respective distinct chromatograms were shown in Fig. 3. During the degradation study, VRT was been degraded higher in oxidation condition and was found to be stable in thermal, acid, base, and photolytic conditions.

Chromatograms of VRT and CAF in mixture (b: acid, c: base, d: thermal, e: oxidation, f: photolytic) (a: non-degradation)
On the other side, the first derivative spectrophotometric method, 0.02 M acetate buffer was chosen as a solvent system on account of its lack of interference, dissolves easily, suitability for routine quantitative determination of VRT. The zero-order UV spectrum and the first derivative spectrum of VRT in 0.02 M acetate buffer are shown in Fig. 4. In the zero-order UV spectra of VRT show a broad peak around 228.0 nm and shoulder around 210.0 nm. Vortioxetine derivative spectra of different degrees were plotted and the best peaks at 237.7 and 257.2 nm in the first derivative curve were obtained with convenient precision.

Direct UV and first derivative UV spectrum of (a) 6.0 μg mL−1, (b) 8.0 μg mL−1, and (c) 10.0 μg mL−1, (d) 14.0 μg mL−1, (e) 18.0 μg mL−1, (f) 24.0 μg mL−1, and (g) 30.0 μg mL−1 VRT in 0.02 M acetate buffer
The proposed methods were validated, and the validation characteristics containing precision, linearity, accuracy, limit of detection (LOD), limit of quantification (LOQ), specificity, and applicability in pharmaceutical formulations were evaluated. To test the linear working curves for both proposed methods chromatograms and spectrums were analyzed in triplicate in seven different concentrations of VRT. The characteristics of the calibration curves are shown in Table 3. Under the experimental conditions, LOD and LOQ were determined to evaluate the detectivity of the proposed methods.
The precision of the repeatability (intra-day) and intermediate precision (inter-day) of the method was evaluated for VRT at three different independent concentrations by using HPLC and derivative method. From the results obtained, % RSD was calculated and was found to be between 0.19 and 0.53% for intra-day variability and 0.34 and 0.57 % for inter-day. Further % RSD was calculated for derivative spectrophotometry. Then, the obtained values for the precision were shown in Table 4.
Assay application
The suggested methods were successfully implemented of VRT in marketed pharmaceutical preparation (Brintellix®). No pretreatment like time-consuming extraction or evaporation step was performed for specimen preparation. Experiments were realized under the same conditions as employed for the VRT. There is no chromatographic interference found for tablet dosage forms with internal standard, formulation components. The results given in Table 5 indicate good accuracy for pharmaceutical preparations. The accuracy of the described methods was quantified by its recovery results.
To check the validity, the accuracy of the proposed methods is represented and determined by recovering VRT from the placebo. The recovery test was performed by adding different amounts of VRT with a concentration range of 5.0-40.0 μg mL−1 to the previously analyzed tablets and finally analyzing the amount recovered or VRT during studies. Recovery of the VRT was calculated as 97.7-102.9% for added drugs. The data obtained by the first derivative spectrophotometric method and RP-HPLC data were compared by using Student’s t test and the F test at 95% confidence level. As can be seen from Table 6, the estimated t and F values (0.80 and 1.30 respectively) did not exceed the theoretical values, indicating that there was no substantial difference between the described methods on account of accuracy. These results indicating that there is no substantial difference that exists between the performances of the two proposed methods regarding their accuracy and precision.
Discussion
In this study, chromatographic and spectrophotometric methods were optimized for the quantification of vortioxetine active substance in Brintellix® preparation.
RP-HPLC and spectrophotometric methods were optimized. Various experiments have been conducted to optimize the chromatographic method. In order to optimize the RP-HPLC assay parameters, various variable effects on peak shape, retention, and resolution as well as suitable internal standard were investigated. The column was chosen, which gave the best results as shown in Table 1.
Forced degradation experiments were conducted to analyze stability-indicating properties and specificity of the suggested method. The degradation of sample solutions under stress conditions was determined. VRT and CAF were exposed to thermal, acid, base, oxidative, and photolytic degraded solutions passes the purity test (Table 2). The respective distinct chromatograms were given in Fig. 3. During the degradation study, VRT was been degraded higher in oxidation condition and was found to be stable in thermal, acid, base, and photolytic conditions.
In order to prove the validity of the developed methods, the parameters reported in the sources were selected for the method validity tests, and the relevant validity criteria were accepted [22]. For this purpose, validation studies, linearity, range, sensitivity, precision, recovery, repeatability, etc. parameters were investigated and statistical evaluations were made. Although the study with UPLC-MS/MS method is more sensitive in the literature, it is an expensive method. Our RP-HPLC and spectrophotometric studies are simple to be used in routine laboratories [6, 7]. MS/MS method was used for the degradation products in the reported study. However, in our study, degradation products were determined by the DAD detector. Chromatographic separation was finished within 2.0 min in our study. The system suitability parameters for VRT were found to be more sensitive compared to the studies in the literature. Additionally, it is an expensive method. Our study is simple to be used in routine laboratories and its analysis time is short and suitable for green chemistry compared to the studies in the reported literature. Our applied method is very sensitive and found LOQ value low [15].
The vortioxetine was analyzed as simple, fast, and suitable for green chemistry which has no required expensive equipments and software for the derivative method. LOD and LOQ values indicate that both methods are sensitive. Correlation coefficients of the methods are close to 1.0 which supports the sensitivity. Intra- and inter-day analyses were performed, and the results that are obtained showed that the purposed methods are precise.
Conclusion
The newly RP-HPLC and first derivative spectrophotometric methods have been proven to be sensitive, precise, accurate, and rapid. These described methods were also compared statistically via calculating t and f values and no difference was found between methods. Purposed methods were applied to the tablet formulation successfully. Since the proposed methods can be used in many control laboratories for the quantification of VRT in pharmaceutical formulations.
Availability of data and materials
All data and materials are available upon request.
Abbreviations
- RP-HPLC:
-
Reverse phase-high performance liquid chromatography
- VRT:
-
Vortioxetine
- CAF:
-
Caffeine
- UV:
-
Ultraviolet
- ICH:
-
International Conference on Harmonization
- LC-MS/MS:
-
Liquid chromatography-mass spectrometry/mass spectrometry
- UPLC-MS/MS:
-
Ultra performance liquid chromatography-mass spectrometry/mass spectrometry
- DAD:
-
Diode array detector
- 5-HT:
-
5-Hydroxytryptamine
- SD:
-
Standard deviation
- RSD:
-
Relative standard deviation
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
- IS:
-
Internal standard
References
D’Agostino A, English CD, JA R (2015) Vortioxetine (Brintellix): a new serotonergic antidepressant. Drug Forecast P&T 40(1):36–40
Deerfield, Illinois: Takeda Pharmaceuticals (2013) America. Brintellix (vortioxetine) pack insert. https://www.lundbeck.com/upload/us/files/pdf/2013_Releases/BrintellixApprovalRelease.pdf.
Wesolowska A, Tatarczynska E, Nikiforuk A, Chojnacka-Wojcik E (2007) Enhancement of the anti-immobility action of antidepressants by a selective 5-HT7 receptor antagonist in the forced swimming test in mice. Eur J Pharmacol 555(1):43–47
Brintellix. In: Drugdex (2016) Greenwood Village, Colorado: Thompson Micromedex; 1974–2008. www.micromedex.com. Accessed 2016.
Bang-Andersen B, Ruhland T, Jorgensen M, Smith G, Frederiksen K, Jensen KG, Zhong H, Nielsen SM, Hogg S, Mork A, Stensbol TB (2011) Discovery of 1-[2-(2,4-dimethylphenyl sulfanyl) phenyl]piperazine (Lu AA21004): a novel multimodal compound for the treatment of major depressive disorder. J Med Chem 54(9):3206–3221
Gu EM, Huang C, Liang B, Yuan L, Lan T, Hu G, Zhou H (2015) An UPLC–MS/MS method for the quantitation of vortioxetine in rat plasma: application to a pharmacokinetic study. J Chromatogr B 997:70–74
Yi Y, Ren G, Zheng M, Zhao D, Li N, Chen X, Lu Y (2020) Simultaneous determination of deuterated vortioxetine and its major metabolite in human plasma by UPLC-MS/MS and application to a pharmacokinetic study in healthy volunteers. J Chromatogr B 1138:121955
Dong SB, Yan Z, Yang H (2016) A sensitive precolumn derivatization method for determination of piperazine in vortioxetine hydrobromide using a C8 column and high-performance liquid chromatography-mass spectrometry. Anal Sci 32(12):1333–1338.
Henriette KU, Martin J, Henrik P, Lars D (2011) Biosynthesis and identification of an N-oxide/N-glucuronide metabolite and first synthesis of an N-O-glucuronide metabolite of Lu AA21004. Drug Metab.Dispos. 39(12):2264–2274
Areberg J, Luntang-Jensen M, Søgaard B, Nilausen DØ (2012) Occupancy of the serotonin transporter after administration of Lu AA21004 and its relation to plasma concentration in healthy subjects. Basic Clin Pharmacol Toxicol 110:401–404
Wroblewski K, Petruczynik A, Buszewski B, Szultka-Mlynska M, Karakula-Juchnowicz H, Waksmundzka-Hajnos M (2017) Determination of vortioxetine in human serum and saliva samples by HPLC-DAD and HPLC-MS. Acta Chromatographica 29(3):325–344
Lei L, Na C, Xingling M, Kaihe X, Lili S, Qiaogen Z, Lili Y (2016) Stability-indicating reversed-phase HPLC method development and characterization of impurities in vortioxetine utilizing LC–MS, IR and NMR. J Pharm Biomed Anal 117:325–332
Kall MA, Rohde M, Jorgensen M (2015) Quantitative determination of the antidepressant vortioxetine and its major human metabolite in plasma. Bioanalysis 7(22):2881–2894
Kertys M, Krivosova M, Ondrejka I, Hrtanek I, Tonhajzerova I, Mokry J (2020) Simultaneous determination of fluoxetine, venlafaxine, vortioxetine and their active metabolites in human plasma by LC-MS/MS using one-step sample preparation procedure. J Pharm Biomed Anal 181:113098
De Diego M, Correa D, Mennickent S, Godoy R, Vergara C (2018) Determination of vortioxetine and its degradation product in bulk and tablets, by LC-DAD and MS/MS methods. Biomed Chromatogr 32(11):e4340
Qin M, Qiao H, Yuan Y, Shao Q (2018) A quantitative LC-MS/MS method for simultaneous determination of deuvortioxetine, vortioxetine and their carboxylic acid metabolite in rat plasma, and its application to a toxicokinetic study. Anal Methods 10(9):1023–1031
Petruczynik A, Wroblewski K, Wojtanowski K, Mroczek T, Juchnowicz D, Karakula-Juchnowicz H, Tuzimki T (2020) Comparison of various chromatographic systems for identification of vortioxetine in bulk drug substance, human serum, saliva, and urine samples by HPLC-DAD and LC-QTOF-MS. Molecules 25(11):2483
Wroblewski K, Petruczynik A, Tuzimki T, Prajsnar K, Przygodzk D, Buszewicz G, Karakula-Juchnowicz H, Rog J, Morylowska-Hajnos J, Waksmundzka-Hajnos M (2019) Optimization of chromatographic systems for analysis of selected psychotropic drugs and their metabolites in serum and saliva by HPLC in order to monitor therapeutic drugs. Open Chem 17(1):1361–1373
Chen Y, Zhu YL, Qiu CZ, Pan QM, Liu WY, Qiu XJ (2019) A simple HPLC method for the determination of vortioxetine in rabbit plasma and its application to a pharmacokinetic study. Latin Am J Pharm 38(6):1111–1115
Dousa M, Klvana R, Doubsky J, Srbek J, Richter J, Exner M, Gibala P (2016) HILIC-MS determination of denotoxic impurity of 2-chloro-N-(2-chloroethyl) ethanamine in the vortioxetine manufacturing process. J Chromatogr Sci 54(2):119–124
Meloun M, Pilarova L, Capova A, Pekarek T (2018) The overlapping thermodynamic dissociation constants of the antidepressant vortioxetine using UV-VIS multiwavelength pH-titration data. J Solution Chem 47(5):806–826
The International Conference on Harmonisation ICH Technical Requirements for Registration of Pharmaceuticals for Human Use on Validation of Analytical Procedures (2005).
Acknowledgements
This work is produced from the master thesis of Cansu ALVER (Ankara University, Health Science Institute). This research has been supported by Ankara University Scientific Research Projects Coordination Unit (Project Numbers: 19 L0237001; 15 L0237007).
Funding
No funding was received.
Author information
Authors and Affiliations
Contributions
NE designed, developed, and supervised experiments, analyzed data, and wrote the paper. CA performed experiments. GT helped to draft the final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tiris, G., Alver, C. & Erk, N. A novel stability-indicating method for determination of a new antidepressant effect of vortioxetine in a pharmaceutical formulation by using RP-HPLC. Futur J Pharm Sci 6, 118 (2020). https://doi.org/10.1186/s43094-020-00135-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-020-00135-w