
Abstract
Background
Two genes are called synthetic lethal (SL) if mutation of either alone is not lethal, but mutation of both leads to death or a significant decrease in organism's fitness. The detection of SL gene pairs constitutes a promising alternative for anti-cancer therapy. As cancer cells exhibit a large number of mutations, the identification of these mutated genes' SL partners may provide specific anti-cancer drug candidates, with minor perturbations to the healthy cells. Since existent SL data is mainly restricted to yeast screenings, the road towards human SL candidates is limited to inference methods.
Results
In the present work, we use phylogenetic analysis and database manipulation (BioGRID for interactions, Ensembl and NCBI for homology, Gene Ontology for GO attributes) in order to reconstruct the phylogenetically-inferred SL gene network for human. In addition, available data on cancer mutated genes (COSMIC and Cancer Gene Census databases) as well as on existent approved drugs (DrugBank database) supports our selection of cancer-therapy candidates.
Conclusions
Our work provides a complementary alternative to the current methods for drug discovering and gene target identification in anti-cancer research. Novel SL screening analysis and the use of highly curated databases would contribute to improve the results of this methodology.
Similar content being viewed by others
Background
High-throughput analyses have provided a tremendous boost to massive drug screening [1]. However, these improved techniques are still blind to biological or structural knowledge. In this sense, chemogenomics provides a complementary strategy for a rational screening that includes structural information of chemical compounds for gene targets [2, 3]. Computational approaches in this so-called virtual screening allow the matching of compounds to their specific gene-product targets, completing the experimental screening [4]. However, the computational approach is still limited by the huge combinatorics represented by the chemical space of possibilities associated to the compounds and their possible targets. As a consequence, all these experimental and computational approaches require the use of the cumulative biological knowledge. For this purpose, database integration into an ontological organization of the current biological knowledge has been suggested as a way to reduce the combinatorics either in virtual or experimental screenings [5]. The work presented here belongs to this last framework, intended as a tool for identifying potential targets for anti-cancer therapy. Cancer is a heterogeneous disease with numerous causes and typologies [6]. One of the essential traits of cancer progression is the underlying high mutational capacity of tumor cells [7–9], having as a consequence the rapid adaptive capacity of the disease. It has been suggested that these ingredients define cancer progression as a Darwinian micro-evolutionary process [10]. As a consequence, cancer cells which have lost essential genes by a mutation are eliminated from the tumor population. Therefore, it is expected that essential genes are conserved in cancer. Under this perspective, targeting essential genes in anti-cancer therapy could kill malignant cells, but might result to be lethal for healthy cells too. This is the case of the anti-proliferative drugs that also damage high turnover tissues, such as epithelium.
The problems reported from the failure of most single-target drug treatments [11] suggest that a new perspective is needed. In this context, a different conceptual framework related with synthetic lethality has been suggested for anti-cancer research [12–14]. Two genes are called synthetic lethal (SL) if mutation of either alone is not lethal, but mutation of both leads to death or a significant decrease in organism's fitness. According to screening methodology, two main types of mutations are considered: amorphic and hypormorphic mutations. The former causes a complete loss of gene function, while the latter refers to a mutation leading to a decreased activity in the respective gene function [15]. In genome-wide screenings of genetic interactions, hypomorphs are associated to essential genes such that the decrease of the gene expression does not result in inviable organisms [16].
The rationale of synthetic lethality offers new insights on selective anti-cancer therapy design by exploiting the existence of SL partners of mutated (cancer-related) genes [12, 17, 18]. Accordingly, given a mutated gene causing function deletion (amorphic mutation) or function decrease (hypomorphic mutation) in a cancer cell, an attack using specific drugs to block the activity of one of its SL partners would cause a lethal condition in such tumor cells. Meanwhile, only minor damage in healthy cells would be expected, constituting thus a selective anti-cancer therapy (see Figure 1). And thus, this approximation can help to overcome a dramatic limitation in drug design.

The rationale of synthetic lethality applied to the design of novel anti-cancer therapies. Two linked nodes (blue circles) represent a SL interaction. (A) In cancer disease, one of the SL partners would appear mutated (red triangle) contrasting to healthy cells where no mutation is accounted for. (B) The attack by drugs to the SL partner of the cancer-related mutated gene might cause a selective damage to tumor cells. In this case, the inactivation of both SL partners only happens in tumor cells.
Another relevant aspect in drug screening is that one drug is tested only for a specific disease and related pathologies. Given a SL pair of genes as described above, one cancer mutated and the other non-mutated, conceptually it is possible that an already approved and even commercialized drug might block the activity of the non-mutated gene product. Therefore, SL-partner screening has a special interest for gene-target identification but also for drug repositioning, i.e, the discovering of novel uses for old drugs [19].
Unfortunately, large-scale screenings of SL gene pairs have been performed only in yeast [20–23] and, to a significantly smaller degree, in C. elegans [24–26] and in other model organisms. To overcome this limitation, we propose the use of the phylogenetic inference of SL genes from yeast to human for pharmacological purposes.
Synthetic lethal screens in yeast have been used to identify genes involved in cell polarity, secretion, DNA repair and cell cycle [27, 28]. Due to the high conservation of genome integrity and cell-cycle related genes from yeast to higher organisms and their close relation with cancer disease [29], massive screenings of yeast SL interactions can provide a valuable information for SL inference applied to novel cancer therapy search. We emphasize though that the aim of this work is not to provide a general inference method of SL network from one organism to another. Instead, it adds to the rationale of drug design by supplying a candidate-list of human gene pairs, potentially SL, that could constitute the basis for future pharmacological testing. Additionally, network thinking has provided an excellent framework for the study of very large genetic systems. In particular, gene-disease [30, 31], gene-target [32] and synthetic-lethal [23, 33, 34] network representations have contributed to the understanding of these systems as a whole. Following this approach, we integrate the information in a network framework from several databases in order to provide supporting evidence for candidates' reliability.
Methods
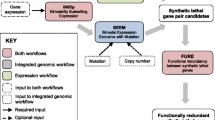
Our identification of potential anti-cancer gene targets proceeds through the integration of the biological information originating from different databases. Figure 2 illustrates the methodology for the data-collection process and for the selection of potential anti-cancer gene targets. The yeast SL network was constructed from the yeast SL interaction list available from BioGRID database [35]. In this network, nodes represent genes and the link between them indicates a SL interaction, i.e. when both are simultaneously mutated, a lethal condition is satisfied. The phylogenetic inference from yeast to human genes was obtained from the Ensembl database noteEnsembl: http://ftp.ensembl.org/pub/current_emf/ensembl_compara/homologies, March 3rd, 2008. The yeast-genes list belonging to SL network was contrasted with this database. The inferred human SL network (in short iHSLN) is then obtained by introducing the yeast SL interactions on their human phylogenetically-conserved counterparts, that is their orthologs (see also [36]). Subsequently, as we detail below, the resulting network was filtered by different biological databases for public use: 1) COSMIC and Cancer Gene Census, 2) Gene ontology and Gene ontology annotation (GO and GOA) and 3) DrugBank database.

Schematic representation of our methodology. Potential anti-cancer gene-targets are proposed for future experimental validation.
Obtaining the yeast synthetic lethal network
The collected data on yeast genetic screens including synthetic lethality is available at BioGRID database (version 2.0.38) [35]. From this data, we retain only those genes having a systematic name as it appears in http://www.yeastgenome.org. The resultant compilation is derived from 1233 articles, with half of the total of 12707 SL relations being contained in five main articles [16, 27, 28, 37, 38]. As commented above, the data consists of knock-out and hypomorph experiments, with the latter corresponding to essential genes [16]. The list of essential genes has been downloaded from the Saccharomyces Genome Deletion Project http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html.
Obtaining the iHSLN
Homologs between H. Sapiens and S. cerevisiae were obtained by similarity measures from the whole-genome multiple alignments by the Ensembl comparative genomics team. Human genes in this study were named according to HUGO gene nomenclature committee (HGNC http://www.genenames.org/). Each entry of the homology data file contains gene's evolutionary history corresponding to a gene tree that diverged from a common ancestor (see Figures 3 and 4). In our study, gene conservation in yeast and human can be summarized in three types of relations. The simplest case corresponds to one-to-one relation between yeast and human genes (orthologous relation). However, duplication events during evolution led to two alternative cases (see Figure 3B), where one yeast gene has more than one human homolog (one-to-m relation), and vice versa (n-to-one relation) [36]. The case of n-to-m was rarely encountered (Figure 3C). A yeast-human inference relation is straightforward for the one-to-one cases, and the human gene inherits the SL neighbors of its yeast homolog. On the contrary, the paralog cases require the introduction of approximate relations. More precisely, we manually curated all the cases where the inference is not one-to-one. For the one-to-m cases, we collapsed the multiple human genes into a single node that inherits the SL links of the yeast ortholog (Figure 4A). This situation does not affect the pattern of interactions derived from yeast SL network. In addition, by checking the biological function of these m human genes, we classify them into subsets of similar functions, and collapse these subsets into separate nodes. Thus a yeast gene having m orthologs might correspond to more than one node in the human SL network. Again, we emphasize the distinction between the single-gene nodes in the yeast SL network and the potentially multiple-gene nodes in the iHSLN. The statistics of the genes vs nodes the from gene-inference process is illustrated in Figure 5.

The orthology relation in the process of yeast-to-human inference method. The one-to-one case of one yeast gene having one human homolog (A). The n-to-one case of several human genes having the same yeast homolog (B). The one-to-m case can be defined analogously. The n-to-m is a very rare case where more than one human gene are homologs of more than one yeast gene (C).

Gene-node convention for the iHSLN construction. Every node in yeast SL network (YSLN) has associated gene. (A) For n-to-one, the corresponding node in the iHSLN contains the human paralogs of the yeast homologous counterpart. (B) In the case that two SL yeast partners are phylogenetically related to a single human gene, an autolink appears in the human SL network. This is and artifact of the method. However, we have kept this information to provide a more detailed picture in case such a node is a suitable candidate.

The statistics of the genes vs nodes the from gene-inference process.
The n-to-one cases merge the SL information from the multiple yeast nodes, and therefore a single human gene inherits the SL interactions from more than one yeast gene. The extreme case is the third one, n-to-m.
Gene function statistical analysis of the iHSLN
Significance of the biological-function representation in iHSLN was evaluated by means of a hypergeometric test using Benjamini & Hochberg false discovery rate with a P-value < 0.05 as a minimal cut-off. All the genes belonging to iHSLN, independent on whether they belong to multi-gene nodes, were considered in the analysis. Only eight genes were not associated to any GOA term. Statistics were performed for biological-function GOA terms using BINGO 2.3 Cytoscape plugging [39]. A detailed report including the list of genes related to specific GOA terms is available at Additional File 1.
Filtering the iHSLN
Biological filtering through the use of databases led to the functional classification of the nodes forming the iHSLN into: (1) cancer-related genes, (2) genes related to the DNA repair mechanism and cell cycle (two relevant functions altered in most cancers) and (3) drug-target genes.
In order to identify cancer-related candidates we use the COSMIC database(COSMIC: http://www.sanger.ac.uk/genetics/CGP/cosmic: 37thversion) that stores the current knowledge on somatic mutations and related details on human cancers. The information was completed by the use of the genes from the Cancer Gene Census http://www.sanger.ac.uk/genetics/CGP/Census that is a project to catalogue those genes for which mutations have been causally implicated in cancer.
Gene Ontology (GO http://www.geneontology.org, April 6th, 2008) database provides the biological description of gene products for a number of predefined functions. To relate one gene with its GO annotation, we used the gene ontology annotation (GO numbers) from GOA http://www.ebi.ac.uk/GOA/ 8th,2008). We also used the available relationships between GO terms [40] to create different filters for different biological processes, in particular for Cell cycle and DNA damage.
In addition to these databases, we have also used the information extracted from the DrugBank (http://www.drugbank.ca, release 2.0). It provides the available studies from the US Food and Drug Administration that relate tested drugs and their gene targets [32, 41, 42]. In this way, we inquire whether clinical studies of drugs acting on our proposed candidates have been previously described in literature. All the representations of the resultant networks were performed with Cytoscape [43]. Networks are provided as additional files.
Results
The iHSLN
Yeast SL network is a graph of 2383 nodes and 12707 links. This network is an extension of the SL network constructed by [28], who remarked that SL interactions yield a giant component with a non-random topology of small-world characteristics.
According to Ensembl database, phylogenetic inference revealed that 52% of yeast SL nodes (1253 of 2383 SL yeast genes) has at least one putative human ortholog (see Figure 5).
Previous analysis of global genomic homology between species estimated that the coverage between S. cerevisiae and H. sapiens is about 20% [44]. The higher percentage observed from the set of the yeast SL genes compared with the global coverage may suggest a possible conservation for SL proteins. However, it is also worth noting that a criterion for gene selection in SL screenings is precisely a high conservation along evolution, and this could be the cause of the high proportion of inferred human genes in the SL set. From the above-mentioned 1253 genes, only 1078 genes have a SL partner, that is those genes among the 1253 that have SL partners without any human ortholog are eliminated (Additional file 2). The final human SL network presented a set of 1002 nodes and 2847 links (Figure 6 and Additional files 4 and 5), where almost all the nodes formed a giant component (933 nodes) with a non-random topology. Considering yeast-to-human inference relations, the nodes in the human network resulted from 766 one-to-one homology cases, 64 human nodes are composed of 138 genes due to the n-to-one cases, and 170 nodes are formed of 386 genes due to one-to-m relations (more precisely, 148 nodes include gene of similar biological function, while 21 nodes contain genes with heterogeneous functionality). Only 2 nodes came from 2 n-to-m situations. We mention that 1.5% (43 nodes) of the links in the human network were autolinks (see Figure 4B for explanation). This is a result of the n-to-one cases, as among these n yeast genes some may be SL partners. This is an artifact of the human SL network. These autolinks in the iHSLN are not eliminated from our network, as the discussion concerning these nodes as candidates needs to be addressed with caution. We stress once more that in our subsequent analysis we refer to nodes rather than to simple genes, as a node may contain several genes.

Statistical analysis of biological-function representation in iHSLN indicated a marked significant overabundance (P-value ≪ 0.05) of genes related to genome integrity and cell cycle among others (see Table 1). This fact is explained by the enrichment of genes related to DNA integrity functions due to the selection criterion applied to yeast SL screenings, enrichment that holds in iHSLN too by the high conservation of these genes. As previously pointed out, such conserved functions in iHSLN are closely related to cancer disease providing a valuable set of potential SL candidates for humans.
Obtaining potential cancer-related SL targets
Cancer-related database approach
In order to evaluate potential candidates, we first identify those human genes in our inference list that are known to be involved in cancer mutations, as detailed in Material and methods. Given this information, the SL partners of these genes are then potential candidates for anti-cancer therapy. In the inset of Figure 7 we represent the 124-nodes sub-network containing those nodes in the human SL network that have been found in the COSMIC and Cancer Gene Census databases. In this network, we observe the SL interactions between cancer-related genes. However, our objective is to obtain possible candidates, that is the SL partners of these 124 that are not described as cancer-related genes. Therefore, we extract from the iHSLN also the first neighbors of these 124 nodes. We illustrate this network in Figure 7 where triangles depict cancer-related nodes and circles indicate their neighbors that are not known to be cancer-related in the used databases. This figure represents a map of the potential SL candidates to be targeted when a given cancer-related mutation is predominant in a tumor. To evaluate the significance of cancer-cancer correlation in iHSLN, we performed a randomization with 5000 runs of the cancer-node attributes maintaining the topology of the network from Figure 7. The statistical test using t-student revealed that cancer-cancer links are significantly overabundant in the iHSLN.(Additional file 6)

Cancer-related genes and their first neighbors from the SL human network (Additional file 6). The inset illustrates the sub-network of cancer-related nodes alone (59 nodes). Nodes without SL partners (65 of 124) were eliminated from this picture. In the large network, both cancer-related (triangles) and their neighbors (circles) in the SL human network. Only the links connecting a cancer-related gene and its neighbor (411 nodes and 694 links) are illustrated.
A word of caution is required before interpreting Figure 7. Cancer-related information is a compendium obtained from many samples, cancers and types of mutations. Given a SL pair conserved in human, if amorph (or even hypomorph) mutations occur in both genes within a tumor cell then a lethal condition should be expected according to our rationale. Following this reasoning, such a combination of mutants should not be observed within a cell. If such an event results in Figure 7, we would state that this combination is not conserved in humans according to SL definition. Therefore, we interpret cancer-cancer SL interactions to occur in different samples.
Notice that the cancer-related databases include both those genes that are also overexpressed in tumor cells. Even though the expression level information is not always available, this information is determinant for a correct selection of SL candidates. The most likely candidates presented in this work are chosen according to an underexpression scenario. In relation with this issue, among the many types of mutations encountered in the databases we wish to single out a few examples of mutations types. Some cancer-related genes in Figure 7 correspond to cases of nonsense mutations, in other words point mutations causing a premature stop codon. Therefore, we expect that such mutations critically damage the function of corresponding proteins. These genes are ATM, NF1, FBXW7, MSH2, BUB1, ERCC2, BLM and MSH6. The first four genes are also documented in the Cancer Gene Census to suffer deletions in different types of cancers.
Providing a proof of the potential predictability of our iHSLN is not straightforward due to the sparseness of data and the lack of a systematic identification of SL in humans. However, a number of examples can be extracted from literature, supporting the utility of our approach. One of them is the above cited BLM gene. It is a highly connected gene with 24 SL partners in the iHSLN that codifies a helicase homolog to SGS1 yeast gene. Its deficiency causes the Bloom syndrome, an autosomal recessive disorder with high disposition to tumorigenesis process. In a work performed in Drosophyla, BLM-MUS81 double mutation is an experimental verification of a SL interaction observed in our iHSLN [45]. This fact has a special interest since BLM helicase facilitates MUS81 endonuclease activity in human cells [46]. More recently, a SL interaction between the above cited NF1 gene and RAD54B, an homologous of the yeast RAD54, was experimentally determined in cancer cells [47]. Although yeast screenings identified this double mutant as a SL pair, due to database incompleteness, the phylogenetic relation of RAD54 was lost during inference process. As we discuss later, the quality of data is a considerable pitfall for a good performance of this methodology.
Another indirect evidence of a potential verification comes from the lethal effect of ATM gene deletion in defective Fanconi anemia (FA) pathway cells. FA pathway inactivation is strongly associated to tumorogenesis process. Interestingly, an activator of this pathway is ATR, a SL partner of ATM in the iHSLN [48].
One of the hubs observed in Figure 7 corresponds with CDC73. It has been observed that mutations in this gene (also known as HRPT2) are associated with malignancy in sporadic parathyroid tumors and hereditary hyperparathyroidism-jaw tumor syndrome [49]. Parafibromin is a tumor suppressor protein encoded by HRPT2 that binds to RNA polymerase II as part of a PAF1 transcriptional regulatory complex. Expression inhibition by HRPT2 RNA interference stimulates cell proliferation and increases the levels of the c-myc proto-oncogene product [50]. According to Figure 7, the CDC73 hub establishes a large number of potential SL candidates. We must stress that, due to the long phylogenetic distance between human and yeast, some of them are likely false positives. Nevertheless, biological arguments in favor of their relevance can be sought in order to strengthen their candidacy. This is the case of the SL link between CDC73 and genes related with the RNA polymerase machinery such as RPAP2 [51], and the transcription elongation factor TCEA3 [52] that capture the common function in RNA processing of these partners. In addition, CDC73 has as SL partner in Figure 7 the JUP (junction plakoglobin or γ-catenin) protein, a component of the catenin complex. JUP is involved in cell adhesion [53], an apparently very distant function regarding RNA processing. However, γ-catenin strongly activates the proto-oncogene c-myc [54]. According to this evidence, it seems reasonable to consider that a biological pathway relates JUP and CDC73 (depicted as SL partners in our study) with c-myc, but with opposite effects. In this case, our methodology reveals a potential anti-cancer strategy: inhibiting γ-catenin expression would compensate the activation of c-myc by the HTRP2 cancer mutation.
GOA filtering approach
In a second approach, and in agreement the to statistical significance of the functional analysis (notably, all GOA terms presented in Table 1 produced a very significant P-value), we added a new layer of information by using the cancer-related functions extracted from GOA. In so doing, we searched for candidates that might not be included in cancer-related databases, but their functions are essential for cancer progression. These functions have an special interest due to, it is well established that mutations in genes involved in DNA damage repair or in cell cycle checkpoints are associated with tumor progression [55]. In this sense, synthetic lethal relationships between DNA-replication genes (such as certain DNA polymerases) and DNA-repair genes (such as mismatch-repair genes) are well documented in model organisms [29, 56]. Moreover, it seems likely that the efficiency of many anti-cancer drugs that interfere with DNA synthesis is in some cases due to the presence of tumor-associated mutations affecting DNA repair or the response to DNA damage [18].
The connected sub-network from Figure 8 (Additional file 7) was obtained by extracting the genes annotated for these functions. Interestingly, the analysis shows that for six SL pairs one gene is directly cancer-related, whereas the other is not known to be so (Table 2). These latter genes constitute preliminary and putative candidates for future experimental validation. It is also worth noting that, as shown in Table 2, we also consider as candidates some SL partners with no known relation to cancer. They are SL partners consistent with a within pathway approach [23] (they act, or are part of the same complex in a pathway). Moreover, we notice that these cancer-unrelated SL pairs (labeled with asterisk in Table 2) consist of essential genes and thus come from hypomorph experiments. Their essential nature strengthens the likelihood of their belonging to the within-pathway model rather than to the between-pathway one (they function in parallel pathways).

SL-inferred human orthologs organized by GO numbers (Additional file 7). Genes' color denotes their biological function (GO number): in blue, cell-cycle related genes, in red, DNA damage related genes and in white, genes related to both processes.
An illustrative case by means of the GOA filtering is again CDC73, also observed in the previous section. In this case, GOA filtering offers evidence of its relation with the DNA damage related gene MMS19 (see Table 2) and helicase component. Interestingly, helicases have been proposed as targets for anti-cancer therapy [57] as they are closely related with the required genetic instability for tumor progression.
Drug association to SL human target genes
DrugBank database information was used here to establish possible relations between existing drugs and inferred SL human genes. We have found that 130 nodes of our iHSLN contain genes associated to one or several drugs in the DrugBank. More specifically, 17% of them are anti-cancer drugs. See Additional file 3 for pattern of interactions for this set of nodes. As we shall see in the next section, the combination of cancer-related and drug-target information into the iHSLN produces a set of genes that are known drug targets and have cancer-related SL partners, i.e. the suitable candidates according to our methodology illustrated in Figure 1.
Finding SL partners of cancer-related genes associated to drugs
As we previously argued, SL partners of cancer-related genes constitute a set of potential targets for anti-cancer treatment. The knowledge about drugs affecting these genes provides, on one hand, supporting evidences of our methodology. This is the case of cancer-unrelated genes for which there exists an anti-cancer drug, genes that are also SL partners of a cancer-related gene. On other hand, when the cancer-unrelated gene is associated to a cancer-unrelated drug, we have a potential new use of a drug that initially has not been conceived for anti-cancer treatment. The integration of this information is included in Figure 9 (Additional file 8). In this network, we eliminated the cancer-unrelated nodes that are not associated to any drug. The remaining set contains 155 nodes of which 124 are cancer-related nodes and 31 are drug-target nodes. A subset of 47 nodes are isolated nodes that match the number of cancer-related nodes having lost all the SL partners during filtering process, and it also indicates that no drug is associated to their SL neighborhood in the current version of DrugBank. Moreover, we notice that 28 nodes of 155 correspond with essential genes in yeast, according to Saccharomyces Genome Deletion Project http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html. Randomization and the subsequent statistical test using t-student revealed that cancer-to-drug-target links are significantly overabundant in the iHSLN.

Drug-cancer SL network representing the set of cancer-related nodes and their SL drug-associated partners (Additional file 8). Triangles represent cancer-related nodes and circles, SL drug-associated partners. Red color indicates that this node has associated a gene which is target of anti-cancer drug. Green color indicates a non-cancer drug associated to the node. Blue color only appears in triangles representing that there is no drug in the DrugBank database associated to these cancer-related genes. Essential genes are overlined. Large nodes are examples of existent anti-cancer drug target with a cancer-related SL pair (PPP6C-PSMB2, BUB1*-TUBB*, ERCC2-DCK7), and also our suggested examples of novel use for existent cancer-unrelated drugs that target a SL partner of a cancer-related gene (RAD50-PRDX2, MRE11A-PRDX2 AND PPP6C-CDC42). The BUB1* node represents BUB1 and BUB1B genes, whereas TUBB* node includes TUBB2B, TUBB3, TUBB2A and TUBB6 tubulin family members.
Figure 9 also singles out (large circles and triangles) three SL pairs that are fundamental to existent anti-cancer treatments consisting in targeting SL partners of the cancer-related genes. In the first example, ERCC2 (also known as XPD) is a cancer-related gene [58]. Studies of its homologous gene in Drosophila show that an excess of XPD produces a titration of CAK complex and reduces CDK7 activity, leading to a cell-cycle arrest [59]. Moreover, CDK7 is a protein of the CAK complex required for cell-cycle progression [60]. It has already been suggested as a potential target for anti-cancer therapies. Pharmacological implications are discussed in [60].
In the second example, PPP6C is a phosphatase involved in cell-cycle progression in human cells through the control of cyclin D1 [61]. Its SL partner, PSMB2, is a proteasome subunit related with protein degradation processes. Bortezomib, a therapeutic proteasome inhibitor is a target of PSMB2 (according to DrugBank information). It is pharmacologically approved for treating relapsed multiple myeloma and mantle cell lymphoma. In addition, the use of Bortezomib in hepathocarcinoma cells reduces the transcriptional levels of cyclin D1, among other effects, leading to cell-cycle arrest. It is postulated that cyclin D1 can act as an oncogene [62, 63]. In this case, we observe that PSMB2 and PPP6C affect the levels of this cell-cycle related gene.
The third example, BUB1* (here the * symbol implies that more than one phylogenetically-related proteins is contained in this node) is a prototype member of a family of genes, some of which encode proteins that bind to the kinetochore and all of which are required for a normal mitotic delay in response to spindle disruption. Mutations in this gene have been associated with aneuploidy and several forms of cancer [64]. Our results reveal that this gene is associated to TUBB* in the SL network. TUBB* represents a number of genes encoding tubulin proteins. Tubulins are targets of the anti-cancer drug Paclitaxel and the vinca alkaloids Vincristine and Vinblastine that also affect the mitotic-spindle assembly process [65].
Table 3 contains a representative list of the assignment of a potential anti-cancer use for existent cancer-unrelated drugs. As examples, two genes (and their drugs) are commented. The first one is PRDX2 (also known as peroxiredoxin-2), a gene encoding a member of the peroxiredoxin family of antioxidant enzymes, which reduce hydrogen peroxide and alkyl hydroperoxides [66]. It is interesting to mention that PRDX2 has been associated to cell proliferation and cell migration by regulation of the PDGF signalling [67]. N-carbamoyl alanine and 3-sulfinoalanine compounds were found to be inhibitors of PRDX2 activity and it has not been yet associated to any anti-cancer treatment. Two SL cancer-related partners of PRDX2, RAD50 and MRE11A are part of MRN protein-complex involved in DNA double-strand break repair, cell-cycle checkpoint activation, telomere maintenance and meiotic recombination [68].
The second example is the SL link between CDC42 and the previously described PPP6C. CDC42 is a gene participating in the rearrangement of actin cytoskeleton, membrane trafficking and cell-cycle progression, and it appears to be involved in cardiovascular diseases, diabetes and neuronal degenerative diseases [69]. This is a target gene of two other compounds (aminophosphonic acid-guanylate ester and guanosine 5'diphosphate) with no relation to cancer treatment in the DrugBank. However, it has been described that this gene is also involved in tumorigenesis and tumor progression, and the aberrant expression of CDC42 has been associated to colorectal tumors [69, 70]. It is worth mentioning that it has been observed that CDC42 controls the cell growth of anaplastic large cell lymphoma through its activation. Pharmacologic inhibition of CDC42 activity by secramine results in a cell-cycle arrest and apoptosis of these cells [71, 72]. We suggest thus that CDC42 inhibition by secramine constitutes a potential anti-cancer treatment, but unfortunately neither CDC42 nor secramine appeared in their respective databases used in this study. This example emphasizes once more that database information is a useful starting point for selecting new candidates. Interestingly, yeast ortholog of CDC42 is an essential gene and thus its mutant in the screening experiments is a hypomorph. Thus it suggests that partial inhibition of CDC42 could be enough to cause a lethal condition in those tumor cells where an amorphic mutation for PPP6C exists. In view of these comments, novel SL screening analysis and the use of highly curated databases would contribute to improve the results of this methodology.
Discussion
By the present study we propose a methodology for providing liable candidates for future experimental validation as drug targets for anti-cancer therapy. The methodology is based on the existence of synthetic-lethal relation between pairs of genes: two genes are synthetically lethal if their simultaneous mutation leads to inviable organism, while their separate mutation has no substantial effect on the organism's fitness. As conceptualized by previous works [17, 18], we used here the extensive experimental data on yeast in order to extend the knowledge to the human genome, and more precisely to anti-cancer therapy.
The rationale behind such approach is that, assuming that there are specific mutations for cancer cells, the identification and artificial mutation (drug action) of their SL partners would result in the death of cancerous cells alone. The mutations would affect also healthy cells, but would not drastically injure them. The combination of different biological databases provides potential filters for reducing the number of false positives. In this work a gene is considered cancer-related if it belongs to COSMIC or Cancer Gene Census databases. This implies certain limitations to our study since it depends on the accuracy and completeness of these databases. In this context, the better annotation of the used database, the more reliable the results. One example of that is the case of CDC42-PPP6C SL pair and the use of secramine drug resulting from a literature search but not from the current DrugBank version.
We have commented that cancer-cancer SL interactions could be interpreted as false positives as we do not expect them to occur in the same cell. We consider that the proof of this statement is an interesting working hypothesis to be tested in future research. Such a future research would aim at providing a quantitative estimation about the likelihood of observing a double mutation in tumor cells as it results from the data analysis. A supporting evidence of this hypothesis would be that SL cancer-related gene pairs are less likely to be observed in the double-mutation dataset. In spite of being related to the current work, we consider such a study to be outside the aim of the current work directed at introducing the potential of this methodology.
In addition, false positives can result also from the long evolutionary distance and different architectures between yeast and human genomes. At this point, the evolutionary conservation of SL pairs is controversial [29, 73–75], even though a recent study inclines the balance towards a significant conservation of synthetic lethal interactions between eukaryotes [75]. In addition, the existence of conserved SL associated to particular functions [29, 74, 76] is promising evidence for the inference methodology presented here. Even though not all yeast SL pairs are expected to be conserved in distant organisms, those associated to essential functions have a higher conservation probability. The identification of only a few of such partners could constitute alone an invaluable information in the strategy of drug design.
As Lawrence Loeb stated with his mutator-phenotype scenario for cancer evolution [7, 9, 77], some genetic instability but not too much is required for cancer progression. An illustrative example, the BUB1-TUBB* SL pair, is closely related with the strategy of forcing instability in order to kill cancer cells. It is reasonable to assume that an attack to tubulins by drugs in tumors where BUB1 appears mutated may drift the tumor population towards extinction by exceeding the limits of mutation tolerance. In this particular case, we speculate that treatments with vinca alkaloids should be more efficient in those cancers where BUB1 is mutated. Analogously, our results reveal the suitability of an attack to MMS19 helicase component when CDC73 is mutated. As argued by [78], compensatory helicase-dependent DNA repair pathways may represent a suitable target for anti-cancer therapy strategies that are designed to introduce DNA damage to tumors with pre-existing defined DNA repair deficiencies. In this context, we provide among our candidates the FEN1-BLM pair and the already confirmed MUS81-BLM [45], both suggested by [78] as potential targets in cancer therapy. However, other anti-cancer strategies such as the attack to protein degradation function by blocking PSMB2 proteasome component has been also uncovered by the presented methodology. In this case, our result suggests that this therapy should be more efficient in tumors where PPP6C is mutated.
Conclusions
We have proposed by the present study a tool for phylogenetic inference of candidates for future experimental validation as drug targets in anti-cancer therapy. Once more, we stress that we do not argue in favor of a methodology of SL-genes inference across distant species, as it has been already discussed in the literature to be a controversial step [73, 75]. Rather our study has a pharmacological utility and constitutes an alternative for massive drug screenings. In addition, the arguments brought forward in favor of the proposed candidates above justify their consideration for future experimental validation.
Furthermore, we provide an additional file on the results discussed above in order to foster the bioinformatic and pharmacological communities towards further analysis of this methodology.
Abbreviations
- SL:
-
Synthetic Lethal
- iHSLN:
-
inferred Human Synthetic Lethal Network
- YSLN:
-
Yeast Synthetic Lethal Network
- GO:
-
Gene Ontology
- HGNC:
-
HUGO gene nomenclature committee.
References
Pereira DA, Williams JA: Origin and evolution of high throughput screening. Br J Pharmacol. 2007, 152: 53-61. 10.1038/sj.bjp.0707373
Bajorath J: Integration of virtual and high-throughput screening. Nat Rev Drug Discov. 2002, 1 (11): 882-894. 10.1038/nrd941
Rognan D: Chemogenomic approaches to rational drug design. Br J Pharmacol. 2007, 152: 38-52. 10.1038/sj.bjp.0707307
Mestres J, Gregori-Puigjané E, Valverde S, Solé RV: Data completeness-the Achilles heel of drug-target networks. Nat Biotechnol. 2008, 26 (9): 983-984. 10.1038/nbt0908-983
Agarwal P, Searls DB: Literature mining in support of drug discovery. Brief Bioinform. 2008, 9 (6): 479-92. 10.1093/bib/bbn035
Weinberg R: The biology of Cancer. 2007, Garland. New York
Loeb LA: A mutator phenotype in cancer. Cancer Res. 2001, 61 (8): 3230-3239.
Loeb LA, Bielas JH, Beckman RA: Cancers exhibit a mutator phenotype: clinical implications. Cancer Res. 2008, 68 (10): 3551-7. discussion 3557. 10.1158/0008-5472.CAN-07-5835
Solé RV, Deisboeck TS: An error catastrophe in cancer?. J Theor Biol. 2004, 228: 47-54. 10.1016/j.jtbi.2003.08.018
Merlo LMF, Pepper JW, Reid BJ, Maley CC: Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006, 6 (12): 924-935. 10.1038/nrc2013
Fernández A, Crespo A, Tiwari A: Is there a case for selectively promiscuous anticancer drugs?. Drug Discov Today. 2009, 14 (1-2): 1-5. 10.1016/j.drudis.2008.10.008
Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH: Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997, 278 (5340): 1064-1068. 10.1126/science.278.5340.1064
Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, Minna JD, Michnoff C, Hao W, Roth MG, Xie XJ, White MA: Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007, 446 (7137): 815-819. 10.1038/nature05697
Meur NL, Gentleman R: Modeling synthetic lethality. Genome Biol. 2008, 9 (9): R135- 10.1186/gb-2008-9-9-r135
Wilkie AOM: The molecular basis of genetic dominance. J Med Genet. 1994, 31: 89-98. 10.1136/jmg.31.2.89
Davierwala AP, Haynes J, Li Z, Brost RL, Robinson MD, Yu L, Mnaimneh S, Ding H, Zhu H, Chen Y, Cheng X, Brown GW, Boone C, Andrews BJ, Hughes TR: The synthetic genetic interaction spectrum of essential genes. Nat Genet. 2005, 37 (10): 1147-1152. 10.1038/ng1640
Kamb A: Mutation load, functional overlap, and synthetic lethality in the evolution and treatment of cancer. J Theor Biol. 2003, 223 (2): 205-213. 10.1016/S0022-5193(03)00087-0
Kaelin WG: The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005, 5 (9): 689-698. 10.1038/nrc1691
Ashburn TT, Thor KB: Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004, 3 (8): 673-683. 10.1038/nrd1468
Forsburg SL: The art and design of genetic screens: yeast. Nat Rev Genet. 2001, 2 (9): 659-668. 10.1038/35088500
Pan X, Yuan DS, Xiang D, Wang X, Sookhai-Mahadeo S, Bader JS, Hieter P, Spencer F, Boeke JD: A robust toolkit for functional profiling of the yeast genome. Mol Cell. 2004, 16 (3): 487-496. 10.1016/j.molcel.2004.09.035
Ooi SL, Pan X, Peyser BD, Ye P, Meluh PB, Yuan DS, Irizarry RA, Bader JS, Spencer FA, Boeke JD: Global synthetic-lethality analysis and yeast functional profiling. Trends Genet. 2006, 22: 56-63. 10.1016/j.tig.2005.11.003
Boone C, Bussey H, Andrews BJ: Exploring genetic interactions and networks with yeast. Nat Rev Genet. 2007, 8 (6): 437-449. 10.1038/nrg2085
Jorgensen EM, Mango SE: The art and design of genetic screens: caenorhabditis elegans. Nat Rev Genet. 2002, 3 (5): 356-369. 10.1038/nrg794
Baugh LR, Wen JC, Hill AA, Slonim DK, Brown EL, Hunter CP: Synthetic lethal analysis of Caenorhabditis elegans posterior embryonic patterning genes identifies conserved genetic interactions. Genome Biol. 2005, 6 (5): R45- 10.1186/gb-2005-6-5-r45
Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG: Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet. 2006, 38 (8): 896-903. 10.1038/ng1844
Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Pagé N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, Andrews B, Tyers M, Boone C: Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001, 294 (5550): 2364-2368. 10.1126/science.1065810
Tong AHY, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, Chen Y, Cheng X, Chua G, Friesen H, Goldberg DS, Haynes J, Humphries C, He G, Hussein S, Ke L, Krogan N, Li Z, Levinson JN, Lu H, Ménard P, Munyana C, Parsons AB, Ryan O, Tonikian R, Roberts T, Sdicu AM, Shapiro J, Sheikh B, Suter B, Wong SL, Zhang LV, Zhu H, Burd CG, Munro S, Sander C, Rine J, Greenblatt J, Peter M, Bretscher A, Bell G, Roth FP, Brown GW, Andrews B, Bussey H, Boone C: Global mapping of the yeast genetic interaction network. Science. 2004, 303 (5659): 808-813. 10.1126/science.1091317
Yuen KWY, Warren CD, Chen O, Kwok T, Hieter P, Spencer FA: Systematic genome instability screens in yeast and their potential relevance to cancer. Proc Natl Acad Sci USA. 2007, 104 (10): 3925-3930. 10.1073/pnas.0610642104
Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabási AL: The human disease network. Proc Natl Acad Sci USA. 2007, 104 (21): 8685-8690. 10.1073/pnas.0701361104
Platzer A, Perco P, Lukas A, Mayer B: Characterization of protein-interaction networks in tumors. BMC Bioinformatics. 2007, 8: 224- 10.1186/1471-2105-8-224
Yildirim MA, Goh KI, Cusick ME, Barabási AL, Vidal M: Drug-target network. Nat Biotechnol. 2007, 25 (10): 1119-1126. 10.1038/nbt1338
Chipman K, Singh A: Predicting genetic interactions with random walks on biological networks. BMC Bioinformatics. 2009, 10: 17- 10.1186/1471-2105-10-17
Paladugu SR, Zhao S, Ray A, Raval A: Mining protein networks for synthetic genetic interactions. BMC Bioinformatics. 2008, 9: 426- 10.1186/1471-2105-9-426
Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M: BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006, D535-D539. 34 Database
Sonnhammer ELL, Koonin EV: Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet. 2002, 18 (12): 619-620. 10.1016/S0168-9525(02)02793-2
Pan X, Ye P, Yuan DS, Wang X, Bader JS, Boeke JD: A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell. 2006, 124 (5): 1069-1081. 10.1016/j.cell.2005.12.036
Krogan NJ, Keogh MC, Datta N, Sawa C, Ryan OW, Ding H, Haw RA, Pootoolal J, Tong A, Canadien V, Richards DP, Wu X, Emili A, Hughes TR, Buratowski S, Greenblatt JF: A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol Cell. 2003, 12 (6): 1565-1576. 10.1016/S1097-2765(03)00497-0
Maere S, Heymans K, Kuiper M: Bingo: a cytoscape plugging to assess over-representation of gene ontology categories in biological networks. Bioinformatics. 2005, 21 (16): 3448-3449. 10.1093/bioinformatics/bti551
, : Gene Ontology: tool for the unification of biology. Nature Genetics. 2000, 25: 25-29. 10.1038/75556
Hopkins AL: Network pharmacology. Nat Biotechnol. 2007, 25 (10): 1110-1111. 10.1038/nbt1007-1110
Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M: DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, D901-D906. 36 Database
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T: Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13 (11): 2498-2504. 10.1101/gr.1239303
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA: The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003, 4: 41- 10.1186/1471-2105-4-41
Trowbridge K, McKim K, Brill SJ, Sekelsky J: Synthetic lethality of Drosophila in the absence of the MUS81 endonuclease and the DmBlm helicase is associated with elevated apoptosis. Genetics. 2007, 176 (4): 1993-2001. 10.1534/genetics.106.070060
Zhang R, Sengupta S, Yang Q, Linke SP, Yanaihara N, Bradsher J, Blais V, McGowan CH, Harris CC: BLM helicase facilitates Mus81 endonuclease activity in human cells. Cancer Res. 2005, 65 (7): 2526-2531. 10.1158/0008-5472.CAN-04-2421
McManus KJ, Barrett IJ, Nouhi Y, Hieter P: Specific synthetic lethal killing of RAD54B-deficient human colorectal cancer cells by FEN1 silencing. Proc Natl Acad Sci USA. 2009, 106 (9): 3276-3281. 10.1073/pnas.0813414106
Kennedy RD, Chen CC, Stuckert P, Archila EM, la Vega MAD, Moreau LA, Shimamura A, D'Andrea AD: Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest. 2007, 117 (5): 1440-1449. 10.1172/JCI31245
Howell VM, Haven CJ, Kahnoski K, Khoo SK, Petillo D, Chen J, Fleuren GJ, Robinson BG, Delbridge LW, Philips J, Nelson AE, Krause U, Hammje K, Dralle H, Hoang-Vu C, Gimm O, Marsh DJ, Morreau H, Teh BT: HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet. 2003, 40 (9): 657-663. 10.1136/jmg.40.9.657
Lin L, Zhang JH, Panicker LM, Simonds WF: The parafibromin tumor suppressor protein inhibits cell proliferation by repression of the c-myc proto-oncogene. Proc Natl Acad Sci USA. 2008, 105 (45): 17420-17425. 10.1073/pnas.0710725105
Jeronimo C, Forget D, Bouchard A, Li Q, Chua G, Poitras C, Thérien C, Bergeron D, Bourassa S, Greenblatt J, Chabot B, Poirier GG, Hughes TR, Blanchette M, Price DH, Coulombe B: Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell. 2007, 27 (2): 262-274. 10.1016/j.molcel.2007.06.027
Labhart P, Morgan GT: Identification of novel genes encoding transcription elongation factor TFIIS (TCEA) in vertebrates: conservation of three distinct TFIIS isoforms in frog, mouse, and human. Genomics. 1998, 52 (3): 278-288. 10.1006/geno.1998.5449
Knudsen KA, Wheelock MJ: Plakoglobin, or an 83-kD homologue distinct from beta-catenin, interacts with E-cadherin and N-cadherin. J Cell Biol. 1992, 118 (3): 671-679. 10.1083/jcb.118.3.671
Kolligs FT, Kolligs B, Hajra KM, Hu G, Tani M, Cho KR, Fearon ER: Gamma-catenin is regulated by the APC tumor suppressor and its oncogenic activity is distinct from that of beta-catenin. Genes Dev. 2000, 14 (11): 1319-1331.
Massagué J: G1 cell-cycle control and cancer. Nature. 2004, 432 (7015): 298-306. 10.1038/nature03094
Tarailo M, Kitagawa R, Rose AM: Suppressors of spindle checkpoint defect (such) mutants identify new mdf-1/MAD1 interactors in Caenorhabditis elegans. Genetics. 2007, 175 (4): 1665-1679. 10.1534/genetics.106.067918
Litman R, Gupta R, Brosh RM, Cantor SB: BRCA-FA pathway as a target for anti-tumor drugs. Anticancer Agents Med Chem. 2008, 8 (4): 426-430.
Kiyohara C, Yoshimasu K: Genetic polymorphisms in the nucleotide excision repair pathway and lung cancer risk: a meta-analysis. Int J Med Sci. 2007, 4 (2): 59-71.
Chen J, Larochelle S, Li X, Suter B: Xpd/Ercc2 regulates CAK activity and mitotic progression. Nature. 2003, 424 (6945): 228-232. 10.1038/nature01746
Lolli G, Johnson LN: CAK-Cyclin-dependent Activating Kinase: a key kinase in cell cycle control and a target for drugs?. Cell Cycle. 2005, 4 (4): 572-577.
Stefansson B, Brautigan DL: Protein phosphatase PP6 N terminal domain restricts G1 to S phase progression in human cancer cells. Cell Cycle. 2007, 6 (11): 1386-1392.
Hinds PW, Dowdy SF, Eaton EN, Arnold A, Weinberg RA: Function of a human cyclin gene as an oncogene. Proc Natl Acad Sci USA. 1994, 91 (2): 709-713. 10.1073/pnas.91.2.709
Sanchez G, Delattre O, Auboeuf D, Dutertre M: Coupled alteration of transcription and splicing by a single oncogene: boosting the effect on cyclin D1 activity. Cell Cycle. 2008, 7 (15): 2299-2305.
Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B: Mutations of mitotic checkpoint genes in human cancers. Nature. 1998, 392 (6673): 300-303. 10.1038/32688
Jordan A, Hadfield JA, Lawrence NJ, McGown AT: Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. Med Res Rev. 1998, 18 (4): 259-296. 10.1002/(SICI)1098-1128(199807)18:4<259::AID-MED3>3.0.CO;2-U
Schäfer M, Werner S: Oxidative stress in normal and impaired wound repair. Pharmacol Res. 2008, 58 (2): 165-171. 10.1016/j.phrs.2008.06.004
Choi MH, Lee IK, Kim GW, Kim BU, Han YH, Yu DY, Park HS, Kim KY, Lee JS, Choi C, Bae YS, Lee BI, Rhee SG, Kang SW: Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature. 2005, 435 (7040): 347-353. 10.1038/nature03587
Williams RS, Williams JS, Tainer JA: Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol. 2007, 85 (4): 509-520. 10.1139/O07-069
Sinha S, Yang W: Cellular signaling for activation of Rho GTPase Cdc42. Cell Signal. 2008, 20 (11): 1927-1934. 10.1016/j.cellsig.2008.05.002
Pulgar TGD, Valdés-Mora F, Bandrés E, Pérez-Palacios R, Espina C, Cejas P, García-Cabezas MA, Nistal M, Casado E, González-Barón M, García-Foncillas J, Lacal JC: Cdc42 is highly expressed in colorectal adenocarcinoma and downregulates ID4 through an epigenetic mechanism. Int J Oncol. 2008, 33: 185-193.
Pelish HE, Peterson JR, Salvarezza SB, Rodriguez-Boulan E, Chen JL, Stamnes M, Macia E, Feng Y, Shair MD, Kirchhausen T: Secramine inhibits Cdc42-dependent functions in cells and Cdc42 activation in vitro. Nat Chem Biol. 2006, 2: 39-46. 10.1038/nchembio751
Ambrogio C, Voena C, Manazza AD, Martinengo C, Costa C, Kirchhausen T, Hirsch E, Inghirami G, Chiarle R: The anaplastic lymphoma kinase controls cell shape and growth of anaplastic large cell lymphoma through Cdc42 activation. Cancer Res. 2008, 68 (21): 8899-8907. 10.1158/0008-5472.CAN-08-2568
Tischler J, Lehner B, Fraser AG: Evolutionary plasticity of genetic interaction networks. Nat Genet. 2008, 40 (4): 390-391. 10.1038/ng.114
Tarailo M, Tarailo S, Rose AM: Synthetic lethal interactions identify phenotypic "interologs" of the spindle assembly checkpoint components. Genetics. 2007, 177 (4): 2525-2530. 10.1534/genetics.107.080408
Dixon SJ, Fedyshyn Y, Koh JLY, Prasad TSK, Chahwan C, Chua G, Toufighi K, Baryshnikova A, Hayles J, Hoe KL, Kim DU, Park HO, Myers CL, Pandey A, Durocher D, Andrews BJ, Boone C: Significant conservation of synthetic lethal genetic interaction networks between distantly related eukaryotes. Proc Natl Acad Sci USA. 2008, 105 (43): 16653-16658. 10.1073/pnas.0806261105
Measday V, Baetz K, Guzzo J, Yuen K, Kwok T, Sheikh B, Ding H, Ueta R, Hoac T, Cheng B, Pot I, Tong A, Yamaguchi-Iwai Y, Boone C, Hieter P, Andrews B: Systematic yeast synthetic lethal and synthetic dosage lethal screens identify genes required for chromosome segregation. Proc Natl Acad Sci USA. 2005, 102 (39): 13956-13961. 10.1073/pnas.0503504102
Cahill DP, Kinzler KW, Vogelstein B, Lengauer C: Genetic instability and darwinian selection in tumours. Trends Cell Biol. 1999, 9 (12): M57-M60. 10.1016/S0962-8924(99)01661-X
Aggarwal M, Brosh RM: Hitting the bull's eye: novel directed cancer therapy through helicase-targeted synthetic lethality. J Cell Biochem. 2009, 106 (5): 758-763. 10.1002/jcb.22048
Acknowledgements
This work was supported by the EU 6th Framework projects SYNLET (NEST-043312, AM and NCP) and ComplexDis (NEST-043241, CRC), NIH (CA 113004, RVS) and Santa Fe Institute (RVS). We are grateful to Bernd Mayer for inspiring this work, and to the SYNLET consortium for useful discussions. We also thank Nuria Lopez Bigas, Jordi Mestres Lopez, Miguel Angel Medina Torres and Josep Conde Colom for providing expertise and advice on this subject.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
NCP performed database manipulation, graph construction and drafted the manuscript. AM and CRC wrote the manuscript, conceived and designed the investigation. AM contributed to computational analysis and CRC performed the biological interpretation. RVS contributed to manuscript preparation and promoted the work. All authors have read and approved the final manuscript.
Electronic supplementary material
12918_2009_384_MOESM1_ESM.XLS
Additional file 1: Detailed information on the statistical analysis of iHSLN biological functions. File in a .xls format. (XLS 282 KB)
12918_2009_384_MOESM2_ESM.XLS
Additional file 2: List of 1078 human genes homologous to the 2383 SL yeast genes from the BioGRID database (Figure 5). File in a .xls format. (XLS 358 KB)
12918_2009_384_MOESM3_ESM.ZIP
Additional file 3: Sub-network of SL nodes that are also drug targets, and their first SL neighbors. File in .cys format. This file must be opened with cytoscape software. (ZIP 54 KB)
12918_2009_384_MOESM4_ESM.SIF
Additional file 4: List of SL relations between the inferred human SL genes (iHSLN). File in .sif format to be loaded in cytoscape software. Edges are represented in two columns separated by the label pp. (SIF 52 KB)
12918_2009_384_MOESM5_ESM.ZIP
Additional file 5: The network of SL relations between the inferred human SL genes (iHSLN) (Figure 6). File in .cys format. This file must be opened with cytoscape software. (ZIP 78 KB)
12918_2009_384_MOESM6_ESM.ZIP
Additional file 6: Cancer-related genes and their first neighbors from the SL human network (Figure 7). File in .cys format. This file must be opened with cytoscape software. (ZIP 40 KB)
12918_2009_384_MOESM7_ESM.ZIP
Additional file 7: An extract of SL-inferred human orthologs organized by GO numbers and related with DNA damage and cell-cycle (Figure 8). File in .cys format. This file must be opened with cytoscape software. (ZIP 16 KB)
12918_2009_384_MOESM8_ESM.ZIP
Additional file 8: Drug-cancer SL network representing the set of cancer-related nodes and their SL drug-associated partners (Figure 9). File in .cys format. This file must be opened with cytoscape software. (ZIP 68 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Conde-Pueyo, N., Munteanu, A., Solé, R.V. et al. Human synthetic lethal inference as potential anti-cancer target gene detection. BMC Syst Biol 3, 116 (2009). https://doi.org/10.1186/1752-0509-3-116
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1752-0509-3-116