
Abstract
Background
Bacteriophage vB_YenP_AP5 is a lytic bacteriophage capable of infecting Yersinia enterocolitica strains of serotype O:3, an epidemiologically significant serotype within this bacterial species that causes yersiniosis in humans. This work describes the complete genome sequence of this phage.
Results
The genome consists of linear double-stranded DNA of 38,646 bp, with direct terminal repeats of 235 bp in length, and a GC content of 50.7%. There are 45 open reading frames which occupy 89.9% of the genome. Most of the proteins encoded by this virus exhibit sequence similarity to Yersinia phage φYeO3-12 and Salmonella phage φSG-JL2 proteins.
Conclusions
Genomic and morphological analyses place the bacteriophage vB_YenP_AP5 in the T7likevirus genus of the subfamily Autographivirinae within the family Podoviridae.
Similar content being viewed by others
Background
Yersinia enterocolitica, a facultatively anaerobic, Gram-negative, non-sporulating, short bacillus, is an important zoonotic pathogen leading to human and animal enteric infection [1]. Among the species of the genus Yersinia, Y. enterocolitica is considered highly heterogeneous and is grouped into a biochemical scheme composed of six biotypes divided into three lineages: avirulent strains belonging to biotype 1A, highly pathogenic strains of biotype 1B, and weakly pathogenic strains of biotypes 2–5 that do not kill mice [2, 3]. Most strains associated with yersiniosis belong to bioserotypes 1B/O:8, 2/O:5,27, 2/O:9, 3/O:3, and 4/O:3, with the latter being the most common in Europe, Japan, Canada, and the USA [1, 4]. Although several yersiniophages have been described for typing Y. enterocolitica[5–8], few have been studied in detail via whole genome sequencing. To date, phage φYeO3-12 displaying specificity for Y. enterocolitica O:3 [9], phage PY54 exhibiting a host range restricted to Y. enterocolitica O:5 and O:5,27 [10], phage φR1-37 with a broader host range within Y. enterocolitica[11], and PY-100 exhibiting a broad host range restricted to the genus Yersinia[12], have been described. Given the considerable interest in bacteriophages because of their potential use as typing, diagnostic, therapeutic, decontaminating, and bio-control agents, our research is aimed at isolating and characterizing novel yersiniophages in order to expand the repertoire of phages available for targeting clinically significant Y. enterocolitica bioserotypes. In this manuscript we report the morphology, genome sequence, and transcriptomic analysis of phage vB_YenP_AP5 (hereafter referred to as AP5).
Results and discussion
Isolation and host range
Analysis of preliminary treated sewage resulted in the initial isolation of 12 phages infecting Y. enterocolitica strains. From these, AP5 was chosen for detailed study because of its ability to infect Y. enterocolitica strains of serotype O:3. The host range of AP5 was determined using 60 strains belonging to ten Yersinia species at 25°C and at 37°C. The results (Table 1) show that AP5 can form plaques only on Y. enterocolitica serotypes O:3, O:2, and O:1 (serotypes with an O antigen known to contain 6-deoxy-L-altropyranose). Other serotypes of Y. enterocolitica as well as other species within the genus Yersinia were unaffected by the presence of phage AP5. Additionally none of the Escherichia coli, Salmonella, or Listeria species strains tested were infected by this bacteriophage (data not shown). Analysis of Y. enterocolitica O:3 rough mutants YeO3-R1 and YeO3-R2 [13] (which are missing the O antigen), were not sensitive to AP5. YeO3-OC, a Y. enterocolitica O:3 deletion mutant (Δwzx-wbcQ) which is missing the entire core operon yet produces O antigen [14], was sensitive to AP5. In contrast, its derivative YeO3-OCR, a rough mutant which is also missing the entire core operon yet is unable to produce O antigen [14] was not sensitive to phage AP5. These results indicate the host receptor for phage AP5 lies within the O antigen of the lipopolysaccharide of Yersinia enterocolitica O:3 strains, and suggests the O side chain of this serotype (6-deoxy-L-altropyranose) is involved.
Morphology
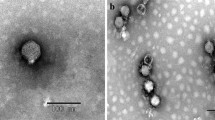
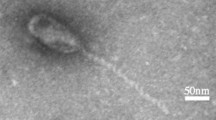
AP5 was negatively stained and examined by transmission electron microscopy (Figure 1). The head is icosahedral in shape exhibiting T7 symmetry of approximately 55.0 nm in diameter. The phage particles are each decorated with a short non-contractile tail of approximately 12.0 nm in length and 8 nm in width. Collectively, these morphological features indicate that this virus belongs to the family Podoviridae.

Electron micrographs of phage AP5. The phage has been negatively stained with 2% potassium phosphotungstate. AP5 is shown at 50,000× magnification or 150,000× magnification. Scale bar indicates size in nm.
General features of the AP5 genome
The DNA sequence of the phage AP5 consists of linear double stranded DNA of 38,646 bp in length. The size of this phage correlates well with other T7-like phage members, which range from 37.4 kb (Pseudomonas phage gh-1) to 45.9 kb (Erwinia phage Era103 [17]. The genomes of T7-like phages typically contain direct terminal repeats (DTRs) that are used during genome replication and packaging [18]. The lengths of the DTRs of AP5 (235 bp) are in agreement with the reported lengths for members of the T7 group, for example phage Salmonella phage φSG-JL2 and Yersinia phage φYeO3-12 have DTRs of 230 bp and 232 bp, respectively [9], whereas Enterobacteria phage T7 has DTRs of 160 bp [19]. Moreover, an alignment of the DTR sequences of phage AP5 and representative members of the T7likevirus genus show a high degree of conservation (Figure 2). Phage AP5 has also an overall genomic guanine plus cytosine (GC) content of 50.7%, compared to 48.5 ± 1.5 mol% for its host [20]. The GC contents of the common representatives of the T7 group, T7 (accession no. V01146.1 [complete sequence of 39,937 bp]) and T3 (accession no. NC_003298.1 [complete sequence of 38,208 bp]), are 48.4% and 49.9%, respectively. The GC content of phage AP5 is in agreement with other T7-like phages which range from 46.2 - 62.3% [21].

Multiple sequence alignment of the direct terminal repeats of phage AP5 and selected members of the T7likevirus genus. Multiple sequence alignment of direct terminal repeat sequences was performed using Clustal Omega [22]. Positions which have a single, fully conserved base pair are indicated by an asterisk (*).
Open reading frames and comparative genomics
The genome of AP5 was scanned for open reading frames (ORF) using computational software. A total of 34,743 nucleotides were involved in the coding of 45 ORFs with sizes ranging from 113 to 3,962 nucleotides (Table 2). The temporal and functional distributions of genes are tightly organized and packed close to each other so that they occupy 89.9% of the genome (Figure 3). The initiation codon ATG is present in 93.3% of the protein-coding genes. Only two other initiation codons occur, TTG and GTG at a frequency of 0.5%, and 0.2%, respectively. All predicted protein-coding genes were screened using BLASTP and Psi-BLAST algorithms against the non-redundant protein database at NCBI. From the 45 coding sequences (CDSs) of AP5, 30 (66.6%) have assigned function, and 15 (33.3%) are similar to proteins of unknown function. While the great majority of the homologs are to proteins of Yersinia phage φYeO3-12 (26), examples of primary sequence similarity to Salmonella phage φSG-JL2 (12), Enterobacteria phages T3 and T7 (6,1) and Klebsiella phage KP32 (1), exist. All of these phages are members of the T7likevirus genus. No function can be speculated about the hypothetical proteins of AP5 without further study. Based upon overall protein homology determined using CoreGenes [23, 24], AP5 shares 42 (76.4%), similar proteins with Enterobacteria phage T7 and Enterobacteria phage T3, 43 (78.2%) similar proteins with Salmonella phage φSG-JL2, and 43 (72.9%) similar proteins with Yersinia phage φYeO3-12. Collectively these results indicate that AP5 is a member of the Autographivirinae, specifically a member of the T7likevirus genus [25]. Thus, the T7 gene nomenclature was adopted for naming the genes of AP5. Since at the protein level phage AP5 showed the greatest sequence identity with Yersinia phage φYeO3-12 proteins, the genomes of the two phages were compared using progressive Mauve [26] (Figure 4). The gene arrangement of essential genes is collinear, highly conserved, and only some genes coding for hypothetical proteins present in φYeO3-12 are dissimilar or absent in AP5. The pairwise % identity of the phage AP5 genome to Yersinia phage φYeO3-12 genome was estimated at 89.6%.

Genetic and physical map of phage AP5. Predicted genes are arranged in the direction of transcription. Genes involved in nucleotide metabolism, DNA replication, and recombination are shown in black. Genes involved in phage assembly are depicted in red. Genes involved in DNA packaging and host lysis are shown in blue. Genes encoding hypothetical proteins with unassigned function are shown in yellow. The genetic map was created using EMBOSS[27].

Progressive mauve alignment of phage AP5 with Yersinia phage φYeO3-12. Inner tracks show regions of DNA sequence similarity (white) interspersed with regions where no sequence similarity exists (black) [26]. The gene arrangement of AP5 (top) and φYeO3-12 (bottom) is displayed in the direction of transcription. Genes depicted in red indicate genes that are dissimilar and or missing from either genome.
Nucleotide metabolism, DNA replication and recombination
In the AP5 genome, at least eleven genes were identified that play a role in nucleotide metabolism, DNA replication, and recombination. The transcribed genes function to overcome host restriction and to convert the metabolism of the host cell to the production of phage proteins. The product of gene 0.3 is a small protein, which mimics B-form DNA, and binds to and inhibits type I restriction endonucleases [28–30], as well as possessing S-adenosyl-L-methionine hydrolase (SAMase) activity acting to degrade the methyl group donor and the methylation activities present in the host [31]. Dam (DNA adenine methyltransferase) methylase modifies GATC, and Dcm (DNA cytosine methyltransferase) methylase modifies CC(A/T)GG sequences [9]. As in Yersinia phage φYeO3-12, the sequences corresponding to restriction enzyme recognition sites GATC and CC(A/T)GG, are underrepresented in phage AP5 DNA, occurring only 4 and 3 times, respectively. Gene 0.7 codes for a protein kinase involved in host transcription shutoff [32] and phosphorylates host elongation factors G and P and ribosomal protein S6 [33]. Other genes include an RNA polymerase (gene 1), a deoxyguanosine triphospho-hydrolase inhibitor (gene 1.2), a DNA ligase (gene 1.3), a host RNA polymerase inhibitor (gene 2), a ssDNA binding protein/helix destabilizing protein (gene 2.5), an endonuclease (gene 3), a primase/helicase (gene 4), a DNA polymerase (gene 5), a host recBCD nuclease inhibitor (gene 5.9), and an exonuclease (gene 6).
DNA packaging and morphogenesis
Several genes were identified that play a role in morphogenesis and DNA packaging. We identified two CDSs which display sequence similarity to the capsid proteins of phages belonging to T7-like viruses. The upstream gene 10A displays homology to Yersinia phage φYeO3-12 major capsid protein 10A [NP_052108], while the downstream gene 10B is similar to the minor capsid protein 10B in Salmonella phage φSG-JL2 [YP_001949782]. Some T7-like phages display two “versions” of the major capsid protein, which are designated as 10A and 10B [34]. The sequences of the amino termini of these proteins are identical, but during translation a -1 ribosomal frameshift allows for alternative reading frames within one mRNA, permitting the elongation of the protein product. The features of this system are a slippery site in the DNA/RNA and a downstream stem-loop structure capable of forming a pseudoknot [35, 36]. Analysis of AP5 using pKiss [37] did not yield evidence for a potential pseudoknot. Gene 9 was identified as the capsid assembly protein required for the formation of procapsids. The structure of this phage is therefore made up of gene 10A and gene 10B (capsid), the head to tail joining protein (gene 9), and an internal core formed by the products of gene 13 (internal virion protein A), gene 14 (internal virion protein B), gene 15 (internal virion protein C), and gene 16 (internal virion protein D). These proteins are homologous to those that form the internal core of the T7 virion. In T7, along with internal virion proteins B and C, the internal virion protein D, is ejected from the phage head and forms part of a putative channel that spans the entire host cell envelope and allows entry of DNA. The N-terminus of this protein has similarity to a lytic transglycosylase and may help form a channel for phage DNA translocation through the peptidoglycan layer of the host envelope [18]. BLASTN analysis of gene 16 (internal virion protein D) confirms the presence of a peptidoglycan hydrolase motif at the N-terminus. Gene 7.3 was identified as the tail assembly protein required for assembly of tail fibers on capsids. Genes 11 and 12 correspond to tail tubular proteins A and B respectively required for assembly of tails. Gene 17, codes for the tail fiber protein or host recognition binding protein and shares 89.3% identity with gp17, the tail fiber protein of Yersinia phage φYeO3-12 [NP_052117], and only 67% identity with gp17 of Salmonella phage φSG-JL2 [YP_001949790]. As with other gp17 homologs, sequence similarity is only found at the N-terminus, the part of the protein that is associated with the tail structure. The C-terminus is involved in ligand interactions and exhibits considerable differences, despite that phage AP5 shares a similar host range with Yersinia phage φYeO3-12 [9]. The large and small terminase subunit homologs were determined to be the products of gene 18 (DNA Packaging Protein A) and gene 19 (DNA Packaging Protein B).
Host cell lysis
The final stage of the phage lytic cycle is degradation of the bacterial cell wall and release of progeny phages. The lysis of the cell wall is typically induced by two phage encoded proteins, a holin and an endolysin [38]. Endolysins are muralytic enzymes produced by dsDNA phages, which hydrolyze the peptidoglycan layer of bacterial cell walls. As in other T7 phages, gene 3.5 of phage AP5 is proposed to be the endolysin protein since it possesses N-acetylmuramoyl-L-alanine amidase activity. Access of endolysins to the cell wall occurs through the presence of a secondary lysis factor, known as a holin. Holins are usually small proteins characterized by the presence of transmembrane domains (TMD) [39]. The predicted proteins of AP5 were scanned for TMDs using TMHMM [40]. TMDs were identified in gene 0.6, gene 6.3, gene 17.5, and gene 19.5, which code for small proteins of 67, 37, 67, and 49 amino acids, respectively. The derived protein from gene 17.5 of AP5 is proposed as a holin since it is a small protein containing an N-terminal TMD and shares sequence similarity to Yersinia phage φYeO3-12 lysis protein [NP_052118]. Phage AP5 has also one more lysis gene (gene 18.5) coding for a phage λ Rz-like lysis protein (PHA00276), an i-spanin of 150 amino acids which presents 98.7% sequence identity to λ Rz-like protein [YP_00194793] in Salmonella phage φSG-JL2. Further inspection of the gene 18.5 sequence, confirms the presence of a nested ORF of 255 bp (in the +1 reading frame) embedded entirely within the sequence coding for an o-spanin with homology to Rz1 (18.7) of bacteriophage T7. Based on these observations, gene 18.5 is proposed as an Rz/Rz1 equivalent lysis gene coding for transmembrane spanins involved in the disruption of the outer membrane of the host [41].
Transcriptional and regulatory sequences
Phage AP5 was not found to contain tRNA genes, which is not an unexpected observation since no T7-like phages have been found to harbour them. A promoter was identified at position 550–580 bp of the genome with sequence similarity to host promoter consensus TTGACA(N15-18)TATAAT with a 2 bp miss-match suggesting the early genes of this type of virus are transcribed by the host RNA polymerase. This is a major dissimilarity between phage AP5 and T3/T7 phages where the latter possess multiple strong promoters recognized by the host RNA polymerase. As with all T7 group phages, the AP5 phage encoded RNA polymerase (RNAP), is responsible for the recognition of phage specific promoters. In phage AP5, we identified 14 phage-specific promoters using PHIRE [42], which are named according to the downstream gene (Table 3). The promoter sequences lie within intergenic regions and show the greatest similarity to those of Yersinia phage φYeO3-12 and bacteriophage T3.
Conclusions
In this manuscript we have reported on the morphology and genome of the phage vB_YenP_AP5. Due to its lytic nature and marked specificity to Y.enterocolitica strains of serotype O:3, this phage is a potential biotechnological tool for diagnostic, therapeutic, and/or bio-control uses, given that O:3 is the most predominant serotype involved in human food-borne infections [4]. Additionally, the genome of this phage does not contain any undesirable laterally transferable genes that are related to bacterial toxins, pathogenicity, antibiotic resistance and/or lysogeny on the basis of homologies with known virulence and resistance genes available in GenBank.
Methods
Bacterial strains and growth media
Tryptic Soy Broth (TSB), Tryptic Soy Agar (TSA), and Tryptic Soft Agar (TSB +0.6% agar) (Difco Laboratories, Detroit, MI) were used to grow the host bacteria and to propagate the phage. In procedures involving phage infection, media were supplemented with filter-sterilized CaCl2.2H2O to a final concentration of 5 mM. Y. enterocolitica strains of serotype O:3 were used as indicator strains for phage isolation (Table 1). Y. enterocolitica 6471/76-c of bioserotype 4/O:3 [43] obtained from the Félix d’Hérelle Reference Center for bacterial viruses (Université Laval, QC, Canada) was used for phage propagation. Y. enterocolitica 6471/76, Y. enterocolitica strains of serotype O:1 and O:2, and Y. enterocolitica O:3 mutants, were acquired from the Haartman Institute, University of Helsinki, Finland. Other strains from the genus Yersinia were obtained from the Ontario Agency for Health Protection and Promotion (OAHPP) (Ontario, Canada), and the American Type Culture Collection (ATCC) (Manassas, Virginia, USA).
Isolation and propagation of phage
A 1 L sample of preliminary treated sewage from a local water treatment plant (Guelph, Ontario) was centrifuged twice at 10,000 g for 20 minutes at 4°C using a Beckman high-speed centrifuge and a JA-10 fixed-angle rotor (Beckman, Palo Alto, CA, USA) and the supernatant passed through a sterile filter membrane of 0.45 μm pore size (Fisher Scientific, Mississauga, ON, Canada). Equal 9 ml volumes of the filtered supernatant and TSB were inoculated with 200 μL of an overnight mixed culture of selected Y. enterocolitica strains of serotype O:3 and incubated for 18–24 h at 30°C with gentle shaking. After incubation, the enrichments were centrifuged at 10,000 g for 20 minutes at 4°C and the supernatant filtered through a sterile disposable filter of 0.45 μm pore size, and the filtrates stored at 4°C. Phages were detected by spot tests [16] on indicator strains incubating for 16–20 h at 25°C. Complete or partial lysis zones were then removed by cutting the soft layer from the plates using a sterile pipette tip and placing them separately in 1 mL of SM buffer (5.8 g of NaCl per liter, 2.0 g of MgSO47H2O per liter, 50 mM Tris–HCl [pH 7.5]), and used in standard double agar overlay plaque assays [43] to identify plaques showing different size and plaque morphology. Three rounds of repeated single plaque isolation were then performed to ensure unique phages were obtained. Purified phages were named following the naming convention of Kropinski et al. [44]. The small drop plaque assay was used to determine the titer of phage preparations [45].
Host range determination
The lytic activity of vB-YenP-AP5 was tested against 60 Yersinia strains as determined by standard spot tests [16]. Briefly, 10 μl from a purified phage suspension containing approximately 108 pfu/mL were spotted in the middle of a lawn of bacteria and left to dry before incubation for 18–24 h. Each strain was tested three times at 25°C and at 37°C. The degree of lysis was recorded using a four-point scale: (+4) complete clearing, (+3) clearing throughout but with a faint hazy background, (+2) substantial turbidity throughout the cleared zone, and (+1) a few individual plaques.
Transmission electron microscopy
The phage was pelleted at 25,000 × g for 1 hour at 4°C, using a Beckman high-speed centrifuge and a JA-18.1 fixed-angle rotor (Beckman, Palo Alto, CA, USA). The phage pellet was washed twice under the same conditions in neutral 0.1 M ammonium acetate [46]. The final phage sediment was re-suspended in 150 μL of SM-buffer supplemented with 5 mM CaCl2. Samples were then deposited onto carbon-coated Formvar films on copper grids, and stained with 2% uranyl acetate (pH 4) or 2% potassium phosphotungstate (PT, pH 7.2), air dried, and examined under a Tecnai G2 F20 transmission electron microscope (FEI, Hillsboro, OR, USA), operating at 120 KEv. Images were collected and analyzed using Digital Micrograph™ Software (Gatan, Pleasanton, CA, USA).
Isolation of phage DNA
To separate phage from bacterial debris, a crude phage lysate was centrifuged at 10,000 × g for 15 min at 4°C and the supernatant filtered through 0.22 μm low protein binding filter (Millipore, USA). Contaminating nucleic acids in the supernatant were digested with pancreatic DNase 1, and RNase A, each added to obtain a final concentration of 10 μg/mL (Sigma-Aldrich Canada Ltd., Oakville, ON), for 15 min at room temperature. DNA isolation was then performed with a commercial Phage DNA Isolation Kit (Norgen BioTek Corp.,Thorold, ON., Canada), as per the manufacturer’s instructions. The DNA was characterized spectrophotometrically.
Genome sequencing and assembly
Phage genomic DNA was fragmented using Ion Xpress™ Plus gDNA Fragment Library kit following the manufacturer’s protocol (Life Technologies, Foster City, CA). The fragmented DNA was collected using Pippin Prep DNA Size Selection System (Sage Science, Beverly, MA) and assessed for concentration and size distribution using a Bioanalyzer 2100 (Agilent Technologies, Mississauga, ON). The DNA fragments were then attached to the surface of Ion Sphere particles (ISPs) using an Ion Xpress Template kit (Life Technologies) according to the manufacturer’s instructions. Template-ISPs were sequenced using 316 micro-chips using an Ion Torrent Personal Genome Machine (PGM) with an Ion PGM Sequencing 400 kit (Life Technologies). The sequence reads were filtered using PGM software to remove low quality sequences, trimmed to remove adaptor sequences and the filtered sequences were assembled. The assembled genome had a coverage of 33.4×. Gaps were identified using the Lasergene® Genomics Suite of DNAStar software (DNAStar Inc., Madison, WI). The gaps were closed by PCR using primers flanking regions adjacent to the gaps and sequencing using a 3730 Genetic Analyzer (Life Technologies). The final assembled genome was manually curated for errors.
Bioinformatics analysis
The phage genome was analyzed for coding sequences using Kodon version 2.0 (Applied Maths Inc., Austin, TX, USA). Genes were identified from among the predicted coding sequences based on the presence of ATG, GTG, CTG or TTG start codons, followed by at least 30 additional codons, and an upstream sequence resembling the following ribosome-binding site, GGAGGT [47, 48]. A search for phage-encoded tRNA genes was performed with tRNAScan-SE and Aragorn, using default parameters [49, 50]. Preliminary annotation of genes was performed using myRAST [51]. Additional manual functional annotation was performed using the Geneious software version 7.1.5 (Biomatters) [52, 53]. Phage-specific promoters were discovered using PHIRE [42] using a length (L) of 22 bp and a degeneracy (D) of 4 bp. Determination of theoretical molecular weight and isoelectric point employed ExPASy via http://web.expasy.org/compute_pi/[54–56]. BLASTP and Psi-BLAST algorithms were used to determine the similarity to described proteins in the National Center for Biotechnology Information [NCBI] database (http://www.ncbi.nlm.nih.gov). Whole genome comparisons were carried out using Mauve [26], and CoreGenes [24].
Genome sequence
The annotated genome sequence for the phage vB_YenP_AP5 was deposited in the NCBI nucleotide database under the accession number KM253764.
References
Bottone EJ: Yersinia enterocolitica: Overview and epidemiologic correlates. Microbes Infect. 1999, 1 (4): 323-333. 10.1016/S1286-4579(99)80028-8.
Wauters G, Kandolo K, Janssens M: Revised biogrouping scheme of Yersinia enterocolitica. Contrib Microbiol Immunol. 1987, 9: 14-21.
Wren BW: The yersiniae-a model genus to study the rapid evolution of bacterial pathogens. Nat Rev Microbiol. 2003, 1 (1): 55-64. 10.1038/nrmicro730.
Fredriksson-Ahomaa M, Stolle A, Korkeala H: Molecular epidemiology of yersinia enterocolitica infections. FEMS Immunol Med Microbiol. 2006, 47 (3): 315-329. 10.1111/j.1574-695X.2006.00095.x.
Baker PM, Farmer JJ: New bacteriophage typing system for Yersinia enterocolitica, Yersinia kristensenii, Yersinia frederiksenii, and Yersinia intermedia: Correlation with serotyping, biotyping, and antibiotic susceptibility. J Clin Microbiol. 1982, 15 (3): 491-502.
Bergan T, Norris JR: Bacteriophage typing of Yersinia enterocolitica. Methods Microbiol. 1978, 1: 225-236.
Kawaoka Y, Mitani T, Otsuki K, Tsubokura M: Isolation and use of eight phages for typing Yersinia enterocolitica O3. J Med Microbiol. 1987, 23 (4): 349-352. 10.1099/00222615-23-4-349.
Nicolle P, Mollaret H, Hamon Y, Vieu JF, Brault J, Brault G: Lysogenic, bacteriocinogenic and phage-typing study of species Yersinia enterocolitica. Ann Inst Pasteur (Paris). 1967, 112 (1): 86-92.
Pajunen MI, Kiljunen SJ, Soderholm ME, Skurnik M: Complete genomic sequence of the lytic bacteriophage phiYeO3-12 of Yersinia enterocolitica serotype O:3. J Bacteriol. 2001, 183 (6): 1928-1937. 10.1128/JB.183.6.1928-1937.2001.
Hertwig S, Klein I, Schmidt V, Beck S, Hammerl JA, Appel B: Sequence analysis of the genome of the temperate Yersinia enterocolitica phage PY54. J Mol Biol. 2003, 15 (3): 605-622.
Kiljunen S, Hakala K, Pinta E, Huttunen S, Pluta P, Gador A, Lonnberg H, Skurnik M: Yersiniophage phiR1-37 is a tailed bacteriophage having a 270 kb DNA genome with thymidine replaced by deoxyuridine. Microbiology. 2005, 151 (12): 4093-4102. 10.1099/mic.0.28265-0.
Schwudke D, Ergin A, Michael K, Volkmar S, Appel B, Knabner D, Konietzny A, Strauch E: Broad-host-range yersinia phage PY100: Genome sequence, proteome analysis of virions, and DNA packaging strategy. J Bacteriol. 2008, 190 (1): 332-342. 10.1128/JB.01402-07.
Al-Hendy A, Toivanen P, Skurnik M: Lipopolysaccharide O side chain of Yersinia enterocolitica O:3 is an essential virulence factor in an orally infected murine model. Infect Immun. 1992, 60: 870-875.
Biedzka-Sarek M, Venho R, Skurnik M: Role of YadA, Ail, and Lipopolysaccharide in serum resistance of Yersinia enterocolitica Serotype O:3. Infect Immun. 2005, 73: 2232-2244. 10.1128/IAI.73.4.2232-2244.2005.
Skurnik M: Lack of correlation between the presence of plasmid and fimbriae in Yersinia enterocolitica and Yersinia pseudotuberculosis. J Appl Bacteriol. 1984, 56: 355-363. 10.1111/j.1365-2672.1984.tb01362.x.
Kutter E: Phage Host Range and Efficiency of Plating. Bacteriophages: Methods and protocols, Volume 1: Isolation, characterization, and interactions. Edited by: Clokie MRJ, Kropinski AM. 2009, New York: Humana Press, 141-149. 1
Kovalyova IV, Kropinski AM: The complete genomic sequence of lytic bacteriophage gh-1 infecting Pseudomonas putida–evidence for close relationship to the T7 group. Virology. 2003, 311: 305-315. 10.1016/S0042-6822(03)00124-7.
Molineux IJ: The T7 Group. The bacteriophages. Edited by: Calendar RL. 2005, Oxford: Oxford University Press
Dunn JJ, Studier FW: Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J Mol Biol. 1983, 166: 477-535. 10.1016/S0022-2836(83)80282-4.
Brenner DJ, Ursing J, Bercovier H, Steigerwalt AG, Fanning GR, Alonso JM, Mollaret HH: Deoxyribonucleic acid relatedness in Yersinia enterocolitica and Yersinia enterocolitica-like organisms. Curr Microbiol. 1980, 4: 195-200. 10.1007/BF02605856.
Lavigne R, Burkal’tseva MV, Robben J, Sykilinda NN, Kurochkina LP, Grymonprez B, Jonckx B, Krylov VN, Mesyanzhinov VV, Volckaert G: The genome of bacteriophage ϕKMV, a T7-like virus infecting Pseudomonas aeruginosa. Virology. 2008, 312: 49-59.
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG: Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011, 7: 539-
Kropinski AM, Borodovsky M, Carver TJ, Cerdeño-Tárraga AM, Darling A, Lomsadze A, Mahadevan P, Stothard P, Seto D, Van Domselaar G, Wishart DS: In silico identification of genes in bacteriophage DNA. Methods Mol Biol. 2009, 502: 57-89. 10.1007/978-1-60327-565-1_6.
Zafar N, Mazumder R, Seto D: CoreGenes: A computational tool for identifying and cataloging “core” genes in a set of small genomes. BMC Bioinformatics. 2002, 3: 12-10.1186/1471-2105-3-12.
Lavigne R, Seto D, Mahadevan P, Ackerman HW, Kropinski AM: Unifying classical and molecular taxonomic classification: analysis of the Podoviridae using BLASTP-based tools. Res Microbiol. 2008, 159: 406-414. 10.1016/j.resmic.2008.03.005.
Darling AC, Mau B, Blattner FR, Perna NT: Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome. 2004, 14: 1394-1403. 10.1101/gr.2289704.
Rice P, Longden I, Bleasby A: EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16 (6): 276-277. 10.1016/S0168-9525(00)02024-2.
Spoerel N, Herrlich P, Bickle TA: A novel bacteriophage defence mechanism: the anti-restriction protein. Nature. 1979, 278: 30-34. 10.1038/278030a0.
Sturrock SS, Dryden DT, Atanasiu C, Dornan J, Bruce S, Cronshaw A, Taylor P, Walkinshaw MD: Crystallization and preliminary X-ray analysis of ocr, the product of gene 0.3 of bacteriophage T7. Acta Crystallogr D Biol Crystallogr. 2001, 57: 1652-1654. 10.1107/S0907444901011623.
Walkinshaw MD, Taylor P, Sturrock SS, Atanasiu C, Berge T, Henderson RM, Edwardson JM, Dryden DT: Structure of Ocr from bacteriophage T7, a protein that mimics B-form DNA. Mol Cell. 2002, 9: 187-194. 10.1016/S1097-2765(02)00435-5.
Studier FW, Movva NR: SAMase gene of bacteriophage T3 is responsible for overcoming host restriction. J Virol. 1976, 19: 136-145.
Marchand I, Nicholson AW, Dreyfus M: High-level autoenhanced expression of a single-copy gene in Escherichia coli: overproduction of bacteriophage T7 protein kinase directed by T7 late genetic elements. Gene. 2001, 262: 231-238. 10.1016/S0378-1119(00)00526-6.
Robertson ES, Aggison LA, Nicholson AW: Phosphorylation of elongation factor G and ribosomal protein S6 in bacteriophage T7-infected Escherichia coli. Mol Microbiol. 1994, 11: 1045-1057. 10.1111/j.1365-2958.1994.tb00382.x.
Condron BG, Atkins JF, Gesteland RF: Frameshifting in gene 10 of bacteriophage T7. J Bacteriol. 1991, 173: 6998-7003.
Alam SL, Atkins JF, Gesteland RF: Programmed ribosomal frameshifting: much ado about knotting!. Proc Natl Acad Sci U S A. 1999, 96: 14177-14179. 10.1073/pnas.96.25.14177.
Chandler M, Fayet O: Translational frameshifting in the control of transposition in bacteria. Mol Microbiol. 1993, 7: 497-503. 10.1111/j.1365-2958.1993.tb01140.x.
Theis C, Janssen S, Giegerich R: Prediction of RNA secondary structure including kissing hairpin motifs. WABI′10 Proceedings of the 10th international conference on Algorithms in bioinformatics. 2010, Heidelberg: Springer-Verlag
Hanlon GW: Bacteriophages: an appraisal of their role in the treatment of bacterial infections. Int J Antimicrob Agents. 2007, 30: 118-128.
Kutter E, Sulakvelidze A: Bacteriophages: biology and applications. 2005, Boca Raton, FL: CRC Press
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL: Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol. 2001, 305 (3): 567-580. 10.1006/jmbi.2000.4315.
Summer EJ, Berry J, Tran TA, Niu L, Struck DK, Young R: Rz/Rz1 Lysis Gene Equivalents in Phages of Gram Negative Hosts. J Mol Biol. 2007, 373: 1098-1112. 10.1016/j.jmb.2007.08.045.
Lavigne R, Sun WD, Volckaert G: PHIRE, a deterministic approach to reveal regulatory elements in bacteriophage genomes. Bioinformatics. 2004, 20: 629-635. 10.1093/bioinformatics/btg456.
Sambrook J, Russel DW: Molecular cloning: A laboratory manual. 2001, New York: Cold Spring Harbor Laboratory Press, 3
Kropinski AM, Prangishvili D, Lavigne R: Position paper: The creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ Microbiol. 2009, 11: 2775-2777. 10.1111/j.1462-2920.2009.01970.x.
Mazzoco A, Waddell TE, Lingohr E, Johnson RP: Enumeration of bacteriophages using the small drop plaque assay system. Bacteriophages: Methods and protocols, Volume 1: Isolation, characterization, and interactions. Edited by: Clokie MRJ, Kropinski AM. 2009, New York: Humana Press, 81-86. 1
Ackerman HW: Basic phage electron microscopy. Bacteriophages: Methods and protocols, Volume 1: Isolation, characterization, and interactions. Edited by: Clokie MRJ, Kropinski AM. 2009, New York: Humana Press, 113-126. 1
Shine J, Dalgarno L: The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc Natl Acad Sci U S A. 1974, 71 (4): 1342-1346. 10.1073/pnas.71.4.1342.
Shine J, Dalgarno L: Terminal-sequence analysis of bacterial ribosomal RNA. Eur J Biochem. 1975, 57 (1): 221-230. 10.1111/j.1432-1033.1975.tb02294.x.
Laslett D, Canback B: ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32 (1): 11-16. 10.1093/nar/gkh152.
Schattner P, Brooks AN, Lowe TM: The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33 (2): 686-689.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O: The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008, 9: 75-10.1186/1471-2164-9-75.
Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, Field M, Heled J, Kearse M, Markowitz S, Moir R, Stones-Havas S, Sturrock S, Thierer T, Wilson A: Geneious v 7.9.1. 2011
Koski LB, Gray MW, Lang BF, Burger G: AutoFACT: an automatic functional annotation and classification tool. BMC Bioinformatics. 2005, 6: 151-10.1186/1471-2105-6-151.
Bjellqvist B, Hughes GJ, Pasquali CH, Paquet N, Ravier F, Sanchez J, Frutiger S, Hochstrasser DF: The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis. 1993, 14: 1023-1031. 10.1002/elps.11501401163.
Bjellqvist B, Basse B, Olsen E, Celis JE: Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions. Electrophoresis. 1994, 15: 529-539. 10.1002/elps.1150150171.
Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A: Protein Identification and Analysis Tools on the ExPASy Server. The Proteomics Protocols Handbook. Edited by: Walker JM. 2005, Totowa, New Jersey: Humana Press
Acknowledgements
This research was supported by the Ontario Ministry of Agriculture Food and Rural Affairs (OMAFRA) Food Safety Research Program (research grant SF6075). The authors would like to thank Dr. Jiping Lee, Agriculture and Food Laboratory, Laboratory Services Division, University of Guelph for performing the genome sequencing. The authors would like to acknowledge the technical assistance with TEM imaging of Mr. Robert Harris, Molecular and Cellular Imaging Facility, at the University of Guelph, and the assistance of Dr. Mikail Skurnik, Haartman Institute, University of Helsinki, Finland for providing selected bacterial strains.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
CLV and AMK contributed to the writing of this manuscript. AMK assembled the genome assisted by CLV. CLV finalized the annotation. AA isolated the phage and performed host range determination experiments. JAO was the principal investigator and MG and SC provided all facilities to complete this work. All the authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Leon-Velarde, C.G., Kropinski, A.M., Chen, S. et al. Complete genome sequence of bacteriophage vB_YenP_AP5 which infects Yersinia enterocoliticaof serotype O:3. Virol J 11, 188 (2014). https://doi.org/10.1186/1743-422X-11-188
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-11-188