
Abstract
Background
Multi-resistant Achromobacter xylosoxidans has been recognized as an emerging pathogen causing nosocomially acquired infections during the last years. Phages as natural opponents could be an alternative to fight such infections. Bacteriophages against this opportunistic pathogen were isolated in a recent study. This study shows a molecular analysis of two podoviruses and reveals first insights into the genomic structure of Achromobacter phages so far.
Methods
Growth curve experiments and adsorption kinetics were performed for both phages. Adsorption and propagation in cells were visualized by electron microscopy. Both phage genomes were sequenced with the PacBio RS II system based on single molecule, real-time (SMRT) technology and annotated with several bioinformatic tools. To further elucidate the evolutionary relationships between the phage genomes, a phylogenomic analysis was conducted using the genome Blast Distance Phylogeny approach (GBDP).
Results
In this study, we present the first detailed analysis of genome sequences of two Achromobacter phages so far. Phages JWAlpha and JWDelta were isolated from two different waste water treatment plants in Germany. Both phages belong to the Podoviridae and contain linear, double-stranded DNA with a length of 72329 bp and 73659 bp, respectively. 92 and 89 putative open reading frames were identified for JWAlpha and JWDelta, respectively, by bioinformatic analysis with several tools. The genomes have nearly the same organization and could be divided into different clusters for transcription, replication, host interaction, head and tail structure and lysis. Detailed annotation via protein comparisons with BLASTP revealed strong similarities to N4-like phages.
Conclusions
Analysis of the genomes of Achromobacter phages JWAlpha and JWDelta and comparisons of different gene clusters with other phages revealed that they might be strongly related to other N4-like phages, especially of the Escherichia group. Although all these phages show a highly conserved genomic structure and partially strong similarities at the amino acid level, some differences could be identified. Those differences, e.g. the existence of specific genes for replication or host interaction in some N4-like phages, seem to be interesting targets for further examination of function and specific mechanisms, which might enlighten the mechanism of phage establishment in the host cell after infection.
Similar content being viewed by others


Background
During the last years, the number of nosocomially acquired infections caused by multi-resistant Achromobacter strains has rapidly increased and become clinically relevant. Achromobacter xylosoxidans is a motile Gram-negative rod [1] that apart from being widely distributed in natural environments, mainly in soil [2] or different water sources [3], has been recognized as an emerging nosocomial pathogen potentially causing different human infections, including endocarditis [4, 5], bacteremia [6, 7], meningitis [8], ocular infections [9, 10] or urinary tract infections [11]. Recent studies on antibiotic resistances in Achromobacter strains [12] showed a high number of resistances against antimicrobial substances from different classification groups, such as penicillins, lincosamides or cephalosporines. Other reports on beta-lactamases [13, 14] and resistances against aminoglycosides [15] in A. xylosoxidans also stress the urgent need to search for new alternatives to fight this opportunistic pathogen. Bacteriophages as natural opponents of bacteria have become an interesting option for treatment of microbial infections in plants, animals and also humans. However, phages against Achromobacter species were nearly unknown, except for some old publications [16, 17]. Recently, the study by Wittmann et al. [12] showed that bacteriophages against Achromobacter species can be easily found in the environment as a diverse set of different phages against Achromobacter could be isolated from soil and sewage samples. The majority of these bacteriophages belong to the Siphoviridae with long and flexible tails, but two members of the Podoviridae, phages JWAlpha and JWDelta, could be identified. Both are lytic phages that contain double-stranded DNA genomes, which are very similar, but not identical. Here we present the first genomic data on Achromobacter phages so far and further physiological analysis of both N4-like phages. Escherichia phage N4 was known as a genetic orphan for a long time [18], having no similarities to any other phages in regard to its conserved genomic structure that includes a large virion-encapsulated RNA polymerase which plays an important role in its transcription mechanism with three different RNA polymerases. Due to the increasing number of sequenced phage genomes, we now have numerous N4-like phages for members of the γ-Proteobacteria [19–21] and α-Proteobacteria [18]. In this study we present the first phages of the N4-like genus from Achromobacter, a member of the β-Proteobacteria.
Results and discussion
General phage characteristics
Morphology
Achromobacter phages JWAlpha and JWDelta were isolated from waste water treatment plants in Werl and Braunschweig, respectively [12]. Morphological analysis by transmission electron microscopy revealed that both phages belong to the Podoviridae and seem to be quite similar. JWAlpha consists of an icosahedral head with a diameter of about 59 nm, whereas the head of JWDelta with a length of about 72 nm and a width of 67 nm is slightly longer. Both phages possess a short tail with an approximate length of 22 nm and several short tail fibers (Figure 1A and B).

Transmission electron micrographs of JWAlpha and JWDelta (A and B) and of an ultrathin section of A. xylosoxidans DSM 11852 cells, 90 min after infection with phage JWAlpha (C). Scale bars represent 100 nm.
Adsorption parameters
Experiments investigating the adsorption kinetics of the two phages showed that 5 min after adding phages to the bacterial host, about 65-70% of the phages have already adsorbed to their host cells (Figure 2). Whereas only about 1% of the originally applied JWAlpha phages were still detectable after 20 min, nearly 25% of the applied JWDelta phages could still be detected. According to the practical method presented by Kropinski [22], adsorption rate constant k was calculated for JWAlpha (k = 1.91 × 10-9 ml/min) and JWDelta (k = 1.56 × 10-9 ml/min). Scanning electron micrographs showed that adsorption of the phages takes place all around the cell surface (Figure 3). Further analyses revealed that no divalent cations like Mg2+ or Ca2+ were necessary for successful adsorption and penetration, as for example it was shown for phages PL-1 [23] or T5 [24] (data not shown).

Adsorption kinetics of JWAlpha and JWDelta to Achromobacter host cells (DSM 11852). Bacterial cultures were infected with a multiplicity of infection of 0.1. Samples were taken at different time points to determine adsorption kinetics. Filtrates were diluted to determine titer of free phage particles in the sample. This experiment was performed in triplicate.

Adsorption of phage JWAlpha to Achromobacter DSM 11852 cells. (Scanning electron micrograph). Sample was taken 5 min after phage application. Adsorbed phages are coloured in red.
Burst size
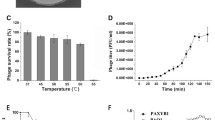
In general, both phages form clear plaques (1–2 mm diameter) on bacterial lawns and lysates of both phages reveal high titers (~1011 pfu/ml). Electron micrographs of ultrathin sections of Achromobacter cells infected with both phages revealed a burst size of about 200 new phages per infected cell (Figure 1C). One-step growth curve experiments showed that both phages have latent periods about 1.5-2 h, after which an increasing number of free phages could be detected (Figure 4). Both curve plateaus with a titer of about 2×1011 pfu per ml were reached after 5 h. The determined burst size of about 180 correlates with the TEM ultra-thin section analysis.

One-step growth curve experiments with JWAlpha and JWDelta. DSM 11852 cells were infected with a multiplicity of infection of 0.1.
Stability
Incubation at different pHs ranging from pH 4-pH 10 for 1 hour and heating at different temperatures (30–60°C) for 10 min did not have any effect on the titer (data not shown) Storage of lysates over months at 4°C did not result in loss of titer, the phages seem to be stable.
Genomics
First endonuclease restriction digestions of their DNA gave a first hint that they are not identical [12]. JWAlpha and JWDelta both contain linear, double-stranded DNA genomes with a length of 72329 bp and 73659 bp, respectively, comparable to all N4-like phages known so far. Both genomes reveal terminal redundant ends of 365 bp for JWAlpha and 420 bp for JWDelta. After analysis of the genomic DNA by pulsed field gel electrophoresis only one single band, but no concatemers could be identified (data not shown) which suggests that both genomes possess no cohesive ends as also previously described for phage N4 [25, 26]. Additionally, a time-limited BAL 31 digestion and a following hydrolysis with restriction endonuclease SmaI revealed that the genomes are not circularly permutated since two distinct bands as representatives of the ends of the genomes were truncated (data not shown). The G + C contents of JWAlpha and JWDelta are 54.4% and 54.2%, respectively. In comparison with complete Achromobacter genomes sequenced so far (65-66% GC content) [27, 28], the G + C contents of the phage genomes are significantly lower than those of possible hosts, which also occurs in other N4-like phage genomes [18]. Open reading frames (ORFs) were determined using MyRAST [29] and ARTEMIS based on three different start codons (ATG, GTG and TTG) and putative Shine Dalgarno sequences upstream the coding regions. Thus, we identified 92 ORFs for JWAlpha (88 ATG, 3 GTG and 1 TTG) and 89 ORFs for JWDelta (86 ATG, 2 GTG and 1 TTG), resulting in a coding percentage of 95.9% and 94.1%, respectively. Analysis of the genome sequences using tRNAscan-SE [30] did not reveal any genes for tRNAs, which leads to the assumption that both phages are adapted ideally to their host organism in regard to codon usage and do not require additional tRNAs of their own. Comparing the number of encoded tRNAs among all N4-like phages, it could be shown that most N4-like phage harbor between 1–3 genes for different additional tRNAs with codons that the host cell does not provide. Whereas Sulfitobacter phage pCB2047-B (Genbank accession number HQ317387, unpublished) and N4-like phages from Salmonella harbor 10 or more genes for different tRNAs, the genomes of the N4-like phages from the Pseudomonas group did not reveal any tRNA genes. Based on a work of de Paepe et al., Bailly-Bechet et al. suggested that virulent phages have more tRNA genes than temperate phages to ensure optimal translation and therefore might replicate faster [31, 32]. In this context, it would be interesting to compare the number of tRNAs with latency times of all N4-like phages.
Though the two phages were isolated from different waste water treatment plants 250 km apart, they reveal identities of 98% at the nucleotide level and their complete genomic structure seems to be highly conserved. In general, the two phages share 87 genes with an amino acid identity range from 67 up to 100%, including 24 identical genes. BLASTP analysis identified 49 genes with strong similarities to phage N4 with an identity range between 25 and 84% at the amino acid level. Overall, compared to other members of the N4 family most similarities could be determined in phages from Escherichia (42–49 genes). Fewer genes with similarities to JWAlpha and JWDelta could be identified, for instance, in Erwinia phage vB_EamP-S6 (31), Pseudomonas phages (25–28) or Vibrio phages (20–21). Both genomes show nearly the same organization with two large gene clusters located on different strands and a small cluster at the end of the genome that is transcribed towards a different direction again (Figure 5). These large clusters can be divided into different subclusters for so-called early genes, DNA metabolism, host interaction, transcription, replication, structural genes, lysis and DNA packaging.

Genome structures of phages JWAlpha and JWDelta and comparison with Escherichia phage N4. Proposed functional clusters are marked by the same colour. Figure was generated using Easyfig with amino acid sequence comparison [33]. Level of amino acid identity range is shown via the gradient scale.
Genes for transcription
E. coli phage N4 has been recognized to be a genetic orphan for a long time, being the only known phage that did not depend on the RNA polymerase (RNAP) of its host to transcribe its early genes. N4-like phages that have been described before harbor at least three genes for RNAPs for the transcription of genes in different stages of their life-cycle [34]. The most striking and highly conserved characteristic of a N4-like phage is the gene for a large RNAP with around 3500 amino acids that is encapsulated in the virion and is injected into the host cell together with the phage DNA for immediate start of early gene transcription [35, 36]. Both Achromobacter phages also harbor three different RNAPs, suggesting the same transcription organization as N4 and its other relatives. The large virion RNAPs consist of 3425 amino acids in both phages, only differing in 10 amino acid residues and containing no cysteine residues as also reported by Kazmierczak et al. [37]. Cysteine residues in a protein might form disulfide bonds and lead to a change of protein folding and tertiary structure, which would make it difficult or even impossible for such a large protein to be injected through the phage tail into the host cell. Like other N4-like phages JWAlpha and JWDelta also carry genes that encode two different RNA polymerase subunits, RNAP1 and RNAP2, which play a role in the transcription of N4 middle genes [38]. Similar to phage N4 the gene for RNAP1 is directly followed by RNAP2, whereas other N4-like phage genomes show insertions of small genes in this area [18, 20, 39]. In contrast to other N4-like phages the gene for RNAP2 is interrupted by a gene similar to gp8 of Celeribacter phage P12053L in both Achromobacter phages, dividing it into two parts and raising the question whether it is still functional.
Gene products involved in host interaction
The so-called early genes of a phage genome often encode proteins that somehow interact with the host, for example to protect the newly-injected phage DNA molecule from degradation in the host cytoplasma and see to its establishment, to shut off the host metabolism or to modify host polymerases for transcription in favor of phage propagation. Phages can shut off their host metabolism in different ways, e.g. by host DNA degradation [40, 41]. Lavigne et al. [42] recently identified gp13 of Pseudomonas phage LUZ19 as an acetyltransferase that hinders both replication and transcription in Pseudomonas posttranslationally by acetylation. In regard to DNA protection, for example, phages use different strategies to avoid degradation by host nucleases. Phage T4 e.g. protects its free genomic ends from degradation by exonucleases with the help of small DNA-binding proteins [43], phage T7 possesses an anti-restriction system that inhibits the activity of the bacterial restriction endonuclease by prevention of its binding to the phage DNA [44, 45]. Phage JWAlpha also harbors a gene (gp24) that might play a role in a putative anti-restriction system. Its deduced amino acid sequence reveals a conserved domain with homologies to restriction alleviation protein Lar from the defective prophage Rac [46] which is able to alleviate restriction and to enhance modification by the restriction and modification system in E. coli at the same time. This is apparently accomplished by altering the activity of a type I methyltransferase to efficiently methylate the phage DNA and protecting it against degradation [47]. Though the two phages have a quite similar genome organization, we could not identify this gene in JWDelta. The deduced amino acid sequence of a putative candidate at the same genomic position as in JWAlpha was comparatively much shorter and did not reveal any homologies to proteins of JWAlpha or Lar-like proteins from the database. Whether JWDelta just lacks this gene or whether the gene product of this putative candidate has a similar function, but a yet unknown structure stays to be examined. In general, the different mechanisms of host takeover after infection are worth closer examination in the future.
Another interesting example for a host interaction system could also be found in both Achromobacter phage genomes. Upstream the replication cluster two genes were assigned that share 99% and 98% identities at the nucleotide level with the homologous gene in the other phage, respectively. Their deduced amino acid sequences reveal homologs to a rIIA-like and a rIIB-like protein from phage N4. This kind of proteins were first described in phage T4 and might play a role in lysis inhibition among other functions that are still not completely clear, but they seem to have an influence on cell energetics, including Mg2+ transport and ATP biosynthesis [48]. Examining all N4-like phage genomes known so far for these genes, we discovered that homologs of both genes are widely distributed among N4-like genomes in different host genera. They could also be identified in N4-like phage genomes from the Roseobacter clade, from E. coli and Pseudomonas, but neither in Erwinia phage vB_EamP_S6 nor in Salmonella or Vibrio phages. Generally, both phage genomes are quite similar in their structure and number of genes. The only differences between them could be found in the area between the early genes for RNA polymerases and the genes for rIIa- and rIIB-like proteins. Apart from the already mentioned gene for a Lar-like protein, JWDelta lacks homologous genes for orf29, orf37 and orf41 from JWAlpha. In comparison with JWDelta, the genome of JWAlpha showed two deletions which resulted in a much shorter orf18 with an unclear function and a missing gap of about 1,3 kb that contains a gene for a HNH endonuclease in JWDelta.
Replication cluster and genes for nucleotide metabolism
Both phage genomes of this study harbor genes for a complete replication cluster in the same order as N4 and other N4-like phages, including a gene for a nucleoside triphosphate pyrophosphohydrolase (Superfamily cl16941) that might provide the helicase with energy by hydrolysis of NTPs, and a gene for a DNA helicase containing a conserved domain UvrD_C_2 (pfam13538) which resembles a AAA-like structural fold that is often found at the C-terminus of helicases. Moreover, this cluster contains a gene that codes for a DNA polymerase with a 3'-5' exonuclease proofreading domain (35EXOc (smart00474)) and a second conserved domain at the C-terminus, DNA_pol_A (pfam00476). The N4 gp43-like gene for a primase was identified based on its similarities to other primase genes from several phages at the amino acid level and the C-terminal Primase C domain PriCT_1 (pfam08708). At the end of the replication cluster we assigned a gene for a putative single-stranded binding protein (Ssb) that though lacking a conserved domain showed a characteristic glycine- and proline-rich C-terminus. The ssb gene is followed by two genes for putative endonucleases, their deduced amino acid sequences revealed characteristic conserved domains NUMOD4 (pfam07463) and HNH_3 (pfam13392) for putative HNH homing endonucleases. Both phages also possess genes involved in the DNA metabolism, to be specific genes for a deoxycytidine triphosphate deaminase with a conserved dcd (PRK00416) domain, a thymidylate synthase (Thy1 (pfam02511)), a homing endonuclease (HNH_3 (pfam13392)) and a thioredoxin with a conserved TRX family domain (cd02947) that might play a role in the regulation of some enzymes involved in DNA metabolism or replication. In contrast to N4-like phages from the Roseobacter clade that are the only other members that also carry genes for thioredoxin, the Achromobacter phages do not harbor a gene for a ribonucleotide-diphosphate reductase, an enzyme involved in formation of deoxyribonucleotides. In general, the genes of the replication cluster are highly conserved and show strong similarities to genes from other N4-like phages at the amino acid level, e.g. the genes for the helicase, DNA polymerase or Ssb. However, besides these genes, there are other genes (orf 52, orf 55, orf 57 and orf 58) with a yet unclear function that reveal similarities only to N4-like phages from Escherichia and Erwinia.
DNA packaging proteins and structural proteins
In general, phages have developed different strategies for packaging DNA into their capsids, depending on their mechanism of replication, head size and its subsequent ability to hold a little bit more than one complete phage genome, which often leads to circularly permutated genomes. For instance, in phage T4 the head full of DNA is about 3% longer than the genome size [49]. Usually, two main proteins are used for DNA packaging, called terminases. These proteins display a hetero-multimeric structure with a small subunit for DNA binding and a large subunit with an ATPase domain and an endonuclease function. In most phage genomes, the terminase genes can be identified upstream the cluster for structural genes. In our study, the genes for the large terminase subunits could be clearly identified in both Achromobacter phage genomes. A phylogenetic analysis with other N4-like phages and several phages from a reduced set used by Fouts et al. [21] with different known DNA packaging strategies using a neighbor-joining method revealed that the Achromobacter terminases cluster together with all other terminases from N4-like phages and probably use the same mechanism for packaging based on direct repeats (Figure 6).

Phylogenetic analysis of terminase large subunits of JWAlpha and JWDelta compared to other N4-like phages and phages with other known DNA packaging strategies. Neighbor-joining tree was constructed based on ClustalW alignment of terminase subunit amino acid sequences with 1000 bootstrap replicates (MEGA5).
After separation of denatured phage particles by SDS-PAGE, the protein profiles of JWAlpha and JWDelta were not distinguishable (Figure 7). Both profiles displayed eight distinct protein bands with nearly the same molecular weight that were further assigned by peptide mass fingerprinting, all bands corresponded to coding sequences from our DNA annotation. In bothprotein profiles, bands with approximately the same molecular weight also correspond to homologous genes with the same function. Peptides from the most prominent band (band 5) in both phages matched to a predicted major coat protein with a molecular mass of about 44 kDa that appears to make the most important structural protein of the phage head. With one exception (band 6), all examined protein bands correspond to deduced amino acid sequences of genes that were identified on the complement strand of the genome and encode structural proteins, such as a putative portal protein (band 2) or putative tail proteins (bands 3 and 7). Protein band 6 is the only example that is encoded by a gene that is not located in the gene cluster for structural proteins. Nevertheless its deduced amino acid sequence revealed a conserved bacterial Ig-like domain (Big_2 [pfam02368]) that occurs in phage surface proteins and might decorate the phage capsid after assembly. Besides all these structural proteins, one non-structural protein could be assigned by peptide mass fingerprinting. The largest band of both profiles (band 1) corresponds to the annotated RNA polymerase with an approximate molecular weight of about 369 kDa in both phages. As it is encapsulated in the virion to be injected into the host cell together with the phage genome, it was no surprise to detect this protein after complete denaturation of the phage capsid.

Identification of structural proteins of phages JWAlpha and JWDelta by SDS-PAGE analysis. Protein profiles of JWDelta and JWAlpha. Proteins were separated on a 17.5% SDS-PAGE. After extraction of gel slices from corresponding bands and tryptic digestion, samples were analyzed via peptide mass fingerprinting. Proteins identified by peptide mass fingerprinting analysis are listed aside along with their approximate molecular weight.
Analysis and comparison of protein profiles from different N4-like phages [20, 21, 39] with JWAlpha and JWDelta showed that in all cases the most prominent protein band was the major head protein with a molecular mass of about 44 kDa. Despite their similar morphology, the protein profiles of these phages differ in number and molecular mass of prominent bands. However, there is a series of ten genes starting with the gene for the virion RNA polymerase and ending with the gene for the portal protein that can be determined in the same order in other N4-like phages (Figure 8). In general, based on the area upstream of this conserved gene cluster, the N4-like phages can be divided into three different groups. The Achromobacter phages JWAlpha and JWDelta, phage N4 and other N4-like Escherichia phages, Erwinia phage vB_EamP-S6 and N4-like phages from the Roseobacter clade reveal genes of the lysis cluster upstream the gene for the portal protein. In case of the N4-like Pseudomonas and Salmonella phages and the Vibrio phages vB_VchP_VchO139-I and vB_VchP_JA1, the described cluster is preceded by genes which are transcribed towards a different direction. In Vibrio phages VBP32 and VBP47 the cluster consists of 11 genes and shows differences in regard to the length of some genes. Most of the structural genes are highly conserved, e.g. the genes for the major coat protein or the portal protein (Table 1). Some genes of JWAlpha and JWDelta, however, reveal only similarities to genes from N4-like phages from Escherichia, as described above for genes of the replication cluster.

Comparison of structural gene clusters of different N4-like phages displaying homologous genes identified by peptide mass fingerprinting. All genes upstream the gene for the virion RNA polymerase are shown. Homologous genes for proteins that could also be identified by peptide mass fingerprinting in other examined phages are marked in dark blue, genes for proteins that could be identified just in one phage are marked in pale blue.
Gene products for host cell lysis
After assembly of new phage progeny, most phages lyse their bacterial host cell in order to release the new phages into the environment. For that purpose, they often use a dual lysis system that consists of a holin that forms a pore in the cytoplasmic membrane to release the endolysin into the periplasmic space where it enzymatically degrades the peptidoglycan [50]. In general, due to the much higher complexity of the Gram-positive cell wall with a lot of different peptidoglycan subtypes [51], endolysins of phages with Gram-positive hosts possess a lot of highly specific enzymatic activities. However, most of the phages with Gram-negative hosts use unspecific N-acetylmuramidases for degradation of the peptidoglycan as there is just one peptidoglycan layer to overcome. The lysis clusters of both Achromobacter phages consist of three genes (orf 78-orf 80 in JWAlpha, orf 75-orf 77 in JWDelta) and show the same organization as lysis clusters from other phages with Gram-negative hosts. Analysis with TMHMM Server v. 2.0 [52] revealed that the deduced amino acid sequences of orf80 from phage JWAlpha and orf77 from JWDelta both contain two transmembrane regions and show similarities to putative holins of Escherichia phages EC1-UPM and vB_EcoP_G7C, hence both proteins could be assigned as class II holins [53]. The gene for the holin is followed by the endolysin gene, BLASTP analysis of the amino acid sequences identified them both as N-acetylmuramidases. Downstream the endolysin genes, one small ORF for a protein could be assigned that also revealed one transmembrane region and similarities to a Rz-like protein. Besides genes for a holin and an enzymatically active endolysin, phages of gram-negative hosts often harbor genes for proteins Rz and Rz1 that somehow interact with the outer membrane and play a role in the final step of host lysis [54, 55]. Examining all N4-like genomes in regard to their lysis clusters, we could identify five different groups of clusters with apparently genus-specific strategies for host lysis. The largest group of the N4-like phages harbors genes for a class II holin with two transmembrane regions, a N-acetylmuramidase and a Rz protein with one transmembrane region as described above for both Achromobacter phages. Further members of this group are the Escherichia phages N4, EC1-UPM, KBNP21 and vB_EcoP_G7C, Erwinia phage vB_EamP_S6 and Enterobacter phage IME11. The second group comprises the N4-like phages from the Roseobacter clade and Enterobacter phage ECP1 which all encode genes for a lysis protein. Since none of their deduced amino acid sequences revealed homologies to muramidases or to peptidases of the MEROPS database, they were apparently annotated as lysis proteins with yet unclear functions. In all roseophage genomes of this group, this gene is framed by two small orfs with one deduced transmembrane region. Though the Enterobacter phage ECP1 also possesses a gene that encodes a lysis protein, the structure of its lysis cluster is different. The whole cluster is located on the other DNA strand and is not transcribed towards the same direction as the preceding and following genes. In addition to that the gene for the lysis protein is preceded by two genes for proteins with three transmembrane regions each, which is characteristic for class I holins. The N4-like Salmonella phages FSL SP-058 and FSL SP-076 belong to the third group with a different lysis cluster. Like phage ECP1, both Salmonella phages possess two putative genes for class I holins, but in addition to a lysis protein they also harbors a gene for a putative lysozyme and, thus, they are the only N4-like phages with two different enzymes for host lysis. The fourth group consists of three Pseudomonas phages of the N4 family, namely P26, LIT1 and LUZ7. Though two putative holin genes could be identified in all three genomes, no muralytic enzyme could be predicted. The endolysin containing region seems to be completely deleted [20] which raises the question whether there are other muralytic enzymes with so far unknown mechanisms. Genomes of members of the fifth group, mainly Vibrio phages, even do not reveal any sign of lysis cluster at all which makes them interesting candidates for examining their lysis mechanism and potentially identifying new muralytic enzymes. Since all bacteriophages of the N4-family known so far have gram-negative hosts, it was no surprise that nearly all of them encode genes for rather unspecific muramidases and no specific endopeptidases as often found in phages with gram-positive hosts [56]. Although the genomes of N4-like phages are highly conserved both in sequence identity and structure, it is interesting to observe these differences in their lysis clusters. In contrast to the replication cluster or the cluster for structural genes, a broader diversity could be determined which is worth further examination.
Phylogenomic analysis
Figure 9 shows the resulting proteome-based phylogenetic tree of the 24 phage genomes. The group of the newly discovered Achromobacter phages is part of a larger clade that contains phages associated to the family Rhodobacteraceae as well as Vibrio phages JA1 and VchO139-1. But whereas the clades related to the phages of Rhodobacteraceae as well as Achromobacter have both high support, the positioning of the denoted Vibrio phages is not supported. Overall, the tree yields the expected relationships of the different groups (e.g., as previously reported [21]) and, since it provides branch support, dubious positioning (usually due to the lack of data) can now be directly identified and discussed accordingly.

Phylogenetic tree based on the proteomes of 24 N4-like phages. The tree was inferred from pairwise distances, assessed via the GBDP approach [57–59], and using the balanced minimum evolution principle [60]. The tree was rooted via midpoint rooting [61]. GBDP used BLAST + [62] as a local alignment programme (i.e., blastp using an initial word length of 3) and applied the following settings for the pairwise distance calculation: trimming algorithm, formula d5 and no e-value filter.
Conclusions
Analysis of the genomes of Achromobacter phages JWAlpha and JWDelta and comparisons of different gene clusters with other phages revealed strong similarities to other N4-like phages, especially of the Escherichia group. Phages of the N4 family infect a lot of different genera with Achromobacter as a new member. Although all these phages show a highly conserved genomic structure and partially strong similarities at the amino acid level, interestingly, some differences could be identified probably depending on their host genus. These differences, e.g. the existence of a gene for a ribonucleotide-diphosphate reductase in N4-like Roseobacter phages or the completely different structure of the replication cluster in N4-like phages of Pseudomonas compared to all other N4-like phages, seem to be interesting targets for further examination of function and specific mechanisms. In particular, further examination of the gene for a Lar-like protein in JWAlpha might enlighten the mechanism of phage establishment in the host cell after infection. A closer look on the mechanisms for host lysis in those phages that revealed no or yet unknown enzymes for cell wall degradation might also be interesting for the identification of new muralytic enzymes.
Material and methods
Genome sequence accession numbers
Genome sequences of JWDelta and JWAlpha were deposited to Genbank under accession numbers KF787094 and KF787095, respectively. Further genome sequences of N4-like phages for comparison could be obtained under following accession numbers: Enterobacteria phage N4 (EF056009), Escherichia phage vB_EcoP_G7C (HQ259105) [63], Escherichia phage KBNP21 (JX415535) [64], Enterobacter phage EcP1 (HQ641380), Enterobacter phage IME11 (JX880034) [65], Escherichia phage EC1-UPM (KC206276) [66], Erwinia phage vB_EamP-S6 (HQ728266) [39], Pseudoalteromonas phage pYD6-A, Pseudomonas phage LIT1 (FN422399), Pseudomonas phage LUZ7 (FN422398) [20], Pseudomonas phage PA26 (JX194238) [67], Roseovarius sp. 217 phage 1 (FR682616), Roseovarius Plymouth Podovirus 1 (FR719956), Ruegeria phage DSS3φ2 (FJ591093), Sulfitobacter phage EE36φ1 (FJ591094) [18], Sulfitobacter phage pCB2047-B (HQ317387), Salmonella phage FSL SP-058 (KC139517), Salmonella phage FSL SP-076 (KC139520) [19], Vibrio phage VBP32 (HQ634196), Vibrio phage VBP47 (HQ634194), Vibrio phage JA-1 (KC438282), Vibrio phage VCO139 (KC438283) [21].
Bacterial strains and growth
Achromobacter xylosoxidans strain DSM 11852 was used for phage propagation and adsorption tests. It was cultured in liquid tryptic soy broth TSB (30 g-l, pH 7.5, OXOID) medium or on Petri dishes of TSB medium supplemented with 1.5% agar (w/v) for approximately 16 h (overnight) at 28°C.
Purification of phages and phage DNA
Purification of the phages by CsCl gradient centrifugation and of their DNA by phenol extraction was done as previously described [68].
Sample preparation and sequencing
SMRTbell™ template libraries were prepared according to the instructions from Pacific Biosciences, Menlo Park, CA, USA, following the Procedure & Checklist for “Low-Input 10 kb Template Preparation and Sequencing” using C2-C2 Chemistry.
Briefly, for preparation of 10 kb libraries ~15 μg genomic DNA was sheared in an Eppendorf microcentrifuge 5424 2×3 min at 5500 rpm using g-tubes™ from Covaris, Woburn, MA, USA. Size range was monitored on an Agilent 2100 Bioanalyzer from Agilent Technologies, Santa Clara, USA. DNAs were concentrated, end-repaired and ligated to hairpin adapters applying components from the DNA/Polymerase Binding Kit 2.0 from Pacific Biosciences, Menlo Park, CA, USA. Reactions were carried out according to the manufacturer´s instructions with exception of the ligation, which took place 2 h at 25°C and overnight at 4°C. SMRTbell™ templates were exonuclease treated for removal of incompletely formed reaction products. A mixture of Exonuclease III and Exonuclease VII (Affymetrix, High Wycombe, UK) was utilized. Hereby, an additional exonuclease treatment after the second purification took place, followed by three purification steps as recommended by Pacific Biosciences. Conditions for annealing of sequencing primers and binding of polymerase to purified SMRTbell™ templates were assessed with the Calculator in RS Remote, PacificBiosciences, Menlo Park, CA, USA. SMRT sequencing was carried out on the PacBio RS (PacificBiosciences, Menlo Park, CA, USA) using the DNA Sequencing Reagent 2.0. For each 10 kb libraries one 120 minutes movie was taken.
Bioinformatics analyses
Phage genome assemblies were performed using the “RS_Preassembler_Allora.1” protocol included in SMRTPortal version 1.3.3. Within that following parameters were applied. For Filtering: Minimum Readlength 100, Minimum Subreadlength 500, Minimum Read Quality 0.8. For Assembly: Minimum Seed Read Length: 3500 for phage JWAlpha, BLASR Options (Advanced) minReadLength 200 maxScore 1000 bestn 24 maxLCPLength 16 nCandidates 24, Trim FASTQ Output true, Use CCS false, Overlap Permissiveness Least permissive, Expected Genome Size 50000, Maximum iterations 10, Detect Chimeras true. In both cases one final contig could be obtained. A quality check of that given consensus sequences regarding overall coverage as well as SNPs was performed using SMRT View and IGV [69] after application of the “RS_Resequencing.1” protocol with default parameters. The phage genomes were annotated using RAST [29] with subsequent manual curation in Artemis [70].
Phage adsorption test and growth curve experiments
For adsorption assays, exponentially growing cultures of Achromobacter xylosoxidans strain DSM 11852 were infected with phages JWAlpha and JWDelta, respectively, with a multiplicity of infection (moi) of 0.01, for growth curve experiments, cells were infected with a moi of 0.1. Samples for both experiments were taken at different time points and directly filtered (membrane syringe filter 0.45 μm, Sartorius, order number 16555). Filtrates were diluted and spotted on a bacterial lawn of DSM 11852 to determine titer of free phages in the culture.
SDS-PAGE analysis of phage proteins
Treatment of phage samples for SDS-PAGE analysis and SDS-PAGE analysis itself were performed as previously described [71].
MALDI-TOF analysis
Coomassie stained protein bands were excised from the gel. The slices were washed twice in H2O for 5 min and dehydrated by adding 200 μl 50% acetonitrile for 5 min. Gel pieces were then washed in 200 μl 0.1 M NH4HCO3 for 15 min at room temperature. Washing solution was discarded and slices were dehydrated by adding acetonitrile again. Dehydrated gel pieces were completely dried in Speed Vac for 10 min. For the tryptic digestion of the proteins the gel slices were reswelled by adding 50 μl digestion solution containing trypsin and incubated over night at 37°C. For extraction of peptides 50 μl of acetonitrile were added, followed by incubation under vigorous shaking for 15 min at 37°C. Supernatants were removed and gel slices were treated with 50 μl 5% formic acid under vigorous shaking for 15 min at 37°C. After that 50 μl of acetonitrile were added, followed again by incubation under vigorous shaking for 15 min at 37°C. Supernatants were united with the removed supernatants and concentrated via Speed Vac. 40 μl 32% methanol/0.25% HCOOH were added, followed by 3 min treatment with ultrasound and analyzed by MALDI Ultraflex-TOF/TOF. Results were compared with a database of predicted phage proteins using Mascot. MALDI-TOF analysis was done at the Helmholtz Centre for Infection Research, Braunschweig, Germany.
Negative staining of phages
Thin carbon support films were prepared by sublimation of a carbon thread onto a freshly cleaved mica surface. Phages were negatively stained with 0.5% (w/v) aqueous uranyl acetate, pH 5.0, according to the method of Valentine et al. [72]. Samples were examined in a TEM 910 transmission electron microscope (Carl Zeiss, Oberkochen) at an acceleration voltage of 80 kV.
Field emission scanning electron microscopy (FESEM)
Bacteria incubated with phage JWAlpha were fixed with 2% glutaraldehyde and 5% formaldehyde, washed with TE-buffer (20 mM TRIS, 1 mM EDTA, pH 6.9) then dehydrated in a graded series of acetone (10, 30, 50, 70, 90, 100%) on ice for 15 min for each step, critical-point dried with liquid CO2 (CPD 30, Bal-Tec, Liechtenstein) and covered with a gold-palladium film by sputter coating (SCD 500, Bal-Tec, Liechtenstein) before being examinated in a field emission scanning electron microscope (Zeiss Merlin) using the Everhart Thornley SE detector and the inlens detector in a 25:75 ratio at an acceleration voltage of 5 kV.
Transmission electron microscopy (TEM)
Samples were fixed in medium with 5% formaldehyde after 1 min 2% glutaraldehyde was added and kept on ice for 1 hour, washed with cacodylate buffer, and further fixed with 1% osmium in 0.08 M cacodylate buffer for 1 hour at room temperature. Then, samples were dehydrated with a graded series of acetone (10, 30, 50%) on ice. At the 70% dehydration step samples were incubated overnight in 70% acetone containing 2% uranyl acetate at 4°C. On the next day samples were further dehydrated with 90% and 100% acetone on ice, the 100% aceton step was repeated at room temperature. Samples were embedded in the epoxy resin Spurr according to described procedures [73]. Ultrathin sections were cut with a diamond knife, picked up with butvar-coated grids, counterstained with uranyl acetate (3 min) and lead citrate (15 sec), and examined in a TEM910 transmission electron microscope (Carl Zeiss, Oberkochen) at an acceleration voltage of 80 kV. Images were recorded digitally at calibrated magnifications with a Slow-Scan CCD-Camera (ProScan, 1024×1024, Scheuring, Germany) with ITEM-Software (Olympus Soft Imaging Solutions, Münster, Germany). Contrast and brightness were adjusted with Adobe Photoshop CS3.
Phylogenomic analysis
The phage dataset comprised of 24 genomes which were compared pairwise on the proteome level using the Genome Blast Distance Phylogeny approach (GBDP) [57–59]. During distance calculation GBDP considers and corrects for several factors that could potentially bias the results such as overlapping HSPs. In general, GBDP was shown to yield robust results even in the presence of a significant amount of paralogous genes, large repeats and partially incomplete genomes. Further, GBDP does not use arbitrary selections of marker genes but rather detects all available matches (i.e., high-scoring segment pairs, HSPs) between pairs of genomes (e.g., via BLAST + [62]). The resulting set of pairwise distances was subsequently used to infer a phylogenetic tree under the balanced minimum evolution criterion [60] as shown previously [74]. If distance values could not be obtained due to the lack of HSPs between the respective proteomes, twice the maximum distance observed in the matrix was used as a substitute (as in [75]). Since GBDP is capable of calculating a set of bootstrap replicates for each pairwise distance [59], the inferred tree was augmented with branch support values to assess its statistical significance.
References
Yabuuchi E, Oyama A: Achromobacter xylosoxidans n. sp. from human ear discharge. Jpn J Microbiol 1971, 15: 477-481.
Busse HJ, Auling G: Genus II. Achromobacter Yabuuchi and Yano 1981, 477VP emend. Yabuuchi, Kawamura, Kosako and Ezaki 1998a, 1083. In Bergey’s manual of systematic bacteriology. Edited by: Brenner DJ, Krieg NR, Staley JT. New York: Springer Verlag; 2005:658-662.
Spear JB, Fuhrer J, Kirby BD: Achromobacter xylosoxidans (Alcaligenes xylosoxidans subsp. xylosoxidans) bacteremia associated with a well-water source: case report and review of the literature. J Clin Microbiol 1988, 26: 598-599.
Ahmed MS, Nistal C, Jayan R, Kuduvalli M, Anijeet HK: Achromobacter xylosoxidans, an emerging pathogen in catheter-related infection in dialysis population causing prosthetic valve endocarditis: a case report and review of literature. Clin Nephrol 2009, 71: 350-354.
van Hal S, Stark D, Marriott D, Harkness J: Achromobacter xylosoxidans subsp. xylosoxidans prosthetic aortic valve infective endocarditis and aortic root abscesses. J Med Microbiol 2008, 57: 525-527.
Behrens-Muller B, Conway J, Yoder J, Conover CS: Investigation and control of an outbreak of Achromobacter xylosoxidans bacteremia. Infect Control Hosp Epidemiol 2012, 33: 180-184.
Tena D, Carranza R, Barbera JR, Valdezate S, Garrancho JM, et al.: Outbreak of long-term intravascular catheter-related bacteremia due to Achromobacter xylosoxidans subspecies xylosoxidans in a hemodialysis unit. Eur J Clin Microbiol Infect Dis 2005, 24: 727-732.
Manckoundia P, Mazen E, Coste AS, Somana S, Marilier S, et al.: A case of meningitis due to Achromobacter xylosoxidans denitrificans 60 years after a cranial trauma. Med Sci Monit 2011, 17: CS63-CS65.
Park JH, Song NH, Koh JW: Achromobacter xylosoxidans keratitis after contact lens usage. Korean J Ophthalmol 2012, 26: 49-53.
Reddy AK, Garg P, Shah V, Gopinathan U: Clinical, microbiological profile and treatment outcome of ocular infections caused by Achromobacter xylosoxidans. Cornea 2009, 28: 1100-1103.
Tena D, Gonzalez-Praetorius A, Perez-Balsalobre M, Sancho O, Bisquert J: Urinary tract infection due to Achromobacter xylosoxidans: report of 9 cases. Scand J Infect Dis 2008, 40: 84-87.
Wittmann J, Dreiseikelmann B, Rohde C, Rohde M, Sikorski J: Isolation and characterization of numerous novel phages targeting diverse strains of the ubiquitous and opportunistic pathogen Achromobacter xylosoxidans. Plos ONE 9(1):e86935. doi:10.1371/journal.pone.0086935
Levesque R, Letarte R, Pechere JC: Comparative study of the beta-lactamase activity found in Achromobacter. Can J Microbiol 1983, 29: 819-826.
Yamamoto M, Nagao M, Hotta G, Matsumura Y, Matsushima A, et al.: Molecular characterization of IMP-type metallo-beta-lactamases among multidrug-resistant Achromobacter xylosoxidans. J Antimicrob Chemother 2012, 67: 2110-2113.
Bador J, Amoureux L, Blanc E, Neuwirth C: Innate aminoglycoside resistance of Achromobacter xylosoxidans is due to AxyXY-OprZ, an RND-type multidrug efflux pump. Antimicrob Agents Chemother 2013, 57: 603-605.
Jones PT, Pretorius GHJ: Achromobacter sp. 2 Phage a3: A Physical Characterization. J gen Virol 1981, 55: 275-281.
Thomson JA, Woods DR: Bacteriophages and cryptic lysogeny in Achromobacter. J Gen Virol 1974, 22: 153-157.
Zhao Y, Wang K, Jiao N, Chen F: Genome sequences of two novel phages infecting marine roseobacters. Environ Microbiol 2009, 11: 2055-2064.
Moreno Switt AI, Orsi RH, den Bakker HC, Vongkamjan K, Altier C, et al.: Genomic characterization provides new insight into Salmonella phage diversity. BMC Genomics 2013, 14: 481.
Ceyssens PJ, Brabban A, Rogge L, Lewis MS, Pickard D, et al.: Molecular and physiological analysis of three Pseudomonas aeruginosa phages belonging to the "N4-like viruses". Virology 2010, 405: 26-30.
Fouts DE, Klumpp J, Bishop-Lilly KA, Rajavel M, Willner KM, et al.: Whole genome sequencing and comparative genomic analyses of two Vibrio cholerae O139 Bengal-specific Podoviruses to other N4-like phages reveal extensive genetic diversity. Virol J 2013, 10: 165.
Kropinski AM: Measurement of the rate of attachment of bacteriophage to cells. Methods Mol Biol 2009, 501: 151-155.
Watanabe K, Takesue S: The requirement for calcium in infection with Lactobacillus phage. J Gen Virol 1972, 17: 19-30.
Luria SE, Steiner DL: The role of calcium in the penetration of bacteriophage T5 into its host. J Bacteriol 1954, 67: 635-639.
Ohmori H, Haynes LL, Rothman-Denes LB: Structure of the ends of the coliphage N4 genome. J Mol Biol 1988, 202: 1-10.
Black LW: DNA packaging in dsDNA bacteriophages. Annu Rev Microbiol 1989, 43: 267-292.
Strnad H, Ridl J, Paces J, Kolar M, Vlcek C, et al.: Complete genome sequence of the haloaromatic acid-degrading bacterium Achromobacter xylosoxidans A8. J Bacteriol 2011, 193: 791-792.
Trimble WL, le Phung T, Meyer F, Silver S, Gilbert JA: Draft genome sequence of Achromobacter piechaudii strain HLE. J Bacteriol 2012, 194: 6355.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, et al.: The RAST Server: rapid annotations using subsystems technology. BMC Genomics 2008, 9: 75.
Schattner P, Brooks AN, Lowe TM: The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Research 2005, 33: W686-W689.
Bailly-Bechet M, Vergassola M, Rocha E: Causes for the intriguing presence of tRNAs in phages. Genome Res 2007, 17: 1486-1495.
De Paepe M, Taddei F: Viruses' life history: towards a mechanistic basis of a trade-off between survival and reproduction among phages. PLoS Biol 2006, 4: e193.
Sullivan MJ, Petty NK, Beatson SA: Easyfig: a genome comparison visualizer. Bioinformatics 2011, 27: 1009-1010.
Zivin R, Zehring W, Rothman-Denes LB: Transcriptional map of bacteriophage N4: location and polarity of N4 RNAs. J Molecul Biol 1981, 152: 335-356.
Falco SC, Laan KV, Rothman-Denes LB: Virion-associated RNA polymerase required for bacteriophage N4 development. Proc Natl Acad Sci USA 1977, 74: 520-523.
Falco SC, Zehring W, Rothman-Denes LB: DNA-dependent RNA polymerase from bacteriophage N4 virions. Purification and characterization. J Biol Chem 1980, 255: 4339-4347.
Kazmierczak KM, Davydova EK, Mustaev AA, Rothman-Denes LB: The phage N4 virion RNA polymerase catalytic domain is related to single-subunit RNA polymerases. EMBO J 2002, 21: 5815-5823.
Willis SH, Kazmierczak KM, Carter RH, Rothman-Denes LB: N4 RNA polymerase II, a heterodimeric RNA polymerase with homology to the single-subunit family of RNA polymerases. J Bacteriol 2002, 184: 4952-4961.
Born Y, Fieseler L, Marazzi J, Lurz R, Duffy B, et al.: Novel virulent and broad-host-range Erwinia amylovora bacteriophages reveal a high degree of mosaicism and a relationship to Enterobacteriaceae phages. Appl Environ Microbiol 2011, 77: 5945-5954.
Parson KA, Snustad DP: Host DNA degradation after infection of Escherichia coli with Bacteriophage T4: dependence of the alternate pathway of degradation which occurs in the absence of both T4 Endonuclease II and nuclear disruption on T4 Endonuclease IV. J Virol 1975, 15: 221-224.
Koerner JF, Snustad DP: Shutoff of host macromolecular synthesis after T-even bacteriophage infection. Microbiol Review 1979, 43: 199-223.
Lavigne R, Lecoutere E, Wagemans J, Cenens W, Aertsen A, et al.: A multifaceted study of Pseudomonas aeruginosa shutdown by virulent podovirus LUZ19. MBio 2013, 4: e00061-13.
Silverstein JL, Goldberg EB: T4 DNA injection II. Protection of entering DNA from host exonuclease V. Virology 1976, 72: 212-223.
Kruger DH, Schroeder C, Hansen S, Rosenthal HA: Active protection by bacteriophages T3 and T7 against E. coli B- and K-specific restriction of their DNA. Mol Gen Genet 1977, 153: 99-106.
Walkinshaw MD, Taylor P, Sturrock SS, Atanasiu C, Berge T, et al.: Structure of Ocr from bacteriophage T7, a protein that mimics B-form DNA. Mol Cell 2002, 9: 187-194.
King G, Murray NE: Restriction alleviation and modification enhancement by the Rac prophage of Escherichia coli K-12. Mol Microbiol 1995, 16: 769-777.
Handa N, Kobayashi I: Type III restriction is alleviated by bacteriophage (RecE) homologous recombination function but enhanced by bacterial (RecBCD) function. J Bacteriol 2005, 187: 7362-7373.
Paddison P, Abedon ST, Dressman HK, Gailbreath K, Tracy J, et al.: The roles of the bacteriophage T4 r genes in lysis inhibition and fine-structure genetics: a new perspective. Genetics 1998, 148: 1539-1550.
Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, et al.: Bacteriophage T4 genome. Microbiol Mol Biol Rev 2003, 67: 86-156.
Young R: Bacteriophage lysis: mechanism and regulation. Microbiol Rev 1992, 56: 430-481.
Schleifer KH, Kandler O: Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev 1972, 36: 407-477.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL: Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 2001, 305: 567-580.
Young I, Wang I, Roof WD: Phages will out: strategies of host cell lysis. Trends Microbiol 2000, 8: 120-128.
Berry J, Summer EJ, Struck DK, Young R: The final step in the phage infection cycle: the Rz and Rz1 lysis proteins link the inner and outer membranes. Mol Microbiol 2008, 70: 341-351.
Zhang N, Young R: Complementation and characterization of the nested Rz and Rz1 reading frames in the genome of bacteriophage lambda. Mol Gen Genet 1999, 262: 659-667.
Wittmann J, Eichenlaub R, Dreiseikelmann B: The endolysins of bacteriophages CMP1 and CN77 are specific for the lysis of Clavibacter michiganensis strains. Microbiology 2010, 156: 2366-2373.
Henz SRR, Huson DHH, Auch AF, Nieselt-Struwe K, Schuster SCC: Whole-genome prokaryotic phylogeny. Bioinformatics 2004, 21: 2329-2335.
Auch AF, Henz S, Holland B, Göker M: Genome BLAST distance phylogenies inferred from whole plastid and whole mitochondrion genome sequences. BMC bioinformatics 2006, 7: 350.
Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M: Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 2013, 14: 60.
Desper R, Gascuel O: Fast and accurate phylogeny minimum-evolution principle. J Comput Biol 2002, 9: 687-705.
Hess PN, De Moraes Russo CA: An empirical test of the midpoint rooting method. Biol J Linnean Soc 2007, 92: 669-674.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, et al.: BLAST+: architecture and applications. BMC Bioinformatics 2009, 10: 421.
Kulikov E, Kropinski AM, Golomidova A, Lingohr E, Govorun V, et al.: Isolation and characterization of a novel indigenous intestinal N4-related coliphage vB_EcoP_G7C. Virology 2012, 426: 93-99.
Nho SW, Ha MA, Kim KS, Kim TH, Jang HB, et al.: Complete genome sequence of the Bacteriophages ECBP1 and ECBP2 isolated from two different Escherichia coli strains. J Virol 2012, 86: 12439-12440.
Fan H, Fan H, An X, Huang Y, Zhang Z, et al.: Complete genome sequence of IME11, a New N4-Like Bacteriophage. J Virol 2012, 86: 13861.
Gan HM, Sieo CC, HoonTang SG, Omar AR, Ho YW: The complete genome sequence of EC1-UPM, a novel N4-like bacteriophage that infects Escherichia coli O78:K80. Virol J 2013, 10: 308.
Kim MS, Cha KE, Myung H: Complete genome of Pseudomonas aeruginosa phage PA26. J Virol 2012, 86: 10244.
Beilstein F, Dreiseikelmann B: Bacteriophages of freshwater Brevundimonas vesicularis isolates. Res Microbiol 2006, 157: 213-219.
Thorvaldsdottir H, Robinson JT, Mesirov JP: Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013,14(2):178-192.
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B: Artemis: sequence visualization and annotation. Bioinformatics 2000,16(10):944-945.
Wittmann J, Gartemann KH, Eichenlaub R, Dreiseikelmann B: Genomic and molecular analysis of phage CMP1 from Clavibacter michiganensis subspecies michiganensis. Bacteriophage 2011, 1: 6-14.
Valentine RC, Shapiro BM, Stadtman ER: Regulation of glutamine synthetase. XII. Electron microscopy of the enzyme from Escherichia coli. Biochemistry 1968, 7: 2143-2152.
Spurr AR: A low-viscosity epoxy resin embedding medium for electron microscopy. J Ultrastruct Res 1969, 26: 31-43.
Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M: Highly parallelized inference of large genome-based phylogenies. Concurr Comp Pract E 2013. doi:10.1002/cpe.3112
Swofford DL: PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Sunderland, Massachusetts: Sinauer Associates; 2003.
Acknowledgments
We would like to thank Simone Severitt for preparing the library and sequencing and Anja Meier (Helmholtz Centre for Infection Research) for performing the peptide mass fingerprinting analysis. Cordial thanks are addressed to Markus Göker (DSMZ) for helpful comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors` contributions
JW, BD and CR designed experiments; JW and BD performed experiments; JPMK conducted phylogenomic analysis; MR performed morphological analysis via transmission electron microscopy, JW and BB did sequence analysis, JW wrote manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Wittmann, J., Dreiseikelmann, B., Rohde, M. et al. First genome sequences of Achromobacter phages reveal new members of the N4 family. Virol J 11, 14 (2014). https://doi.org/10.1186/1743-422X-11-14
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-11-14