
Abstract
Background
The use of food-grade lactococci as bacterial carriers to DNA delivery into epithelial cells is a new strategy to develop live oral DNA vaccine. Our goal was to develop a new plasmid, named pValac, for antigen delivery for use in lactococci. The pValac plasmid was constructed by the fusion of: i) a eukaryotic region, allowing the cloning of an antigen of interest under the control of the pCMV eukaryotic promoter to be expressed by a host cell and ii) a prokaryotic region allowing replication and selection of bacteria. In order to evaluate pValac functionality, the gfp ORF was cloned into pValac (pValac:gfp) and was analysed by transfection in PK15 cells. The applicability of pValac was demonstrated by invasiveness assays of Lactococcus lactis inlA+ strains harbouring pValac:gfp into Caco-2 cells.
Results
After transfection with pValac:gfp, we observed GFP expression in PK15 cells. L. lactis inlA+ were able to invade Caco-2 cells and delivered a functional expression cassette (pCMV:gfp) into epithelial cells.
Conclusion
We showed the potential of an invasive L. lactis harbouring pValac to DNA delivery and subsequent triggering DNA expression by epithelial cells. Further work will be to examine whether these strains are able to deliver DNA in intestinal cells in vivo.
Similar content being viewed by others
Background
Numerous infectious agents invade the host through the mucosa to cause disease. The use of bacterial carriers to deliver DNA vaccine by oral route constitutes a promising vaccination strategy [1–3]. Most of the bacteria used to deliver DNA vaccine into mammalian cells are invasive pathogens such as Shigella flexneri, Yersinia enterocolitica, Listeria monocytogenesis, Salmonella thiphymurium or Mycobaterium [3–7]. Such bacteria are able to invade professional or non-professional phagocytes and deliver eukaryotic expression vectors resulting in cellular expression of the gene of interest [1, 4, 8]. Despite the use of attenuated strains, the risk associated with potential reversion to the wild-type (virulent) phenotype is a major concern [9].
The use of food-grade lactic acid bacteria (LAB) as DNA delivery vehicles represents an attractive alternative to the use of such attenuated pathogens and other mucosal delivery systems such as liposomes or microparticles [10]. LAB is a diverse group of bacteria transforming sugars into lactic acid. These non-pathogenic and non-invasive Gram-positive bacteria occupy different ecological niches, ranging from plant surfaces to the digestive tract (DT) of man and animals [11].
Antigen and cytokine delivery at the mucosal level by food-grade Lactococcus lactis, the model LAB, has been intensively investigated [12–16] (for review see [10]). In contrast to bacteria-mediated delivery of protein antigens, bacteria-mediated delivery of DNA could lead to host expression of post-translational modified antigens and therefore to the presentation of conformational-restricted epitopes to the immune system [17].
We previously developed a strategy using recombinant invasive lactococci to deliver a plasmid containing a eukaryotic expression cassette gene into epithelial cells. We demonstrated that L. lactis expressing Listeria monocytogenes Internalin A (inlA) gene (LL-inlA+) was internalized by human epithelial cells in vitro and enterocytes in vivo after oral administration of guinea pigs [18]. We also showed that green fluorescent protein (gfp) open reading frame (ORF) under the control of a eukaryotic promoter carried by such LL-inlA+ strains could be delivered into and expressed by epithelial cells [18]. These results were obtained with a large plasmid (10 kb) which is a cointegrate between an E. coli and a L. lactis replicons. During further attempts to insert antigens in this plasmid, we verified that its structure and size made difficulty not only cloning strategies but also transformation steps in lactococci.
To improve our delivery DNA strategy, we constructed a new smaller plasmid, named pValac (Vaccination using lactic acid bacteria). The pValac plasmid was constructed by the fusion of: i) a eukaryotic region, containing the CytoMegaloVirus promoter (pCMV), a multiple cloning site, and the polyadenylation signal of Bovine Growth Hormone (BGH polyA) and ii) a prokaryotic region, containing the RepA/RepC replication origin for both E. coli and L. lactis and a chloramphenicol resistance gene for bacteria selection.
Methods
Bacterial strains and growth conditions
The bacterial strains and plasmids used in this work are listed in Table 1[18–21]. Escherichia coli DH5α was grown on Luria-Bertani medium and incubated at 37°C with vigorous shaking. L. lactis MG1363 was grown in M17 medium containing 0.5% glucose (GM17). Bacteria were selected by addition of antibiotics as follows (concentrations in micrograms per milliliter): for E. coli, erythromycin (100) and chloramphenicol (10); for L. lactis, erythromycin (5) and chloramphenicol (10).
DNA manipulations
DNA manipulations were performed as described [22] with the following modifications: for plasmid DNA extraction from L. lactis, TES (25% sucrose, 1 mM EDTA, 50 mM Tris-HCl pH 8) containing lysozyme (10 mg/ml) was added for 10 min at 37°C to prepare protoplasts. Enzymes were used as recommended by suppliers. Electroporation of L. lactis was performed as described [23]. L. lactis transformants were plated on GM17 agar plates containing the required antibiotic and were counted after 2-day incubation at 30°C.
pValac vector design and construction
The eukaryotic region of pValac was obtained from the pVAX1 vector (Table 1). A 860 bp DNA fragment was generated by PCR using a polymerase with proof reading activity (Platinum pfx high fidelity polymerase, Invitrogen, Sao Paulo, Brazil) and the oligonucleotides CMVBglFwd (5' GGAGATCTGCGTTACATAACTTACGG 3') and BGHClaRev (5' GGATCGATTAGAAGCCATAGAGCCC 3') introducing respectively a BglII and a ClaI (underlined) sites in the fragment. The amplified PCR product was cloned into TOPO vector (Table 1) resulting in TOPO:VAX1 and was introduced by transformation in E. coli DH5α (Table 1). The integrity of the insert was confirmed by sequencing [24] using DYEnamic™ ET Dye Terminator Kit in a MEGABACE 1000 apparatus (GE Healthcare, Sao Paulo, Brazil). TOPO:VAX1 was further digested with BglII and ClaI restriction enzymes and gel purified (S.N.A.P. gel purification kit, Invitrogen). The prokaryotic region of pValac was obtained from the pXylT:CYT (Table 1). A 2882 bp DNA fragment was obtained after BglII and ClaI digestion and gel purified (S.N.A.P. gel purification kit, Invitrogen). BglII/ClaI-digested and purified TOPO:VAX1 and pXylT:CYT fragments were ligated using T4 DNA ligase (Invitrogen) to obtain pValac vector (3742 pb) (Table 1). pValac was established by transformation in E. coli DH5α and then in L. lactis MG1363 strains (Table 1). The integrity of the pValac sequence was confirmed by sequencing as described above.
pValac:gfp construction
The gfp ORF was cloned into pValac in order to evaluate its functionality. The 726 bp gfp ORF, obtained from pEGFP-N1 plasmid (Table 1), was digested with XbaI and BamHI restriction enzymes. The gfp ORF fragment obtained was purified (S.N.A.P. gel purification kit, Invitrogen) and then inserted into the pValac MCS using the same restriction enzymes resulting in pValac:gfp (4468 bp). The integrity of the gfp ORF was confirmed by sequencing as described above.
Transfection assays of pValac into porcine epithelial cells
The pValac:gfp plasmid was assayed for GFP expression by transfection into Porcine Kidney cell line (PK15 cells). Fifty to 80% confluent PK15 cells were cultured in Dulbecco modified Eagle medium, 10% fetal calf serum, 2 mM L-glutamine (BioWhittaker, Cambrex Bio Science, Verviers, Belgium), 100 U penicillin and 100 g streptomycin. PK15 cells were transfected with 1.6 μg of pValac:gfp, pEGFP-N1 (positive control) or pIL253 (negative control) previously complexed with Lipofectamine 2000 (Invitrogen). pIL253 was used as a negative control due to the fate that it is an empty lactococcal plasmid; being more suitable for our next step (see below). The GFP-producing cells were visualized 48 hours after transfection with an epifluorescent microscope (Nikon Eclipse TE200 equipped with a digital still camera Nikon DXM1200). Transfection assays were performed in triplicate.
Invasiveness assays of bacteria into human epithelial cells
To demonstrate the efficacy of pValac as a delivery vector, LL-inlA+ strains were transformed with pValac:gfp (LL-inlA+ pValac:gfp+) (Table 1). In vitro invasion assays of bacteria into human cells were performed using the human colon carcinoma cell line Caco-2 as previously described [25] with some modifications [18]. Briefly, eukaryotic cells were cultured in P6 wells plates containing 1 × 106 cells per dish in RPMI supplemented with 2 mM L-glutamine and 10% fetal calf serum. LL-inlA+ pValac:gfp+ or L. lactis pIL253 pValac:gfp+ (LL-pIL253 pValac:gfp+) (negative control) (Table 1) (OD600 = 0.9–1.0) were added to mammalian cells so that the multiplicity of infection (MOI) was about 103 bacteria/cell. After three hours of internalization, cells were treated for two hours with gentamicin (20 mg/ml) to kill extracellular bacteria. Fluorescent cell quantification was evaluated at 24 and 48 hours after gentamicin treatment by flow cytometry on Fluorescent Activated Cell Sorter (FACS, Becton Dickinson, France). The GFP-producing cells were visualized with an epifluorescent microscope (Nikon Eclipse TE200 equipped with a digital still camera Nikon DXM1200). Internalization and FACS assays were performed in triplicate.
Results and discussion
Picturing pValac
In this work, which is part of an ongoing project geared to implement safer strategies for DNA deliver and expression into eukaryotic cells, we reinforce the use of Lactococcus lactis as DNA delivery vehicle [18, 26]. To improve our delivery DNA strategy, we constructed a new expression plasmid, the pValac.
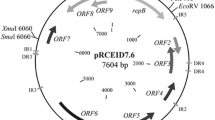
pValac is depicted in Figure 1A. It harbours the eukaryotic region containing the CytoMegaloVirus promoter (pCMV), a multiple cloning site (MCS), and the polyadenylation signal of Bovine Growth Hormone (BGH polyA) needed for a gene expression by eukaryotic host cells. Its prokaryotic region contains the RepA/RepC replication origin for both E. coli and L. lactis and a chloramphenicol resistance gene (Cm) for bacteria selection. The MCS (Figure 1B), inserted between the eukaryotic promoter pCMV and the BGH polyA, carries some potential restriction enzymes that can be used to clone a gene of interest and the T7 primer binding site for its sequencing.

Structure of pValac plasmid. A: Boxes indicate: Multiple cloning site (MCS) and BGH polyadenylation region (polyA). Arrows indicate: cytomegalovirus promoter (pCMV); replication origin of L. lactis (Rep A) and E. coli (Rep C) and chloramphenicol resistance gene (Cm). ClaI and BglII restriction sites used to ligate eukaryotic and prokaryotic regions are showed. B: Multiple cloning site showing the T7 promoter/priming site, different restriction enzyme sites and polyA site.
Transfection assays of pValac into porcine epithelial cells
The pValac:gfp ORF was used for transfection assays into PK15 cells. Forty-eight hours after transfection with pValac:gfp and pEGFP-N1 (positive control), we observed comparable GFP expression in these epithelial cells (Figure 2A and 2B, respectively). No GFP expression was observed after transfection with pIL253 (Figure 2C). This result demonstrates that pValac is functional and it could be used for our further experiment.

Epifluorescent micrograph of 48 hours GFP expression by PK15 cells after transfection. PK15 cells were transfected with pValac:gfp, pEGFP-N1 (positive control) and pIL253 (negative control) plasmids. A: pValac:gfp, B: pEGFP-N1, C: pIL253.
Invasiveness assays of bacteria into human epithelial cells
Internalization of LL-inlA+ pValac:gfp+ into Caco-2 cells led to GFP expression in approximately 1% of the cells 48 hours after cell invasion (Figure 3, Panels A1 and B1). A very low percentage of GFP expression was detected when the non-invasive control strain LL-pIL253 pValac:gfp+ was used (Figure 3, Panels A2 and B2). A MOI of 102 bacteria/cell is generally used for pathogens like L. monocytogenes [25] due to its virulence factors that helps bacteria to escape from vacuoles [27]. Here we used a higher MOI (103bacteria/cell) for L. lactis, a suitable multiplicity required for an efficient internalization for these bacteria [18]. We thus demonstrate that invasive LL-inlA+ pValac:gfp+ were able to invade Caco-2 cells and to deliver a functional expression cassette (pCMV:gfp) into epithelial cells.

Gene expression analysis after invasion assays. A: In vitro gene transfer after 48 hours following invasion of Caco-2 cells with L. lactis strains carrying pValac:gfp assessed by FACS. A1: LL-inlA+ pValac:gfp+; A2: LL-pIL253 pValac:gfp+ (negative control). B: Epifluorescent micrograph of GFP expression by Caco-2 human epithelial cell line after internalization. B1: LL-inlA+ pValac:gfp+; B2: LL-pIL253 pValac:gfp+.
It is worth to note that concerning expression data, it is not surprisingly that we had comparable levels of approximately 1% using pValac:gfp or pVE3890 [18] since both plasmids contain the same eukaryotic genetics components, the pCMV promoter and the BGH polyA. Nevertheless, we observed that pValac is more suitable for cloning and transformation in lactococci. It is easily comprehensible that working with a small plasmid is advantageous to assemble these molecular techniques [28–31] than working with a big size plasmid.
The hypothesis for DNA delivery and expression is based on the infection of host cells by bacterial carriers: following internalization, invasive L. lactis is probably taken up in the vacuoles and target for degradation, thereby releasing pValac:gfp. Then, by an unknown mechanism, the plasmid escapes the vacuoles and reaches the nucleus where the gene (gfp ORF in our case) could be translated by the host cell [32–34]. Questions arise if while non-recombinant lactic acid bacteria are generally regarded as safe they still be viewed as such when invasive. L. lactis inlA+ survival rate was measured and showed a decrease from 4,5 log CFU/ml for 24 hours after internalisation to 2 log CFU/ml after 60 hours (data not shown). In fact, it was already suggested that L. lactis vaccine vectors engineered to access the cytoplasmic antigen presenting pathway are incapable of further growth in this environment [35, 36]. This result suggests that, in vitro, these bacteria could still be regarded as safe when engineered to be invasive.
Conclusion
Mucosal epithelium constitutes the first barrier to be overcome by pathogens during infection. The use of non-invasive bacteria for oral DNA vaccine delivery to induce intestinal mucosal immunity is a promising vaccination strategy used during the last decade. An attractive DNA vaccine strategy is based on the use of the food-grade LAB, Lactococcus lactis, as DNA delivery vehicle at the mucosal level.
In this sense, we constructed the pValac, a new plasmid for DNA delivery. pValac contains eukaryotic genetic elements, allowing cloning and further expression of an antigen of interest by an eukaryotic host cell as well as a prokaryotic region allowing replication and selection of bacteria. After cloning the gfp ORF in pValac we could show that: i) invasive L. lactis strains (inlA+) carrying pValac:gfp were able to enter epithelial cells and ii) after internalization, the host cells expressed the GFP protein. Therefore we could demonstrate the potential application of both plasmid and strain, to implement safer strategies for oral DNA deliver and expression into eukaryotic cells using LAB.
Further experiments have been performed to examine whether these strains are able to release enough DNA to ensure an efficient intestinal cell expression in vivo. In long term, an alternative strategy for DNA vaccine delivery could be achieved based on these recombinant L. lactis carriers.
References
Dietrich G, Bubert A, Gentschev L, Sokolovic Z, Simm A, Catic A, Kaufmann SH, Hess J, Szalay AA, Goebel W: Delivery of antigen-encoding plasmid DNA into the cytosol of macrophages by attenuated suicide Listeria monocytogenes. Nat Biotechnol. 1998, 16: 181-185.
Gentschev I, Dietrich G, Spreng S, Pilgrim S, Stritzker J, Kolb-Mäurer A, Goebel W: Delivery of protein antigens and DNA by attenuated intracellular bacteria. Int J Med Microbiol. 2002, 291: 577-582.
Schoen C, Stritzker J, Goebel W, Pilgrim S: Bacteria as DNA vaccine carriers for genetic immunization. Int J Med Microbiol. 2004, 294: 319-335.
Grillot-Courvalin C, Goussard S, Courvalin P: Bacteria as gene delivery vectors for mammalian cells. Curr Opin Biotechnol. 1999, 10: 477-481.
Xu F, Ulmer JB: Attenuated Salmonella and Shigella as carriers for DNA vaccines. J Drug Target. 2003, 11: 481-488.
Loeffler DI, Schoen CU, Goebel W, Pilgrim S: Comparison of different live vaccine strategies in vivo for delivery of protein antigen or antigen-encoding DNA and mRNA by virulence-attenuated Listeria monocytogenes. Infect Immun. 2006, 74: 3946-3957.
Daudel D, Weidinger G, Spreng S: Use of attenuated bacteria as delivery vectors for DNA vaccines. Expert Rev Vaccines. 2007, 6: 97-110.
Dietrich G, Spreng S, Gentschev I, Goebel W: Bacterial systems for the delivery of eukaryotic antigen expression vectors. Antisense Nucleic Acid Drug Dev. 2000, 10 (5): 391-399.
Dunham SP: The application of nucleic acid vaccines in veterinary medicine. Res Vet Sci. 2002, 73: 9-16.
Wells JM, Mercenier A: Mucosal delivery of therapeutic and prophylactic molecules using lactic acid bacteria. Nat Rev Microbiol. 2008, 6 (5): 349-362.
Bolotin A, Quinquis B, Renault P, Sorokin A, Ehrlich SD, Kulakauskas S, Lapidus A, Goltsman E, Mazur M, Pusch GD, Fonstein M, Overbeek R, Kyprides N, Purnelle B, Prozzi D, Ngui K, Masuy D, Hancy F, Burteau S, Boutry M, Delcour J, Goffeau A, Hols P: Complete sequence and comparative genome analysis of the dairy bacterium Streptococcus thermophilus. Nat Biotechnol. 2004, 22: 1554-1558.
Bermúdez-Humarán LG, Cortes-Perez NG, Le Loir Y, Alcocer-Gonzalez JM, Tamez-Guerra RS, De Oca-Luna RM, Langella P: An inducible surface presentation system improves cellular immunity against human papillomavirus type 16 E7 antigen in mice after nasal administration with recombinant lactococci. J Med Microbiol. 2004, 53: 427-433.
Hanniffy S, Wiedermann U, Repa A, Mercenier A, Daniel C, Fioramonti J, Tlaskolova H, Kozakova H, Israelsen H, Madsen S, Vrang A, Hols P, Delcour J, Bron P, Kleerebezem M, Wells J: Potential and opportunities for use of recombinant lactic acid bacteria in human health. Adv Appl Microbiol. 2004, 56: 1-64.
Mannan P, Jones KF, Geller BL: Mucosal vaccine made from live, recombinant Lactococcus lactis protects mice against pharyngeal infection with Streptococcus pyogenes. Infect Immun. 2004, 72: 3444-3450.
Robinson K, Chamberlain LM, Lopez MC, Rush CM, Marcotte H, Le Page RW, Wells JM: Mucosal and cellular immune responses elicited by recombinant Lactococcus lactis strains expressing tetanus toxin fragment C. Infect Immun. 2004, 72: 2753-2761.
Nouaille S, Ribeiro LA, Miyoshi A, Pontes D, Le Loir Y, Oliveira SC, Langella P, Azevedo V: Heterologous protein production and delivery systems for Lactococcus lactis. Genet Mol Res. 2003, 2 (1): 102-111.
Beláková J, Horynová M, Krupka M, Weigl E, Raska M: DNA vaccines: are they still just a powerful tool for the future?. Arch Immunol Ther Exp (Warsz). 2007, 55 (6): 387-398.
Guimarães VD, Gabriel JE, Lefévre F, Cabanes D, Gruss A, Cossart P, Azevedo V, Langella P: Internalin expressing Lactococcus lactis are able to invade guinea pigs small intestine and deliver DNA into mammalian epithelial cells. Microbes Infect. 2005, 7 (5-6): 836-844.
Gasson MJ: Plasmid complements of Streptococcus lactis NCDO 712 and other lactic streptococci after protoplast-induced curing. J Bacteriol. 1983, 154: 1-9.
Miyoshi A, Jamet E, Commissaire J, Renault P, Langella P, Azevedo V: A xylose-inducible expression system for Lactococcus lactis. Fems Microbiol Lett. 2004, 239: 205-212.
Simon D, Chopin A: Construction of a vector plasmid family and its use for molecular cloning in Streptococcus lactis. Biochimie. 1988, 70 (4): 559-566.
Sambrook J, Fritsch EF, Maniatis T: Molecular Cloning: A Laboratory Manual. 1989, Cold Spring Harbor, Cold Spring Harbor Press
Langella P, Le Loir Y, Ehrlich SD, Gruss A: Efficient plasmid mobilization by pIP501 in Lactococcus lactis subsp. Lactis. J Bacteriol. 1993, 175: 5806-5813.
Sanger F, Nicklen S, Coulson AR: DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977, 74: 5463-5467.
Dramsi S, Biswas I, Maguin E, Braun L, Mastroeni P, Cossart P: Entry of Listeria monocytogenes into hepatocytes requires expression of InlB, a surface protein of internalin multigene family. Mol Microbiol. 1995, 16: 251-261.
Guimarães VD, Innocentin S, Lefèvre F, Azevedo V, Wal JM, Langella P, Chatel JM: Use of native lactococci as vehicles for delivery of DNA into mammalian epithelial cells. Appl Environ Microbiol. 2006, 72: 7091-7.
Dramsi S, Cossart P: Listeriolysin O: a genuine cytolysin optimized for an intracellular parasite. J Cell Biol. 2002, 156: 943-6.
Kondo JK, McKay LL: Plasmid transformation of Streptococcus lactis protoplasts: optimization and use in molecular cloning. Appl Environ Microbiol. 1984, 48: 252-259.
Simon D, Rouault A, Chopin MC: High-efficiency transformation of Streptococcus lactis protoplasts by plasmid DNA. Appl Environ Microbiol. 1986, 52: 394-5.
Fernandez-Castillo R, Vargas C, Nieto JJ, Ventosa A, Ruiz-Berraquero F: Characterization of a plasmid from moderately halophilic eubacteria. J Gen Microbiol. 1992, 138: 1133-7.
Schleyer U, Bringer-Meyer S, Sahm H: An easy cloning and expression vector system for Gluconobacter oxydans. Int J Food Microbiol. 2008, 125: 91-5.
Vassaux G, Nitcheu J, Jezzard S, Lemoine NR: Bacterial gene therapy strategies. J Pathol. 2006, 208: 290-8.
Loessner H, Endmann A, Leschner S, Bauer H, Zelmer A, zur Lage S, Westphal K, Weiss S: Improving live attenuated bacterial carriers for vaccination and therapy. Int J Med Microbiol. 2008, 298: 21-6.
Schoen C, Loeffler DI, Frentzen A, Pilgrim S, Goebel W, Stritzker J: Listeria monocytogenes as novel carrier system for the development of live vaccines. Int J Med Microbiol. 2008, 298: 45-58.
Goetz M, Bubert A, Wang G, Chico-Calero I, Vazquez-Boland JA, Beck M, Slaghuis J, Szalay AA, Goebel W: Microinjection and growth of bacteria in the cytosol of mammalian host cells. Proc Natl Acad Sci USA. 2001, 98: 12221-12226.
Bahey-El-Din MM, Casey PG, Brendan T, Griffin BT, Gahana CGM: Lactococcus lactis-expressing listeriolysin O (LLO) provides protection and specific CD8+ T cells against Listeria monocytogenes in the murine infection model. Vaccine. 2008, 26 (41): 5304-5314.
Acknowledgements
This study was supported by grants from Centro Nacional de Pesquisa e Desenvolvimento Cientifico (CNPq, Brazil) and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG, Brazil). S. Innocentin is a recipient of a European Ph.D. Marie Curie grant from the LABHEALTH program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
VG and SI performed the experiments of the work. VG drafted the manuscript and AM contributed to improve it. JMC and FL coordinated it. PL, VA and AM conceived the study as project leaders. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Guimarães, V., Innocentin, S., Chatel, JM. et al. A new plasmid vector for DNA delivery using lactococci. Genet Vaccines Ther 7, 4 (2009). https://doi.org/10.1186/1479-0556-7-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-0556-7-4