
Abstract
Background
Plasmodium falciparum shares its most recent common ancestor with parasites found in African apes; these species constitute the so-called Laverania clade. In this investigation, the evolutionary history of Plasmodium lineages found in chimpanzees (Pan troglodytes) was explored.
Methods
Here, the remainders of 74 blood samples collected as part of the chimpanzees’ routine health examinations were studied. For all positive samples with parasite lineages belonging to the Laverania clade, the complete mitochondrial genome (mtDNA), the gene encoding dihydrofolate reductase-thymidylate synthase (dhfr-ts), the chloroquine resistance transporter (Pfcrt), the circumsporozoite protein (csp), merozoite surface protein 2 (msp2), and the DBL-1 domain from var2CSA were amplified, cloned, and sequenced. Other Plasmodium species were included in the mtDNA, dhfr-ts, and csp analyses. Phylogenetic and evolutionary genetic analyses were performed, including molecular clock analyses on the mtDNA.
Results/Conclusions
Nine chimpanzees were malaria positive (12.2%); four of those infections were identified as P. falciparum, two as a Plasmodium reichenowi- like parasite or Plasmodium sp., one as Plasmodium gaboni, and two as Plasmodium malariae. All P. falciparum isolates were resistant to chloroquine indicating that the chimpanzees acquired such infections from humans in recent times. Such findings, however, are not sufficient for implicating chimpanzees as an animal reservoir for P. falciparum.
Timing estimates support that the Laverania clade has co-existed with hominids for a long-period of time. The proposed species P. gaboni, Plasmodium billbrayi, and Plasmodium billcollinsi are monophyletic groups supporting that they are indeed different species.
An expanded CSP phylogeny is presented, including all the Laverania species and other malarial parasites. Contrasting with other Plasmodium, the Laverania csp exhibits great conservation at the central tandem repeat region. Msp2 and var 2CSA, however, show extended recent polymorphism in P. falciparum that likely originated after the P. reichenowi-P. falciparum split. The accumulation of such diversity may indicate adaptation to the human host. These examples support the notion that comparative approaches among P. falciparum and its related species will be of great value in understanding the evolution of proteins that are important in parasite invasion of the human red blood cell, as well as those involved in malaria pathogenesis.
Similar content being viewed by others

Background
Despite the extraordinary progress made in malaria control, there are still 200 million clinical cases per year worldwide, with more than 2.6 billion people living at risk of infection[1, 2]. Part of the challenge emerges from the fact that the four Plasmodium species commonly found infecting humans exhibit noticeable differences in their clinical manifestations, geographic distributions, and basic biological characteristics[3]. Such differences are explained, at least in part, by their independent origins as human parasites[4, 5]. Indeed, phylogenetic studies have shown that host switches have been common among primate malarial parasite lineages and that they were important in the origin of human malarias[6–13].
Without a doubt, primate malarial parasites have a broader host range than previously suspected. Evidence of host switches in modern times are found in the case of the macaque parasite Plasmodium knowlesi infecting humans in specific ecological settings[14, 15] and the cases of Plasmodium vivax-Plasmodium simium and Plasmodium malariae-Plasmodium brasilianum that are shared between humans and several species of New World monkeys[12, 16]. In addition, the human parasites P. vivax and P. malariae have been found in African apes[9, 17]. It is worth noting that, whereas P. knowlesi is a zoonosis transmitted from macaques[15], there is no data of malaria transmission from non-human primates to humans in the other cases[18].
The research community had previously known only one species closely related to Plasmodium falciparum, Plasmodium reichenowi found in chimpanzees[5, 19–22]. However, the discovery of new Plasmodium lineages in African apes that share recent common ancestors with P. falciparum and P. reichenowi unveiled a more complex evolutionary history[9–11, 21–23]. Previous studies on these African ape malarias have extensively discussed the possibility of an animal reservoir for P. falciparum malaria[9, 11, 18, 22]. Whereas this is an issue of considerable importance, it is not the only one. Indeed, the value of these non-human falciparum-like parasites for understanding the genetic diversity of P. falciparum has not received adequate attention[19, 20, 24]. In this investigation, data from new isolates of Plasmodium spp. found in chimpanzees (Pan troglodytes) are reported. This expanded dataset allowed exploring the problem of delimiting species in malarial parasites, as this issue relates to the origin of P. falciparum. Then, those lineages were used to investigate the evolution of three malarial antigens: 1) the gene encoding the circumsporozoite protein (CSP), a pre-erythrocytic malaria vaccine antigen; 2) the gene encoding MSP2, an erythrocytic stage malaria vaccine antigen; and 3) the var2CSA gene that has been associated with malaria in pregnancy. Overall, this study reports long-term functional constrains in some of these antigens (e.g. CSP), as well as patterns of high polymorphism within P. falciparum that have a relatively recent origin (msp2 and DBL-1 domain from var2CSA), bringing insights on their importance in the human-falciparum relationship.
Methods
Samples and diagnostic
The chimpanzees are housed at the Jane Goodall Institute’s (JGI) Tchimpounga Chimpanzee Rehabilitation Center in the Republic of Congo (RC). Blood samples were collected throughout 2009-2010 by the veterinary staff as part of the chimpanzees’ routine health examinations following standards approved by the Pan Africa Sanctuary Alliance. The archived remainders of those blood draws were made available by JGI to Arizona State University (ASU) for malaria diagnostics. The samples were exported by the JGI under the RC CITES Export Permit No. 001 (Feb 2, 2010) and imported by ASU under U.S. CITES Import Permit No. 09US094332/9. Other samples used in this investigation were described elsewhere[9, 13, 25].
Genomic DNA was extracted from whole blood (approximately 200 μl) using the QIAamp® DNA Blood Mini kit (Qiagen, GmbH, Hilden, Germany) and each sample was screened for Plasmodium parasites by nested polymerase chain reaction (PCR), using primers for a 1200 bp fragment of the Cytochrome b (cytb) gene[13, 25]. Details about the protocols are shown in the Additional files1 and2.
Species diversity and phylogenetic analyses
Approximately 5,800 bp of the parasites’ mitochondrial genomes (mtDNA) and the gene encoding dihydrofolate reductase-thymidylate synthase (dhfr-ts) were amplified (Additional files1 and2). Independent alignments for nucleotide sequences of the mtDNA and dhfr-ts gene were made using ClustalX v2.0.12 and Muscle as implemented in SeaView v4.3.5 with manual editing. A list of the species included in phylogenetic analyses is provided in the Additional file3; information about the species such as their basic biology, geographic distribution, and host-range can be found elsewhere[3]. Phylogenetic relationships were estimated using Maximum Likelihood (ML) methods as implemented in PhyML v3.0[26] and Bayesian methods using MrBayes v3.1.2[27].
In the case of the mtDNA, both phylogenetic methods used a general time reversible + gamma model (GTR + G) because it best fit the data as estimated by MEGA v5.0[28]. The reliability of the nodes in the ML tree was assessed by the bootstrap method with 200 pseudo-replications. In the case of Bayesian analysis[27], each of the three mitochondrial genes plus the non-coding regions were used as separate partitions[25]. Bayesian support for the nodes was inferred in MrBayes using 8 ×106 Markov Chain Monte Carlo (MCMC) steps, and after convergence was reached, the 50% of the sample as a burn-in was discarded. Sampling was performed every 100 generations. Convergence is reached after the average standard deviation of the posterior probability is below 0.01 and the value of the potential scale reduction factor (PSRF) is between 1.00 and 1.02[27]. In this analysis, all the lineages reported elsewhere were also included (Additional file3)[9, 13, 25]. As a comparison, the genetic divergences in the mtDNA among and within different Plasmodium species were estimated using the Kimura 2-parameter model as implemented in MEGA v5.
Similar procedures were followed when estimating the phylogeny of dhfr-ts. A difference was that Bayesian support for the nodes was inferred in MrBayes using 3 ×106 Markov Chain Monte Carlo (MCMC) steps. In the dhfr-ts analysis, sequences from different Plasmodium species available in GenBank were also included (see Additional file4).
Estimation of divergence times
Time trees were estimated using BEAST v1.6[29] on the nucleotide mtDNA sequences (Additional file3) using four categories: each gene (cox1, cox3, cytb) plus the non-coding regions as a separate partition. Relaxed clock methods were applied with a GTR + G model of substitution with heterogeneity among sites and in the four partitions, optimizing the parameters specifically for each of the partitions. Uniform distributions were used as priors for the calibration intervals and the MCMC was carried out until convergence and good-mixing of the samples were reached[29]. Since time estimates are sensitive to the assumptions and methods utilized[13, 25, 30–35], the exploration of different scenarios is always recommended[25]. In this investigation three events were used as calibration points, one based on a biogeographical landmark and two from fossils. The scenarios explored, based on the calibration points described in Additional file1, were: (1) a combination of the relaxed 6-14.2 Mya calibration for Papio-Macaca divergence with the minimum of 20 Mya for the origin of lemur parasites[30, 31]; (2) a combination of the 6-14.2 Mya calibration with the two the minimums: 23.5 Mya for the human/Macaca split[35] and 20 Mya; and (3) the most conservative time period of 6-8 Mya narrowly defined around the fossils of the Papio-Macaca divergence[32, 34] with a minimum of 20 Mya for the origin of the lemur lineage[25].
Plasmodium falciparum chloroquine resistance transporter
In order to ascertain whether the P. falciparum strains found in chimpanzees were acquired from humans, the chloroquine resistance transporter (Pfcrt) was sequenced so that mutations conferring resistance to the drug could be detected[36]. A fragment of approximately 250 bp was amplified by PCR. This fragment contained all the mutations that have been associated previously with chloroquine resistance (C72S, M74I, N75(E/K) and K76T)[36]. Details about the PCR protocol are provided in the Additional files1 and2.
Malaria antigens
Circumsporozoite protein (CSP)
CSP is the predominant protein found on the surface of the sporozoite[37–39], the haploid stage that is inoculated by the mosquito vector into the vertebrate host. In this investigation, the orthologous genes encoding the CSP protein in malarial parasites from chimpanzees and other non-human primate malarias were amplified (Additional files1 and2) and compared against those sequences available in the GenBank (see Additional file4)[13, 16]. The alignment of csp sequences was done using only the N and C-terminal regions since those can be accurately aligned[16], and the phylogenetic relationships were estimated following the methodology described for mtDNA with a general time reversible + invariant model (GTR + I). Bayesian support for the nodes was inferred in MrBayes using 2 ×106 Markov Chain Monte Carlo (MCMC) steps.
Merozoite surface protein 2
Merozoite surface protein 2 (MSP2) is a highly abundant GPI-anchored protein of P. falciparum that is found exclusively in the Laverania clade[9, 40–42]. The msp2 alleles found in the extant P. falciparum populations have been grouped into two groups; so-called allele families 3D7 and FC27. Previous studies have suggested that these allele families, like others such as msp1, are maintained by balancing selection driven by the host immune responses[42–44]. This investigation compared the available msp2 alleles in the GenBank (Additional file4) from the two families against orthologs found in malarial parasites from chimpanzees (see Additional files1 and2). The alignment and phylogenetic analyses followed the same methodology as above; however, the data fitted a Hasegawa-Kishino-Yano + gamma model (HKY + G) using MEGA v5.0[28]. Bayesian support for the nodes was inferred in MrBayes using 10 × 106 Markov Chain Monte Carlo (MCMC) steps.
Var2CSA protein
Pregnancy-associated malaria (PAM) is related to the expression of a var gene, known as var 2CSA[45–48]. Unlike others members of the var gene family, it is found in all P. falciparum parasites isolates[49]. Here, approximately 1,500 bp (out of 10,000 bp) of the var 2CSA gene containing the complete Duffy binding-like 1 (DBL1) were amplified from seven malaria chimpanzee isolates. The PCR protocol and primers are described in the Additional files1 and2. Alignment of DBL1 var 2CSA nucleotide sequences, obtained from chimpanzees and those available in the GenBank (Additional file4), was done using ClustalX v2.0.12 and Muscle as implemented in SeaView v4.3.5 with manual editing. The phylogenetic relationship among DBL1 var 2CSA alleles was estimated as explained above with a general time reversible + gamma model (GTR + G). In this case, Bayesian support for the nodes was inferred using 20 ×106 MCMC steps.
The genetic polymorphism within each species (P. falciparum and the non-falciparum chimpanzee lineages) was studied by using the statistic π, which is the average number of substitutions per site between any two sequences. In addition, in order to explore the putative effect of natural selection, the average numbers of synonymous substitutions per synonymous site (dS) and non-synonymous substitutions per nonsynonymous site (dN) between a pair of sequences were calculated using Nei and Gojobori’s method[50] with the Jukes and Cantor correction. The difference between dS and dN was estimated, and the standard deviation was calculated using 1,000 pseudo-replications for dS and dN, as well as a two tailed Z-test on the difference between dS and dN also with 1000 bootstrap pseudo-replications[51], as implemented in MEGA v5.0[28]. The null hypothesis was that dS = dN; or selectively neutral.
In addition, the mean (relative) evolutionary rate for each amino acid residue on the same alignment of 918 bp (306 aa) was estimated as implemented in MEGA v5.0[28, 51]. These rates were scaled such that the average evolutionary rate across all sites is 1. This means that sites showing a rate < 1 are evolving slower than average and those with a rate > 1 are evolving faster. These relative rates were estimated separating P. falciparum from Plasmodium sp. using the Jones-Taylor-Thornton model with a discrete Gamma + G distribution (4 categories). The maximum likelihood estimate of the gamma shape parameter was 0.3637 and the maximum Log likelihood for this computation was−4,469.104.
GeneBank accession numbers published in this study
The sequences reported in this study were deposited in GenBank under the following accession numbers: (1) from JX893150 to JX893155 and KF040083 for mtDNA; (2) from JX893156 to JX893168 for dhfr-ts genes; (3) from JX899656 to JX899667 for csp genes; (4) from JX899668 to JX899677 for the DBL1 var 2CSA genes, and (5) from JX893169 to JX893171, and JX899678 for msp2 (see Additional file4).
Results
Species diversity and phylogenetic analyses
Seventy-four chimpanzee (47 males and 27 females) blood samples were screened for Plasmodium parasites. Blood smears were collected, however, none of the samples were positive by microscopy. Nine chimpanzees were found positive (12.2%) by nested PCR using cytochrome b (cytb); seven males (7/47 = 14.8%) and two female (2/27 = 7.41%). Four of the nine positive samples were identified as P. falciparum (isolates 11, 14, 22 and 30) using BLAST, one a mixed infection of P. reichenowi and a Plasmodium reichenowi like parasite that will be further referred as Plamodium sp. (isolate 20), one as Plamodium sp. (isolate 70), one as P. gaboni (isolate 39), and two as P. malariae (isolate 16 and 56). One of the chimpanzees infected with Plamodium sp. was co-infected with P. malariae (isolate 70). Seven parasites’ mitochondrial genomes were generated from these samples: four P. falciparum, one P. reichenowi, one Plasmodium sp. closely related to P. reichenowi, and one Plamodium gaboni (Additional file 3). These new sequences were analysed together with comparable data from African ape malarias that were previously reported[9, 21]; those included P. gaboni and a so-called Plasmodium sp. parasite lineage, as well as Plamodium billcollinsi and Plamodium billbrayi. The clade that contains P. falciparum and P. reichenowi and their sister lineages (P. gaboni, P. billbrayi, P. billcollinsi and Plasmodium sp.) will be collectively referred to as Laverania, a sub-genus used in classical taxonomy for just the species P. falciparum and P. reichenowi[52].
Genetic divergences among the mitochondrial genome of the Plasmodium species found in chimpanzees are shown in Table 1. As expected, the average genetic distance between the P. gaboni isolate sequence reported here and the one previously reported (FJ895307) was just 0.003 ± 0.001 with a total of 18/5,800 differences between the two isolates. This falls within the level of polymorphism observed in recognizable species such as P. vivax and P. falciparum. However, the average genetic distance between P. billbrayi and P. gaboni (0.0181 ± 0.0015 for the complete mtDNA, see Table 1), was comparable to the divergence observed between P. vivax and Plasmodium cynomolgi (0.0118 ± 0.0016, Table 1) and the one found among rodent malarias[25, 53]. Furthermore, the average divergence estimated between the so-called Plasmodium sp. and P. reichenowi lineages, as well as to P. billcollinsi (see Table 1), were also comparable to those between well-recognized Plasmodium species found in mammals (Table 1).
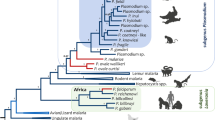
Phylogenetic analyses were performed separately on the complete mtDNA (Figure 1) and dhfr-ts gene (Figure 2). Overall, both phylogenies were consistent, however, they differ in where the root is placed by using Plasmodium gallinaceum as an out-group. In the dhfr-ts phylogeny, the Laverania clade seems to share a recent common ancestor with rodent parasites. In both cases, maximum likelihood and Bayesian methods yield comparable results, indicating that the different Laverania lineages diverged following the same consistent pattern as one expected for different species (Figures 1 and2). Plasmodium gaboni and P. billbrayi form a monophyletic group that share a common ancestor with the clade of P. falciparum-P. reichenowi (Figure 2). Thus, both phylogenies reproduce almost identical evolutionary histories and all the Laverania lineages reported here. Furthermore, these lineages show divergences from their closest sister taxa that are comparable to those observed among well-established Plasmodium species. Overall, these results are consistent with previous studies that have identified multiple Plasmodium species in chimpanzees[9–11, 21].

Phylogenetic tree of Plasmodium spp. based on complete mitochondrial genomes. Bayesian and Maximum Likelihood (ML) methods yielded identical topologies, only the Bayesian tree is shown. The values above branches are posterior probabilities together with bootstrap values (in bold) as a percentage obtained for the maximum likelihood tree (see methods). Human malaria parasites are labelled in red and the chimpanzee identification is labelled in green. Branches indicated in green correspond to the African apes malarias, branches in blue correspond to the other mammals Plasmodium parasites (rodents, lemurs, Asian primates, etc.), and black braches mean the out-group used in this study.

Phylogenetic tree of Plasmodium spp. based on the gene encoding dhfr-ts . Bayesian and Maximum Likelihood (ML) methods yielded identical topologies, only the Bayesian tree is shown. The values above branches are posterior probabilities together with bootstrap values (in bold) as a percentage obtained for the maximum likelihood tree (see methods). Human malaria parasites are labelled in red and the chimpanzee identification is labelled in green. * indicates a clade without support in the maximum likelihood tree.
Estimation of divergence times
The mtDNA phylogeny was used to estimate the time of origin of the African ape parasites (Figure 3). Table 2 shows the divergence times of Plasmodium splits in the Laverania clades as estimated by BEAST v1.6[54] considering three different scenarios (see also Additional file 5 for the node numbers). As expected, the credibility intervals obtained using these scenarios overlap (Table 2). Under the first scenario, the estimates of the divergence times for the radiation of African ape malarias suggest that all five lineages might have originated between 13.09 and 22.93 million years ago (Mya); therefore, African ape parasites may have started to diverge long before the divergence of Pan and Homo (2.7-13.0 Mya)[55, 56]. The P. billbrayi-gaboni split is estimated between 1.92-4.69 Mya and for P. reichenowi-P. falciparum between 4.02 and 7.84 Mya. Those estimates are older than those reported previously[9], but similar to those times estimated by Pacheco et al.[13, 25], using fewer sequences but the same calibration points. However, regardless of the wide confidence interval, the time frames for the reichenowi-falciparum split are still consistent with the divergence of the genera Pan and Homo. Overall, these time estimates suggest that the Laverania species likely have been associated with hominids for a long time[9, 13, 25].

Timetree of the divergence of malarial parasites. Divergence times were estimated with BEAST using the most conservative scenario based on the minimum divergence of Macaca and Papio using fossils (6-8 Mya) and a minimum of 20 Mya for the origin of lemur parasites. Times are shown in millions of years ago (Mya).
Plasmodium falciparum chloroquine resistance transporter
The P. falciparum isolates found in the chimpanzees were acquired recently from humans. The mtDNA genome sequences clearly belong to P. falciparum[57] and all the isolates had Pfcrt mutations (allele CVIET from Africa/Southeast Asia)[36] associated with chloroquine resistance. It is also worth noting that all the other Laverania species lack those Pfcrt mutations associated with chloroquine resistance. This result was expected since those species are not exposed to this anti-malarial drug.
Evolution of malaria antigens
Circumsporozoite protein
A phylogeny for the csp gene using the non-repetitive regions is depicted in Figure 4. It follows the trend evidenced by the mtDNA and dhfr-ts phylogenies in terms of showing that P. falciparum and all the Plasmodium sp. found in chimpanzees are part of a monophyletic group. However, there were differences in the relationship of the major clades when the csp gene tree was compared with the mtDNA and dhfr-ts phylogenies. Since only the non-repetitive region can be properly aligned[16], and not all phylogenetic analyses include the same number of species, those differences are likely the result of the limited phylogenetic information found in relatively short sequences.

Bayesian phylogenetic tree of chimpanzee Plasmodium based on the csp gene. The values above branches are posterior probabilities (see methods). Human malaria parasites are labelled in red and the chimpanzee identification is labelled in green.
Usually, the central tandem repeat regions of the CS proteins exhibit extensive diversity in Plasmodium species[13, 37–39]. Such diversity is evidenced in the number of repeats within Plasmodium species and diverging motifs, even among closely related species or within a single species[13, 16, 37–39, 58–61]. However, unlike other malarial parasites[13, 16, 61], there is a strong conservation of the basic asparagine rich tandem motif among the species in the Laverania clade (Table 3). Specifically, the motifs PNAN and PNVD are present not only in P. falciparum[44, 62], but also in all Laverania species. There are, however, a few differences. The motif PNVN is only found in P. reichenowi and Plasmodium sp. but not in P. falciparum. Likewise PNAD, a motif found in low number in P. billcollinsi, P. billbrayi, and P. gaboni is not present in P. falciparum[44, 62]. Finally, two different asparagine rich motifs could be identified in P. billcollinsi; (PNANPN)17 and the less common (PNANPNPN)2. The frequency of the repeats appears in Table 3. Interestingly, Plasmodium yoelii and P. billcollinsi are the only species with repeats between the regions identified as Pf1 and Pf2 (Figure 5, Table 3) that can bind heparin/heparin sulphate binding sites (see below). In the case of P. billcollinsi, the repeat also involves an asparagine rich motif.

Alignment of the N-terminal region for orthologous genes that encode CS protein in different Plasmodium spp. Functional regions are described elsewhere[63, 64]. The species names are abbreviated as follow: Pvi-P. vivax (Thai), Pcy-P. cynomolgi (Gombak), Pin-P. inui (Hawking), Por-Plasmodium sp. from orangutans (VS28), Phy-P. hylobati, Pco, P. coatneyi, Pkn-P. knowlesi, Pfr-P. fragile, Pmn-Plasmodium sp. from mandrill, Pgo-P. gonderi, Pma-P. malariae, Pbe-P. berghei, Pyo-P. yoelli, Pbillc-P. billcollinsi, Pbillb-P. billbrayi, Pga-P. gaboni, Psp-Plasmodium sp., PreA-P. reichenowi (Sanger), PreB-P. reichenowi from chimp 20, Pf-ch-P. falciparum from chimp 22, Pf3D7-P. falciparum strain 3D7, and Pgall-P. gallinaceum.
The alignment of the N and C-terminal domains of the csp gene is illustrated in Figures 5 and6, respectively, incorporating information about functionally relevant parts of the protein[37, 63, 64]. The signal peptides (Figure 5) were predicted using the SignalP 4.0 Server[65]. The CS proteins from P. billcollinsi, P. gaboni, P. billrayi, and P. reichenowi show differences in a region of low complexity that is found prior to the so-called region I (see Figure 5). The region I (KLKQP)[64, 66], contained in Pf2[63], is a protease cleavage site that plays a critical role in the processing of the CS protein in the mammalian host and exhibits high affinity to heparin sulphate found on the surface of liver cells or hepatocytes[37, 38, 63, 64, 66]. The motif KLKQP is conserved between P. reichenowi and P. falciparum, as is the case in many of the other Plasmodium species. However, three of the Laverania lineages differ, having the variant KLR QP instead of KLK QP, (specifically P. billcollinsi (Pbillc), P. billbrayi (Pbillb), and P. gaboni (Pgab)) (Figure 5). Previously, the only known CSP sequences with variations in this region were the avian parasite P. gallinaceum, with the motif NLNQP, and the rodent parasite P. yoelii, with KLNQP[66, 67]. Surprisingly, the two African Cercopithecidae parasites, P. gonderi (identified as Pgo in Figures 5 and6) and the one from mandrills (Pmn) lack the region I (KLKQP). These results were reproduced not only by independent PCRs, but also by a perfect match in Plasmodium gonderi genomic data[68].

Alignment of the C-terminal region for orthologous genes that encode CS protein in different Plasmodium spp. Functional regions are described elsewhere[63, 64]. The species names are abbreviated as follow: Pvi-P. vivax (Thai), Pcy-P. cynomolgi (Gombak), Pin-P. inui (Hawking), Por-Plasmodium sp. from orangutans (VS28), Phy-P. hylobati, Pco, P. coatneyi, Pkn-P. knowlesi, Pfr-P. fragile, Pmn-Plasmodium sp. from mandrill, Pgo-P. gonderi, Pma-P. malariae, Pbe-P. berghei, Pyo-P. yoelli, Pbillc-P. billcollinsi, Pbillb-P. billbrayi, Pga-P. gaboni, Psp-Plasmodium sp., PreA-P. reichenowi (Sanger), PreB-P. reichenowi from chimp 20, Pf-ch-P. falciparum from chimp 22, Pf3D7-P. falciparum strain 3D7, and Pgall-P. gallinaceum.
The other two regions in the CSP with capacity to bind heparin/heparin sulphate (HS)[63, 64], in addition to Pf2-region I+, are a peptide in the N-terminal or Pf1 and one in the C-terminal domain or Pf4-Region II+, see Figure 5 and6[37, 38, 63, 64, 66]. Pf1 is not considered critical[63] and is not conserved in P. vivax and related parasites[5, 6], however, it is highly conserved among Laverania and the only avian CS protein sequence available[67] setting them apart from the other Plasmodium found in mammals (Figure 5). The Pf4 with the so-called region II +[63, 64] (Figure 6) is conserved among Plasmodium species and almost identical in the Laverania clade (EWSPCSVTCGNGIQVRIK) with few synonymous substitutions (dS = 0.1504 vs. dN = 0.0). The CS.T3, a T-cell epitope responsible for a CD4+ T-cell response that correlates with protection[64], is also identical among P. falciparum and the Plasmodium species from chimpanzees (Figure 6). Thus, with the exception of the specific changes described above, the CS in Laverania is highly conserved, including the observed motifs in the tandem repeat region, the latest contrast with other Plasmodium spp. found in mammals.[13, 16, 61, 67].
Merozoite surface protein 2
The two msp2 allele families in P. falciparum are almost identical at nucleotide sites encoding the N and C termini[42], but they differ in a central low complexity region that clearly separates the two allele families into the so-called 3D7 and FC27 types, named after the strains from which they were originally sequenced[69]. It has been proposed that these two allele families originated after the P. reichenowi-P. falciparum split[44, 70]. One line of evidence for this came from the so-called repeat homology regions or RHRs[44, 70], where repeats observed in P. reichenowi showed similarities with one allele family or the other. Figure 7 depicts the amino acid alignment of the msp2 allele families and the new sequences of P. reichenowi (isolate 20, mixed infection) and Plasmodium sp. (isolates Shegue and 20) parasites; these four were the only complete sequences obtained in this investigation. Plasmodium reichenowi shares with the 3D7 the so-called RHR1 marked in yellow[44] but it was not found in the Plasmodium sp. parasite or in the FC27 family. Attempts were made to identify the second RHR[44, 70] (in blue) that is shared among P. reichenowi and P. falciparum. However, the patterns do not seem clear and whether it is actually homologous was difficult to determine. A conserved motif (in pink) is shared between FC27 alleles and P. reichenowi.

Alignment of the N and C-terminal region for orthologous genes that encode MSP2 in the Laverania species. The repeat homology regions 1 or RHR1 described by Rich and Ayala 2000[44] are shown in yellow.
The second line of evidence comes from the conserved flanking regions (Block 1 and 5 sensu[42]) where P. reichenowi seems separated from the two-allele families. However, no formal phylogenetic analyses were performed. Here a phylogeny of the newly sequenced msp2 from chimpanzee parasites together with all complete P. falciparum msp2 sequences was inferred (Figure 8). The chimpanzee lineages are different from the two-allele families. Indeed the P. reichenowi and the Plasmodium sp. alleles form a monophyletic group clearly different from the two P. falciparum allele families. Given the host switches observed, the time of origin for these two families cannot be estimated by assuming that P. reichenowi and P. falciparum diverged with their host as previously done[42].

Bayesian phylogenetic tree of the Laverania Plasmodium species based on the msp2 gene. The values above branches are posterior probabilities (see methods). The two groups that are called allele families, 3D7 (in red) and FC27 (in blue) are shown.
Evolution of the var2CSA DBL1 domain
The var 2CSA DBL1 domain was not successfully amplified from the all the chimpanzee isolates; however, data was obtained from P. billbrayi, P. gaboni, the Plasmodium sp. lineage, P. reichenowi and three of the P. falciparum parasites. A Bayesian phylogeny is depicted in Figure 9; it includes all the sequences obtained in this investigation together with those available at the GenBank. As expected, the P. falciparum parasites found in chimpanzees clearly group within the known sequences from human infections. Although there are different malarial parasites present in chimpanzees, the var 2CSA sequences did not show much divergence among them. In some cases, there is an apparent gene duplication as evidenced by the two sequences obtained from some of the isolates, e.g. the isolates that yield P. gaboni mtDNA (isolate 39) and Plasmodium sp. (isolate 70) (Figure 9). Nevertheless, given that these are field isolates, there is still the possibility of mixed infections even when those were no detected using other genes.

Bayesian phylogenetic tree of the Laverania Plasmodium species based on the var2CSA DBL1 gene. The values above branches are posterior probabilities (see methods).
Table 4 shows estimates of the genetic diversity for the alleles reported here. The diversity of the DBL1 domain from var 2CSA DBL1 in chimpanzees, even when all the sequences are considered as one “species”, was lower than the diversity found in P. falciparum (0.0939 ± 0.0062 vs. 0.1173 ± 0.0064 respectively). It is also worth noting that in almost all comparisons there were significantly more synonymous than non-synonymous substitutions (Table 4) suggesting functional constraint. The diversity within P. falciparum and those lineages found in chimpanzees was further explored using codon-based analyses. Figure 10 shows the evolutionary rate per codon. The estimate of evolutionary rates per codon shows that many P. falciparum codons (in blue) vary more than in the chimpanzee lineages (in red) (Figure 10). Overall, the results indicate that there is extensive variation within P. falciparum that is not observed among distinct ape malaria lineages. Thus, like in msp2, such divergence seems to be originated after the P. falciparum-P. reichenowi split.

Mean (relative) evolutionary rates for the VAR2CSA protein. These rates are scaled such that the average evolutionary rate across all sites is 1. This means that sites showing a rate < 1 are evolving slower than average and those with a rate > 1 are evolving faster than average. These relative rates were estimated under the Jones-Taylor-Thornton model (+G). A discrete Gamma (+G) distribution was used to model evolutionary rate differences among sites (4 categories). Blue dots correspond with P. falciparum and red dots are chimpanzee parasites. The graph shows that P. falciparum sequences are by far more divergent that those lineages found in chimpanzees.
Discussion
Defining Plasmodium species
The molecular evidence indicates that there are several species within the Laverania clade. These lineages have been independently reported[9–11, 21] indicating that they are actively transmitted. Regardless of the absence of morphological evidence, these are well-defined phylogenetic species since they show a level of divergence comparable with those observed between related taxa such as P. vivax and Plamodium cynomolgi. The use of molecular methods for defining species in malarial parasites is not free of controversy[71], however, such debates are not new in biology[72].
There are several considerations favouring the use of molecular criteria to delimit species in Plasmodium. First, several infections appear to be sub-microscopic making the use of morphological evidence difficult if not impossible, in many cases. Second, in the case of Laverania parasites, whenever blood stages have been observed, they seem to be indistinguishable from P. falciparum[3, 21, 73]. Such limitations on the use of morphological traits are relatively common in malarial parasites. For example, trained microscopists could not detect P. knowlesi when observed in humans and misidentified these zoonotic infections as P. malariae or P. falciparum[14, 74]. Furthermore, there is clear genetic divergence in the mtDNA among parasites morphologically identified as Plamodium relictum in birds[75, 76]. Finally, there are many non-human primates that are endangered species, so studies on parasite biodiversity likely will use opportunistic samples that may only allow for DNA-based approaches[9–11, 21, 22]. It is worth noting that unlike malarial parasites in Asian apes or lemurs[3, 13, 25, 52, 73], there were not multiple P. falciparum-like morphologically described species in African apes. The only recognized species was P. reichenowi[3, 52, 73], a parasite from which there is molecular data, and these lineages are related, but clearly not the same parasite[9–11, 21, 22]. Thus, it is not necessary to establish whether these new species correspond with others previously described. Overall, the DNA evidence allows a stable taxonomy that facilitates comparisons among studies.
Separate discussions are what is considered a level of divergence that could indicate different species or subspecies[22, 25, 53, 76, 77] and what kind of genetic data should be used. For example, the use of 400-600 bp segments of the mitochondrial gene cytb, as has been widely used in avian malaria studies, seems insufficient. However, a suitable fragment of the mtDNA that includes the cytb and cox1[10] provides valuable phylogenetic signal that is consistent with the complete mtDNA and other genes[5, 10, 13, 25]. In the context of this investigation, P. gaboni and P. billbrayi are a good case study[22]. The available evidence, at least at the level of complete mtDNA sequences, indicates that the divergence between P. gaboni and P. billrayi is comparable to that between P. vivax and P. cynomolgi. Accordingly, if P. billbrayi were considered a synonymy of P. gaboni, and consequently studied as part of a single taxon, then the observed mtDNA “polymorphism” would exceed that found within any other known Plasmodium species with worldwide distributions (Table 1). Such pattern will require an ad hoc explanation that cannot be supported on what is known about the mitochondrial genome diversity in Plasmodium. Thus, it seems more parsimonious to hypothesize the existence of two different species, rather than explaining how such a high level of polymorphism can be harboured within one species. The P. gaboni-P. billbrayi separation is supported by the dhfr and csp genes but some of those differences are hard to interpret in the absence of mtDNA data (e.g. differences in low complexity regions in the csp, see Figure 5). However, taking together the observed differences in the dhfr and csp genes and the mtDNA divergence, a recent but clear divergence between P. gaboni and P. billbrayi was found. Thus, the hypothesis that these two evolutionary lineages are different species seems to hold.
Likewise, the so-called Plasmodium sp. appears to be a distinct lineage that is closely related to P. reichenowi; whether these are species or sub-species require some additional considerations. The Plasmodium sp. lineage corresponds with mitochondrial haplotypes identified as P. reichenowi that were previously isolated from chimpanzees in the same region and reported as part of clade C1[10], as well as with a previous study using complete mtDNA[9]. The differences in terms of the csp and the var 2CSA are mostly qualitative and difficult to interpret. The dhfr as nuclear gene, however, separates P. reichenowi from Plasmodium sp. by only 11 SNPs (d = 0.007 versus d = 0.016 between P. reichenowi and P. falciparum using Kimura 2-parameter). This difference is hard to interpret since this gene is seldom sequenced in non-human primates so we lack a good database for comparison. Although the msp2 actually separates these two lineages (P. reichenowi from Plasmodium sp.) in terms of its low complexity regions and SNPs in the 3′ and 5′ termini, it is an antigen under balancing selection[42]. Nevertheless, the differences on dhfr and msp2 establish a linkage between nuclear and mitochondrial genes[77] suggesting that these two lineages are at least different sub-species. The divergence of these two P. reichenowi s.l. lineages seems to be the result, at least in part, of the geographic differentiation of the host populations (P. troglodytes)[10]. However, in order to set a high standard, additional data is needed in order to determine whether these should be considered sub-species or species. The reference mitochondrial haplotypes for this lineage called as Plasmodium sp. in this investigation are identified with the accession numbers GQ355476 and KF040083. At this point, it is worth noting that identifying species or sub-species is not only of taxonomic interest. Indeed, using “pooled data” from otherwise different species could affect comparative analyses aiming to ascertain the processes shaping the genetic diversity of P. falciparum. Furthermore, having delimited molecular lineages is epidemiologically critical in determining whether or not apes are a reservoir for P. falciparum[18].
Apes as potential reservoirs for human malarias
The finding of chloroquine resistant P. falciparum in chimpanzees provides evidence of a host switch from humans at a time scale that is epidemiologically relevant. However, whether this indicates that chimpanzees could act as malaria reservoirs is a different matter. Indeed, there are several criteria that need to be fulfilled, such as evidence that apes can sustain P. falciparum infections and produce gametocytes[73, 78], as well as understanding the ecological factors that may allow vectors to feed on apes and humans. It is worth noting that P. falciparum does not yield high parasitemias at least in chimpanzees[73, 78], so it may be possible that the same phenomenon happens in gorillas. Thus, there is no evidence that currently confirms or categorically rules out apes as reservoirs for P. falciparum.
Evolution of gene encoding malarial antigens
This investigation shows how ape malarias provide new insights on the evolution of important antigens. In the case of the CS protein, unlike other Plasmodium from mammals[13, 16, 59, 61], the central tandem repeat region is conserved among the Laverania lineages. Whether this pattern is the result of immune pressures or functional constrains[62] from genomes with a trend toward accumulating asparagine rich low complexity regions[79], are issues that will require additional studies. Another observation is the conservation of peptides binding heparin sulphate (HS). Specifically, the two cell surface receptors involved in the sporozoite attachment to the hepatocytes (highly sulphated HS associated with region I + or Pf2 and Pf4 region II +, see Figure 1[63, 64]) are almost identical among the Laverania lineages. These similarities suggest that the Laverania sporozoites can bind comparable receptors in the human and apes livers[59] providing additional support to the observation that these parasites can potentially infect several species of hominids.
Although Laverania parasites are the scope of this study, the lack of region I in Plamodium gonderi and the Plasmodium spp. should be highlighted. These are the only known Plasmodium species with a CS protein missing this functionally important region[16, 38, 63]. In the absence of additional information, any interpretation is speculative. However, this CS protein “natural knockout” of the region I (see[38, 66]) suggests that these parasites may have some unique processing of their CS proteins and may provide new insights on the Plasmodium invasion of the liver.
Regarding the msp2, it has been proposed that these two major groups of alleles (also referred as allele families) originated soon after the P. reichenowi-P. falciparum split[42, 44, 70]. This contrasts with the allele families in msp1 that are considered an old polymorphism[43]. The data presented here indicates that the diversity of msp2 alleles observed in P. falciparum originated after the P. falciparum-P. reichenowi split. This hypothesis was originally proposed by the discovery of the so-called mosaic structure of P. reichenowi MSP2[44, 70], however, it was still possible that additional data from other Laverania species could provide evidence of an older polymorphism in msp2. Indeed, some motifs are conserved among the different allele families, and P. reichenowi and Plasmodium sp. parasites (Figure 7), suggesting that such convergence/recombination events predate the P. reichenowi-P. falciparum divergence. Here, a phylogeny (Figure 8) shows that the two-allele families are part of a monophyletic group separated from the chimpanzee parasites. Considering that msp2 is only found in the Laverania clade, the fast divergence of these two alleles families may had been driven by allele-specific immunity[41, 80] that could lead to a pattern consistent with balancing selection[42]. Recent high genetic diversity within extant P. falciparum populations seems to be also the case for var 2CSA.
As described earlier, var 2CSA is critical in pregnancy-associated malaria and is one of the few members of the var gene family that is found in all P. falciparum isolates[45–47]. This preliminary study shows that var 2CSA can be found in relatively divergent Laverania lineages (Figure 9). Considering the fact that other var genes are seldom shared among even P. falciparum isolates[48, 49], this observation reinforces the importance that this particular var gene has in P. falciparum and the other species of the Laverania clade. The second observation that emerges is that var 2CSA accumulated extraordinary genetic polymorphism after the P. reichenowi and P. falciparum split. Whereas these observations should be considered preliminary, lineages that are highly divergent in the mtDNA (e.g. P. gaboni) show relatively low divergence in the DBL1 var 2CSA. The results presented here also indicated that it is possible that var 2CSA is duplicated in some chimpanzee lineages, a phenomenon that has been observed in some P. falciparum strains (e.g. HB3, see Figure 9). Taken together, this study shows that the presence of var 2CSA is relatively “ancient” (e.g. shared between ape lineages and humans), a situation contrasting that seen with other var genes[49]. This indicates some functional importance for this var 2CSA gene. Such evidence allows hypothesizing that natural selection has maintained this var gene in the Laverania group; thus, it should confer some adaptive advantage for the parasite. If acquired immunity that results from malaria exposure affects transmission in humans (parasite fitness), then var 2CSA may increase the parasite transmissibility in a second segment of the population that otherwise will be immune, such as primigravid women. Unfortunately, there is no information on gametocytaemia in primigravid women or whether parasite transmissibility increases for that malaria high risk group. Thus, in the absence of such data, this hypothesis of a selective advantage for var 2CSA cannot be formally tested.
Conclusions
The Laverania clade consists of a highly diverse group of species. All the species described are actively transmitted; evidence of this is that they have been independently found in different studies. Thus, they are stable evolutionary and ecological units as expected for any Plasmodium species. This investigation provides additional evidence that chimpanzees can acquire P. falciparum from humans. However, whether they can sustain transmission and act as a reservoir is a matter that requires additional studies.
This study emphasizes the value of comparative studies on genes involved in the infection of humans by malarial parasites. Such studies improve our understanding of the molecular bases of these parasites pathogenesis and provide information on the rate and mode of evolution of antigens that are considered vaccine targets. As specific examples, this investigation provides evidence of constraints in the evolution of the CS protein among the Laverania species that have not been observed in other Plasmodium lineages; specifically, the conservation of its tandem repeat region. In contrast, antigens such as MSP2 and VAR2CSA likely accumulated genetic variation very recently, suggesting the role of natural selection and highlighting their importance as adaptations to the human host.
Studying the evolution of the P. falciparum clade will require an extensive sampling of parasites from African apes; sampling that is difficult due to ethical concerns and regulations limiting research on apes. In addition, collecting viable isolates of these falciparum-like parasites in order to use them for comparative studies may prove to be even more complex. Nevertheless, it is worthwhile to consider such studies if they will not expose African apes to any significant risk.
Abbreviations
- CIDRpam:
-
A cysteine-rich inter-domain region
- cox1:
-
cytochrome oxidase subunit 1 gene
- cox3:
-
cytochrome oxidase subunit 3 gene
- CSA:
-
Chondroitin sulphate A binding in the placenta
- CSP:
-
Circumsporozoite protein
- cytb:
-
Cytochrome b gene
- DBL:
-
Duffy binding-like domains
- dhfr-ts:
-
Gene encoding dihydrofolate reductase-thymidylate synthase
- GTR:
-
General time reversible + gamma (G) or invariants (I)
- HKY + G:
-
Hasegawa-Kishino-Yano + gamma model
- HS:
-
Heparan sulphate
- ML:
-
Maximum Likelihood
- MCMC:
-
Markov Chain Monte Carlo
- Mya:
-
Million years ago
- MSP1:
-
Merozoite Surface Proteins 1, MSP2, Merozoite surface proteins 2
- MSP2:
-
Merozoite surface proteins 2
- mtDNA:
-
Mitochondrial genomes
- PAM:
-
Pregnancy-associated malaria
- PCR:
-
Nested polymerase chain reaction
- PfCRT:
-
Chloroquine resistance transporter
- PSRF:
-
Potential scale reduction factor.
References
Cibulskis RE, Aregawi M, Williams R, Otten M, Dye C: Worldwide incidence of malaria in 2009: estimates, time trends, and a critique of methods. PLoS Med. 2011, 8: e1001142-10.1371/journal.pmed.1001142.
WHO: World malaria report. 2011, Geneva: World Health Organization, http://www.who.int/malaria/world_malaria_report_2011/ (accessed July 24, 2012)
Coatney RG, Collins WE, Warren M, Contacos PG: The primate malaria. 1971, Washington DC: US Government Printing Office
Escalante AA, Ayala FJ: Phylogeny of the malarial genus Plasmodium derived from rRNA gene sequences. Proc Natl Acad Sci USA. 1994, 91: 11373-11377. 10.1073/pnas.91.24.11373.
Escalante AA, Freeland DE, Collins WE, Lal AA: The evolution of primate malaria parasites based on the gene encoding cytochrome b from the linear mitochondrial genome. Proc Nat Acad Sci USA. 1998, 95: 8124-8129. 10.1073/pnas.95.14.8124.
Escalante AA, Cornejo OE, Freeland DE, Poe AC, Durrego E, Collins WE, Lal AA: A monkey’s tale: the origin of Plasmodium vivax as a human malaria parasite. Proc Natl Acad Sci USA. 2005, 102: 1980-1985. 10.1073/pnas.0409652102.
Rich SM, Leendertz FH, Xu G, LeBreton M, Djoko CF, Aminake MN, Takang EE, Diffo JL, Pike BL, Rosenthal BM, Formenty P, Boesch C, Ayala FJ, Wolfe ND: The origin of malignant malaria. Proc Natl Acad Sci USA. 2009, 106: 14902-14907. 10.1073/pnas.0907740106.
Duval L, Fourment M, Nerrienet E, Rousset D, Sadeuh SA, Goodman SM, Andriaholinirina NV, Randrianarivelojosia M, Paul RE, Robert V, Ayala FJ, Ariey F: African apes as reservoirs of Plasmodium falciparum and the origin and diversification of the Laverania subgenus. Proc Natl Acad Sci USA. 2010, 107: 10561-10566. 10.1073/pnas.1005435107.
Krief S, Escalante AA, Pacheco MA, Mugisha L, André C, Halbwax M, Fischer A, Krief JM, Kasenene JM, Crandfield M, Cornejo OE, Chavatte JM, Lin C, Letourneur F, Grüner AC, McCutchan TF, Rénia L, Snounou G: On the diversity of malaria parasites in African apes and the origin of Plasmodium falciparum from Bonobos. PLoS Pathog. 2010, 6: e1000765-10.1371/journal.ppat.1000765.
Liu W, Li Y, Learn GH, Rudicell RS, Robertson JD, Keele BF, Ndjango JB, Sanz CM, Morgan DB, Locatelli S, Gonder MK, Kranzusch PJ, Walsh PD, Delaporte E, Mpoudi-Ngole E, Georgiev AV, Muller MN, Shaw GM, Peeters M, Sharp PM, Rayner JC, Hahn BH: Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature. 2010, 467: 420-425. 10.1038/nature09442.
Prugnolle F, Durand P, Ollomo B, Duval L, Ariey F, Arnathau C, Gonzalez JP, Leroy E, Renaud F: A fresh look at the origin of Plasmodium falciparum, the most malignant malaria agent. PLoS Pathog. 2011, 7: e1001283-10.1371/journal.ppat.1001283.
Tazi L, Ayala FJ: Unresolved direction of host transfer of Plasmodium vivax v. P. simium and P. malariae v. P. brasilianum. Infect Genet Evol. 2011, 11: 209-221. 10.1016/j.meegid.2010.08.007.
Pacheco MA, Reid MJ, Schillaci MA, Lowenberger CA, Galdikas BM, Jones-Engel L, Escalante AA: The origin of malarial parasites in orangutans. PLoS One. 2012, 7: e34990-10.1371/journal.pone.0034990.
Singh B, Kim Sung L, Matusop A, Radhakrishnan A, Shamsul SS, Cox-Singh J, Thomas A, Conway DJ: A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004, 363: 1017-1024. 10.1016/S0140-6736(04)15836-4.
Lee KS, Divis PC, Zakaria SK, Matusop A, Julin RA, Conway DJ, Cox-Singh J, Singh B: Plasmodium knowlesi: reservoir hosts and tracking the emergence in humans and macaques. PLoS Pathog. 2011, 7: e1002015-10.1371/journal.ppat.1002015.
Escalante AA, Barrio E, Ayala FJ: Evolutionary origin of human and primate malarias: evidence from the circumsporozoite protein gene. Mol Biol Evol. 1995, 12: 616-626.
Kaiser M, Löwa A, Ulrich M, Ellerbrok H, Goffe AS, Blasse A, Zommers Z, Couacy-Hymann E, Babweteera F, Zuberbühler K, Metzger S, Geidel S, Boesch C, Gillespie TR, Leendertz FH: Wild chimpanzees infected with 5 Plasmodium species. Emerg Infect Dis. 2010, 16: 1956-1959. 10.3201/eid1612.100424.
Sundararaman SA, Liu W, Keele BF, Learn GH, Bittinger K, Mouacha F, Ahuka-Mundeke S, Manske M, Sherrill-Mix S, Li Y, Malenke JA, Delaporte E, Laurent C, Mpoudi Ngole E, Kwiatkowski DP, Shaw GM, Rayner JC, Peeters M, Sharp PM, Bushman FD, Hahn BH: Plasmodium falciparum-like parasites infecting wild apes in southern Cameroon do not represent a recurrent source of human malaria. Proc Natl Acad Sci USA. 2013, 110: 7020-7025. 10.1073/pnas.1305201110.
Jeffares DC, Pain A, Berry A, Cox AV, Stalker J, Ingle CE, Thomas A, Quail MA, Siebenthall K, Uhlemann AC, Kyes S, Krishna S, Newbold C, Dermitzakis ET, Berriman M: Genome variation and evolution of the malaria parasite Plasmodium falciparum. Nat Genet. 2007, 39: 120-125. 10.1038/ng1931.
Weedall GD, Polley SD, Conway DJ: Gene-specific signatures of elevated non-synonymous substitution rates correlate poorly across the Plasmodium genus. PLoS One. 2008, 3: e2281-10.1371/journal.pone.0002281.
Ollomo B, Durand P, Prugnolle F, Douzery E, Arnathau C, Nkoghe D, Leroy E, Renaud F: A new malaria agent in African hominids. PLoS Pathog. 2009, 5: e1000446-10.1371/journal.ppat.1000446.
Rayner JC, Liu W, Peeters M, Sharp PM, Hahn BH: A plethora of Plasmodium species in wild apes: a source of human infection?. Trends Parasitol. 2011, 27: 222-229. 10.1016/j.pt.2011.01.006.
Escalante AA, Ayala FJ: Malaria: host range, diversity and speciation. Evolution of virulence in eukaryotic microbes. Edited by: Sibley LD, Howlett BJ, Heitman J. 2012, New Jersey: Wiley-Blackwell, 93-110.
Escalante AA, Lal AA, Ayala FJ: Genetic polymorphism and natural selection in the malaria parasite Plasmodium falciparum. Genetics. 1998, 149: 189-202.
Pacheco MA, Battistuzzi FU, Junge RE, Cornejo OE, Williams CV, Landau I, Rabetafika L, Snounou G, Jones-Engel L, Escalante AA: Timing the origin of human malarias: the lemur puzzle. BMC Evol Biol. 2011, 11: 299-10.1186/1471-2148-11-299.
Guindon S, Gascuel O: A simple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52: 696-704. 10.1080/10635150390235520.
Ronquist F, Huelsenbeck JP, MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19: 1572-1574. 10.1093/bioinformatics/btg180.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739. 10.1093/molbev/msr121.
Drummond AJ, Rambaut A: BEAST version 1.3 [computer program]. 2003, Available at http://beast.bio.ed.ac.uk/Main_Page
Poux C, Madsen O, Marquard E, Vieites DR, De Jong WW, Vences M: Asynchronous colonization of Madagascar by the four endemic clades of primates, tenrecs, carnivores, and rodents as inferred from nuclear genes. Syst Biol. 2005, 54: 719-730. 10.1080/10635150500234534.
Ali JR, Huber M: Mammalian biodiversity on Madagascar controlled by ocean currents. Nature. 2010, 463: 653-656. 10.1038/nature08706.
Delson E: Fossil macaques, phyletic relationships and a scenario of deployment. The macaques—studies in ecology, behavior and evolution. Edited by: Lindburg DG. 1980, New York: Van Nostrand Reinhold Co, 10-29.
Mu J, Joy DA, Duan J, Huang Y, Carlton J, Walker J, Barnwell J, Beerli P, Charleston MA, Pybus OG, Su XZ: Host switch leads to emergence of Plasmodium vivax malaria in humans. Mol Biol Evol. 2005, 22: 1686-1693. 10.1093/molbev/msi160.
Hayakawa T, Culleton R, Otani H, Horii T, Tanabe K: Big bang in the evolution of extant malaria parasites. Mol Biol Evol. 2008, 25: 2233-2239. 10.1093/molbev/msn171.
Benton MJ, Donoghue PC: Paleontological evidence to date the tree of life. Mol Biol Evol. 2007, 24: 26-53.
Djimde A, Doumbo OK, Steketee RW, Plowe CV: Application of a molecular marker for surveillance of chloroquine-resistant falciparum malaria. Lancet. 2001, 358: 890-891. 10.1016/S0140-6736(01)06040-8.
Rathore D, Nagarkatti R, Jani D, Chattopadhyay R, de la Vega P, Kumar S, McCutchan TF: An immunologically cryptic epitope of Plasmodium falciparum circumsporozoite protein facilitates liver cell recognition and induces protective antibodies that block liver cell invasion. J Biol Chem. 2005, 280: 20524-20529. 10.1074/jbc.M414254200.
Coppi A, Pinzon-Ortiz C, Hutter C, Sinnis P: The Plasmodium circunsporozoite protein is proteolytically processed during cell invasion. J Exp Med. 2005, 201: 27-33. 10.1084/jem.20040989.
Reece WH, Pinder M, Gothard PK, Milligan P, Bojang K, Doherty T, Plebanski M, Akinwunmi P, Everaere S, Watkins KR, Voss G, Tornieporth N, Alloueche A, Greenwood BM, Kester KE, McAdam KP, Cohen J, Hill AV: A CD4(+) T-cell immune response to a conserved epitope in the circumsporozoite protein correlates with protection from natural Plasmodium falciparum infection and disease. Nat Med. 2004, 10: 406-410. 10.1038/nm1009.
Felger I, Tavul L, Kabintik S, Marshall V, Genton B, Alpers M, Beck HP: Plasmodium falciparum: extensive polymorphism in merozoite surface antigen 2 alleles in an area with endemic malaria in Papua New Guinea. Exp Parasitol. 1994, 79: 106-116. 10.1006/expr.1994.1070.
Adda CG, MacRaild CA, Reiling L, Wycherley K, Boyle MJ, Kienzle V, Masendycz P, Foley M, Beeson JG, Norton RS, Anders RF: Antigenic characterization of an intrinsically unstructured protein, Plasmodium falciparum merozoite surface protein 2. Infect Immun. 2012, 80: 4177-4185. 10.1128/IAI.00665-12.
Ferreira MU, Hartl DL: Plasmodium falciparum: worldwide sequence diversity and evolution of the malaria vaccine candidate merozoite surface protein-2 (MSP-2). Exp Parasitol. 2007, 115: 32-40. 10.1016/j.exppara.2006.05.003.
Hughes AL: Positive selection and interallelic recombination at the merozoite surface antigen-1 (MSA-1) locus of Plasmodium falciparum. Mol Biol Evol. 1992, 9: 381-393.
Rich SM, Ayala FJ: Population structure and recent evolution of Plasmodium falciparum. Proc Natl Acad Sci USA. 2000, 97: 6994-7001. 10.1073/pnas.97.13.6994.
Viebig NK, Gamain B, Scheidig C, Lépolard C, Przyborski J, Lanzer M, Gysin J, Scherf A: A single member of the Plasmodium falciparum var multigene family determines cytoadhesion to the placental receptor chondroitin sulphate A. EMBO Rep. 2005, 6: 775-781. 10.1038/sj.embor.7400466.
Duffy MF, Byrne TJ, Elliott SR, Wilson DW, Rogerson SJ, Beeson JG, Noviyanti R, Brown GV: Broad analysis reveals a consistent pattern of var gene transcription in Plasmodium falciparum repeatedly selected for a defined adhesion phenotype. Mol Microbiol. 2005, 56: 774-788. 10.1111/j.1365-2958.2005.04577.x.
Fried M, Duffy PE: Adherence of Plasmodium falciparum to chondroitin sulfate A in the human placenta. Science. 1996, 272: 1502-15044. 10.1126/science.272.5267.1502.
Bockhorst J, Lu F, Janes JH, Keebler J, Gamain B, Awadalla P, Su XZ, Samudrala R, Jojic N, Smith JD: Structural polymorphism and diversifying selection on the pregnancy malaria vaccine candidate VAR2CSA. Mol Biochem Parasitol. 2007, 155: 103-112. 10.1016/j.molbiopara.2007.06.007.
Rask TS, Hansen DA, Theander TG, Gorm Pedersen A, Lavstsen T: Plasmodium falciparum erythrocyte membrane protein 1 diversity in seven genomes–divide and conquer. PLoS Comput Biol. 2010, 6: e1000933-10.1371/journal.pcbi.1000933.
Nei M, Gojobori T: Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986, 3: 418-426.
Nei M, Kumar S: Molecular evolution and phylogenetics. 2000, New York: Oxford University press
Garnham PCC: Malaria parasites and other haemosporidia. 1966, Oxford: Blackwell Scientific Publications
Ramiro RS, Reece SE, Obbard DJ: Molecular evolution and phylogenetics of rodent malaria parasites. BMC Evol Biol. 2012, 12: 219-10.1186/1471-2148-12-219.
Drummond AJ, Ho SY, Phillips MJ, Rambaut A: Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4: e88-10.1371/journal.pbio.0040088.
Hasegawa M, Kishino H, Yano T: Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985, 22: 160-174. 10.1007/BF02101694.
Arnason U, Gullberg A, Burguete AS, Janke A: Molecular estimates of primate divergences and new hypotheses for primate dispersal and the origin of modern humans. Hereditas. 2000, 133: 217-228.
Joy DA, Feng X, Mu J, Furuya T, Chotivanich K, Krettli AU, Ho M, Wang A, White NJ, Suh E: Early origin and recent expansion of Plasmodium falciparum. Science. 2003, 300: 318-321. 10.1126/science.1081449.
Costa FT, Snounou G, Letourneur F, Lebrun N, Landau I, Rénia L: The primary structure of the circumsporozoite protein of Plasmodium atheruri, a malaria parasite of the African porcupine Atherurus africanus. Mol Biochem Parasitol. 2001, 114: 125-127. 10.1016/S0166-6851(01)00248-1.
Rathore D, Hrstka SC, Sacci JB, De la Vega P, Linhardt RJ, Kumar S, McCutchan TF: Molecular mechanism of host specificity in Plasmodium falciparum infection: role of circumsporozoite protein. J Biol Chem. 2003, 278: 40905-40910. 10.1074/jbc.M306250200.
Patil A, Orjuela-Sánchez P, Da Silva-Nunes M, Ferreira MU: Evolutionary dynamics of the immunodominant repeats of the Plasmodium vivax malaria-vaccine candidate circumsporozoite protein (CSP). Infect Genet Evol. 2010, 10: 298-303. 10.1016/j.meegid.2010.01.006.
Galinski MR, Arnot DE, Cochrane AH, Barnwell JW, Nussenzweig RS, Enea V: The circumsporozoite gene of the Plasmodium cynomolgi complex. Cell. 1987, 48: 311-319. 10.1016/0092-8674(87)90434-X.
Escalante AA, Grebert HM, Isea R, Goldman IF, Basco L, Magris M, Biswas S, Kariuki S, Lal AA: A study of genetic diversity in the gene encoding the circumsporozoite protein (CSP) of Plasmodium falciparum from different transmission areas—XVI: asembo bay cohort project. Mol Biochem Parasitol. 2002, 125: 83-90. 10.1016/S0166-6851(02)00216-5.
Ancsin JB, Kisilevsky R: A binding site for highly sulfated heparan sulfate is identified in the N terminus of the circumsporozoite protein: significance for malarial sporozoite attachment to hepatocytes. J Biol Chem. 2004, 279: 21824-21832. 10.1074/jbc.M401979200.
Doud MB, Koksal AC, Mi LZ, Song G, Lu C, Springer TA: Unexpected fold in the circumsporozoite protein target of malaria vaccines. Proc Natl Acad Sci USA. 2012, 109: 7817-7822. 10.1073/pnas.1205737109.
Petersen TN, Brunak S, Von Heijne G, Nielsen H: SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011, 8: 785-786. 10.1038/nmeth.1701.
Coppi A, Natarajan R, Pradel G, Bennett BL, James ER, Roggero MA, Corradin G, Persson C, Tewari R, Sinnis P: The malaria circumsporozoite protein has two functional domains, each with distinct roles as sporozoites journey from mosquito to mammalian host. J Exp Med. 2011, 208: 341-356. 10.1084/jem.20101488.
McCutchan TF, Kissinger JC, Touray MG, Rogers MJ, Li J, Sullivan M, Braga EM, Krettli AU, Miller LH: Comparison of circumsporozoite proteins from avian and mammalian malarias: biological and phylogenetic implications. Proc Natl Acad Sci USA. 1996, 93: 11889-11894. 10.1073/pnas.93.21.11889.
Carlton JM, Das A, Escalante AA: Genomics, population genetics and evolutionary history of Plasmodium vivax. Adv Parasitol. 2013, 81: 203-222.
Fenton B, Clark JT, Khan CM, Robinson JV, Walliker D, Ridley R, Scaife JG, McBride JS: Structural and antigenic polymorphism of the 35-to 48-kilodalton merozoite surface antigen (MSA-2) of the malaria parasite Plasmodium falciparum. Mol Cell Biol. 1991, 11: 963-971.
Dubbeld MA, Kocken CH, Thomas AW: Merozoite surface protein 2 of Plasmodium reichenowi is a unique mosaic of Plasmodium falciparum allelic forms and species-specific elements. Mol Biochem Parasitol. 1998, 92: 187-192. 10.1016/S0166-6851(97)00244-2.
Valkiunas G, Ashford RW, Bensch S, Killick-Kendrick R, Perkins S: A cautionary note concerning Plasmodium in apes. Trends Parasitol. 2011, 27: 231-232. 10.1016/j.pt.2011.02.008.
De Queiroz K: Species concepts and species delimitation. Syst Biol. 2007, 56: 879-886. 10.1080/10635150701701083.
Bray RS: Studies on malaria in chimpanzees: VI laverania falciparum. Am J Trop Med Hyg. 1958, 7: 20-24.
Obare P, Ogutu B, Adams M, Odera JS, Lilley K, Dosoo D, Adhiambo C, Owusu-Agyei S, Binka F, Wanja E, Johnson J: Misclassification of Plasmodium infections by conventional microscopy and the impact of remedial training on the proficiency of laboratory technicians in species identification. Malar J. 2013, 12: 113-10.1186/1475-2875-12-113.
Beadell JS, Fleischer RC: A restriction enzyme–based assay to distinguish between avian hemosporidians. J Parasitol. 2005, 91: 683-685. 10.1645/GE-3412RN.
Mantilla Granados JS, Matta NE, Pacheco MA, Escalante A, Gonzalez AD, Moncada LI: Identification of Plasmodium (Haemamoeba) lutzi (Lucena, 1939) from Turdus fuscater (great thrush) in Colombia. J Parasitol. 2013, 99: 662-668. 10.1645/12-138.1.
Sutherland CJ, Tanomsing N, Nolder D, Oguike M, Jennison C, Pukrittayakamee S, Dolecek C, Hien TT, Do Rosário VE, Arez AP, Pinto J, Michon P, Escalante AA, Nosten F, Burke M, Lee R, Blaze M, Otto TD, Barnwell JW, Pain A, Williams J, White NJ, Day NP, Snounou G, Lockhart PJ, Chiodini PL, Imwong M, Polley SD: Two nonrecombining sympatric forms of the human malaria parasite Plasmodium ovale occur globally. J Infect Dis. 2010, 201: 1544-1550. 10.1086/652240.
Martin MJ, Rayner JC, Gagneux P, Barnwell JW, Varki A: Evolution of human-chimpanzee differences in malaria susceptibility: relationship to human genetic loss of N-glycolylneuraminic acid. Proc Natl Acad Sci USA. 2005, 102: 12819-12824. 10.1073/pnas.0503819102.
Zilversmit MM, Volkman SK, DePristo MA, Wirth DF, Awadalla P, Hartl DL: Low-complexity regions in Plasmodium falciparum: missing links in the evolution of an extreme genome. Mol Biol Evol. 2010, 27: 2198-2209. 10.1093/molbev/msq108.
Genton B, Betuela I, Felger I, Al-Yaman F, Anders RF, Saul A, Rare L, Baisor M, Lorry K, Brown GV, Pye D, Irving DO, Smith TA, Beck HP, Alpers MP: A recombinant blood-stage malaria vaccine reduces Plasmodium falciparum density and exerts selective pressure on parasite populations in a phase 1-2b trial in Papua New Guinea. J Infect Dis. 2002, 185: 820-827. 10.1086/339342.
Acknowledgements
This research was supported by a grant from the US National Institutes of Health, R01GM080586 to AAE. We thank the Jane Goodall Institute and the Tchimpounga Chimpanzee Rehabilitation Center in the Republic of Congo for providing access to their archived samples. The authors like to express their gratitude to William E Collins (“Bill”), who has recently retired from the Centers for Disease Control and Prevention after serving 59 years in the US Federal Government. His passion for non-human primate malarias inspired our early investigations and the name of P. billcollinsi given to one of the Laverania species. We thank Benjamin Rice for valuable comments and the DNA laboratory at the School of Life Sciences, ASU for their technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MP conducted the molecular genetic studies and analyzed the data. MC and KC provided the samples and contributed to the editing of the manuscript. AE supervised and directed the research and contributed to the writing and editing of the manuscript. All authors contributed in the project design. All authors read and approved the final manuscript.
Electronic supplementary material
12936_2013_2930_MOESM4_ESM.xlsx
Additional file 4: A complete list of Plasmodium species included in all the analyses (dhfr, csp, msp2 and var 2CSA). (XLSX 24 KB)
12936_2013_2930_MOESM5_ESM.pdf
Additional file 5: Beast node numbers for the Plasmodium phylogeny as used in Table 2. All calibration points used are shown. (PDF 74 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Pacheco, M.A., Cranfield, M., Cameron, K. et al. Malarial parasite diversity in chimpanzees: the value of comparative approaches to ascertain the evolution of Plasmodium falciparum antigens. Malar J 12, 328 (2013). https://doi.org/10.1186/1475-2875-12-328
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-12-328