
Abstract
Background
Regardless of the growing interest in detecting population structures in malarial parasites, there have been limited discussions on how to use this concept in control programmes. In such context, the effects of the parasite population structures will depend on interventions’ spatial or temporal scales. This investigation explores the problem of identifying genetic markers, in this case microsatellites, to unveil Plasmodium genetic structures that could affect decisions in the context of elimination. The study was performed in a low-transmission area, which offers a good proxy to better understand problems associated with surveillance at the final stages of malaria elimination.
Methods
Plasmodium vivax samples collected in Tumeremo, Venezuela, between March 2003 and November 2004 were analysed. Since Plasmodium falciparum also circulates in many low endemic areas, P. falciparum samples from the same locality and time period were included for comparison. Plasmodium vivax samples were assayed for an original set of 25 microsatellites and P. falciparum samples were assayed for 12 microsatellites.
Results
Not all microsatellite loci assayed offered reliable local data. A complex temporal-cluster dynamics is found in both P. vivax and P. falciparum. Such dynamics affect the numbers and the type of microsatellites required for identifying individual parasites or parasite clusters when performing cross-sectional studies. The minimum number of microsatellites required to differentiate circulating P. vivax clusters differs from the minimum number of hyper-variable microsatellites required to distinguish individuals within these clusters. Regardless the extended number of microsatellites used in P. vivax, it was not possible to separate all individual infections.
Conclusions
Molecular surveillance has great potential; however, it requires preliminary local studies in order to properly interpret the emerging patterns in the context of elimination. Clonal expansions and clusters turnovers need to be taken into account when using molecular markers. Those affect the number and type of microsatellite markers, as well as, the expected genetic patterns in the context of operational investigations. By considering the local dynamics, elimination programmes could cost-effectively use molecular markers. However, population level studies need to consider the local limitations of a given set of loci in terms of providing epidemiologically relevant information.
Similar content being viewed by others

Background
There has been a growing interest in the population structure of malarial parasites [1–7]. Population structure (deviation from random mating) is a common phenomenon in nature; it is the result of several processes including inbreeding, epidemic population expansions, and geographic isolation (isolation by distance) [8, 9]. In most cases, identifying the population structure of a given infection agent relies on a blind approach that considers “sufficient” gene sampling and a “good” survey of the natural populations [10], where “good” and “sufficient” depend on the underlying question. However, evaluating the importance of genetic structures for control and elimination programmes requires understanding how they provide information with epidemiologic value. As examples, whereas sometimes the population structure is informative per se (e.g. gene flow or separating domestic from imported cases); in others it becomes a problem, e.g. clonal expansions may hamper the ability of separating a new infection from a recrudescence case in a drug efficacy study. Thus, population structures affect the interpretation of patterns emerging from molecular markers in contexts that are relevant for control and elimination programmes.
It is worth noting that genetic structures are detectable at the time scales determined by the loci mutation rates. Thus, whereas neutral single nucleotide polymorphisms (SNPs) are widely used for genetic analyses in Plasmodium falciparum[11–14] and are currently established in Plasmodium vivax[15], their rates of evolution may identify genetic structures that are not informative for elimination programmes in some contexts; e.g. SNPs may not describe the dynamics of haplotypes carrying drug-resistant associated mutations [4]. Those more programmatic applications require markers evolving at mutation rates that can identify events at relatively recent (epidemiologically relevant) time scales.
Microsatellites are useful markers under such circumstances, they allow detecting genetic structures at recent divergence times [16] and they are abundant in the P. falciparum and P. vivax genomes [17]. In addition, these loci are considered to be selectively neutral, unless they are located near genes under selection (linked), e.g. genes with mutations conferring drug resistance [14, 18–20]. These characteristics make them useful in large-scale population genetic studies (including genome scans looking for mutations under selection) and for answering operational relevant questions such as separating new infections from a recrudescent case [5, 21–24]. Even though this has been the case in P. falciparum, the production of hypnozoites (homologous or heterologous) in P. vivax and the high rate of multiple clone infections represent a more complex dynamics which may alter the results of a study. Moreover, regardless their high mutation rate, epidemiologic uses for microsatellite loci should consider the effect of the population structures due to demographic processes such as clonal expansions (clonal genetic structure) in areas with low transmission [2–4, 25–28]. Since the population genetic structures are usually unknown a priori, this investigation focuses on the importance of pilot studies.
Here, a P. vivax population was characterized in a single low transmission area using isolates collected in two consecutive years. Then, the genetic structure in P. vivax was ascertained by using 25 microsatellite loci. Although such extended number of loci is rarely used in operational research [1, 5], this panel allowed evaluating the variation per locus in the study area and their reproducibility in the laboratory. The data obtained was then used to determine the number and properties of loci required to discriminate different epidemiologic scenarios: identification of individual infections (applicable when trying to separate between recrudescence and new infections) and discrimination between clusters of isolates. In order to better understand the dynamics of genetic structures in P. vivax, a sympatric P. falciparum population sampled during the same time period was included for comparison. Most studies only consider one of the two species [1–4] limiting the comparisons that can be made in areas where both parasites are endemic. Since these two malarial parasite populations come from a low-transmission area, strong linkage disequilibrium was expected [1–4, 27, 28]. However, in the case of P. falciparum, the population underwent strong drug selection [28]. Thus, if the expansion of a resistant lineage determined the clonal structure [4, 28, 29] of P. falciparum, then it was expected a more stable cluster structure through time in this parasite [4] than the one likely to be observed in P. vivax where no drug resistance has been documented.
This investigation found that the number and type of microsatellite loci needed for operational research should be tailored given the objective. Indeed, the number of microsatellite loci required to differentiate circulating P. vivax clusters differs from the minimum number of hyper-variable microsatellites required to distinguish individuals within these clusters. It was also found that the capacity of separating individual infections with a manageable number of microsatellite loci is limited in low transmission areas as the one under investigation; hence, the use of these loci for separating new infections from recrudescent cases need to take into account such limitations. Both parasites follow similar patterns with alternation of clonal lineages at loci non-liked with drug resistant mutations. Thus, whereas drug resistant haplotypes could be fixed in P. falciparum[4, 28], cyclical clonal replacements are still taking place. Finally, the inclusion of a temporal sampling scheme in the pilot investigation allowed detecting changes in the genetic structure due to migration or other demographic processes. Overall, this study emphasizes the need to evaluate the local diversity of loci and demography by using a pilot study before designing a molecular epidemiologic investigation.
Methods
Study area and Plasmodium isolates
Two hundred fifteen blood samples with Plasmodium monoinfections (107 with P. vivax and 108 infected with P. falciparum) were used from a surveillance study in Tumeremo (Bolivar State), Venezuela, between March 2003 and November 2004. Any patient positive for malaria by microscopy was invited to provide a sample for further characterization of the parasite and then treated according to national guidelines. The study protocol was approved by the bioethics commission of the Instituto de Altos Estudios Dr. Arnoldo Gabaldon in Venezuela.
Malaria transmission in Venezuela has fluctuated since 2000, and peaked in 2004, 2005 and 2007. In 2004, the number of cases in the country rose to almost 42,000, primarily as a result of an increase in P. vivax malaria in Bolivar State. The endemic nature of malaria in the State of Bolivar is determined by migration related to gold mining, particularly on the country’s border with Guyana. Bolivar state has the greatest number of reported cases of malaria (85% of the nationally reported cases), mainly among miners, agricultural workers and indigenous groups [28]. Plasmodium vivax is the predominant malaria parasite species in the country followed by P. falciparum and Plasmodium malariae. Anopheles darlingi and Anopheles marajoara have been the main vectors identified in the study area [30].
Microsatellite analysis
Genomic DNA was isolated from whole blood using the QIAamp DNA mini kit (QIAGEN, Valencia, CA). Plasmodium vivax samples were assayed for 25 microsatellites [31, 32] and P. falciparum samples were assayed for 12 microsatellites [33]. Fluorescently labelled PCR products were separated on an Applied Biosystems 3730 capillary sequencer and scored using GeneMarker v1.95 (SoftGenetics LLC). The finding of one or more additional alleles was interpreted as a co-infection with two or more genetically distinct clones in the same isolate (multiple-clone infection, transmitted by one or several mosquitoes) [33]. Additional alleles generating peaks of at least one third the height of the predominant allele were also scored. Missing data (no amplifications) are reported by loci but not considered for defining haplotypes.
Population genetic analysis
Microsatellite data was formatted using the Microsatellite tool kit [34]. The heterozygosity estimate (H E ) was used as a measure of overall genetic diversity. This was defined as , where n is the number of isolates analysed and pi is the frequency of the i-th allele (i=1, …, L) in the population. H E gives the average probability that a pair of alleles randomly selected from the population is different. The sampling variance for H E was calculated as [35]. To calculate allele frequencies per locus, samples with mixed infections were included. (If a sample contained, e.g., three microsatellite alleles, each was weight by 1/3 to calculate frequencies.) H E was calculated using Mathematica 8 (Wolfram Research, Inc.) (code available on request).
To test whether microsatellite haplotypes clustered as a single geographic population, the model-based clustering algorithm implemented in the Structure 2.1 software was applied [36]. This software uses a Bayesian clustering approach to assign isolates to K populations or clusters characterized by a set of allele frequencies at each locus. The number of such populations may be either previously known or unknown. In the context of this investigation, this approach allows for the identification of groups or populations of parasites that could be circulating in this area (Tumeremo, Venezuela). The observed genetic diversity was evaluated at different K values (K =2 to 10). Given that this clustering algorithm incorporates stochastic simulations, each K value was run independently ten times with a burn-in period of 10,000 iterations followed by 50,000 iterations. The admixture model was used in all analysis which allows for the presence of individuals with ancestry in two or more of the K populations [36]. Structure harvester v0.6.8 was used to visualize the output from Structure [37]. To facilitate the interpretation of population-genetic clustering results, CLUMPP (Cluster Matching and Permutation Program) was also used. CLUMPP strips away the ‘label switching’ heterogeneity so that the ‘genuine multimodality’ can be detected and quantified [38]. In addition, distruct 1.1 was used to graphically display the clustering results [39]. The posterior probability for each number of populations or clusters (K) is computed and then the K value that better explains the genetic data is an estimate of the number of circulating clusters or populations circulating. Whereas such genetic structures are a deviation from the expectations under one population undergoing random mating, each cluster cannot be considered a random mating population on its own since such clusters could represent clonal lineages. Moreover, Fstat 2.9.3.2 [40] was used to calculate Fst between clusters.
Evidence of linkage disequilibrium between alleles from different loci in parasite populations was analysed with Arlequin 3.11 [41]. A standardized index of association (ISA) was also used to test for evidence of overall multilocus linkage disequilibrium in the Venezuelan population. This test compares the variance (V D ) of the number of alleles shared between all pairs of haplotypes observed in the population (D) with the variance expected under random association of alleles (V E ) as follows: ISA = (V D / V E − 1) (r − 1), where r is the number of loci analysed. V E is derived from 10,000 simulated data sets in which alleles were randomly reshuffled among haplotypes. Significant linkage disequilibrium is detected if V D is greater than 95% of the values derived from the reshuffled data sets. Data were analysed with LIAN 3.1 [42]. Pairwise linkage disequilibria (LD) were also calculated. In particular, R [43] was calculated. For each pairwise comparison only those samples were included, in which data was not missing and only one allele occurred at one of the two loci. If multiple alleles for a locus were found in a sample, e.g., four alleles, the respective haplotypes were weighted by ¼to calculate haplotype frequencies. Allele frequencies were calculated as described above (p i ) from all included samples. Calculations were implemented in Mathematica 8 (Wolfram Research, Inc.) (code available on request).
Additionally, deviations from pairwise linkage-equilibrium were explored by using Freeman-Halton’s test [44] (generalized Fisher’s exact test for kxm contingency tables) at a significance level of 5%. Of particular note, contingency tables, resulting from pairwise comparison of microsatellite loci, typically have sparse entries, and in many cells the count is less than 5. This renders asymptotic tests (e.g. G-test) inappropriate. Since exact tests require integer-valued cell counts, for each pairwise of microsatellite loci, only samples that had exactly one microsatellite allele at both loci could be included in the analysis. (Therefore, for some pairwise comparisons, fewer samples were included than for calculating R.) As the number of tests performed and size of resulting contingency tables becomes numerically challenging, Monte-Carlo estimates for exact p-values were calculated using 10,000 random iterations. However, for 2x2 tables Fisher’s exact test was always performed. Computations were performed with SAS® (Version 9.2).
Statistical analysis
To test the hypothesis that the probability of sampling parasites from different clusters differs over time, the Kruskal-Wallis test was performed with the sampling date as the dependent variable and the clusters as the independent variable. The null hypothesis was that the probability of sampling from each cluster was the same during the two years. To test if the chronological sequence of clusters would not appear randomly, the asymptotic test of Barton and David [45] was performed (“generalization” of the Wald-Wolfowitz runs test). In addition, associations of time and cluster-membership were further explored. For this purpose, cluster-membership was arranged as a nominal variable, and sample date - grouped in monthly ranges - as an ordinal variable in a k x l contingency table. The χ2 and likelihood-χ2 tests were used. The Pearson’s Ø-coefficient, Pearson’s contingency coefficient (Cp), and Cramer’s V were also obtained. All statistical tests were performed using SAS® (Version 9.2).
Results
Twelve P. falciparum imperfect microsatellites reported by Anderson et al.[33], were tested for their ability to amplify in this population and to demonstrate polymorphism on a set of Venezuelan field isolates from 2003–2004 (Table 1). Suitable markers were considered as those loci that amplified at least 80% of the samples and showed evidence of polymorphism with a clear peak pattern. Microsatellites TA40 and TA87 did not meet the criteria and were not considered in the analysis, but loci TA1, Polyα, TA60, ARA2, Pfg377, PfPK2, TA109, TA81, TA42 and 2490 were included for the population genetic analysis. These markers are distributed across seven chromosomes and no two loci are physically closely linked. Markers TA1 and TA109 are 14.4 cM apart on chromosome 6; markers Pfg377 and PfPk2 are on chromosome 12 and separated by a distance of 45.8 cM; and markers TA81 and TA42 are 68.9 cM apart on chromosome 5. All loci had repeat units of 3bp or more.
For P. vivax, a panel of 11 dinucleotide [31] and 14 markers of 3bp or more [32] was tested. A total of 22 microsatellites that consistently amplified the samples and showed polymorphism were selected (Table 2). These markers were distributed in 13 of the 14 chromosomes and linkage disequilibrium has been previously reported between microsatellites MS4-MS5 in chromosome 6 and MS7-MS8 in chromosome 12 [46].
Multiple infections
In P. falciparum, 2.8% of the 108 samples had multiple infections while in P. vivax 15.9% out of 107 samples were also found with multiple infections harbouring two or three alleles at two or more loci. When grouping samples according to their date of collection, a greater proportion of mixed clone infections were found in P. vivax samples from 2004 (at the peak of malaria transmission in Venezuela), with more than double the amount of mixed infections found in P. vivax samples from 2003.
Genetic diversity
The frequency distribution of alleles at each locus was determined using all samples; however, three samples each were excluded in P. falciparum and P. vivax since they only amplified two or none loci. Most of the P. vivax loci but MS3, MS7 and MS10 followed (almost) a normal distribution (histogram of microsatellite repeats), which indicates an appropriate sampling of the alleles present in Tumeremo. However, many of the P. vivax markers showed a considerable number of (rare) alleles (frequency lower than 5% further referred as Minor Allele Frequency or MAF, see Figure 1 A and B). In P. falciparum, the same trend was not observed since only a small number of alleles per loci were found suggesting low genotypic diversity due to recent strong selection by drug pressure and low recombination [28]. Furthermore, note that P. vivax relapses from hypnozoites occur, which increases the potential to harbor higher genetic diversity as compared with P. falciparum if patients do not comply with the primaquine treatment and as a result it hampers the ability to differentiate between reinfection and recrudesce cases. The expected heterozygosity (H E ) was calculated in both species using all microsatellites (Figure 1 C and D). P.vivax was found to be highly polymorphic (H E = 0.73 ± 0.032) in comparison to P. falciparum (H E = 0.44 ± 0.035). Moreover, the year by year analysis (Additional file 1) revealed higher levels of genetic diversity in samples from 2004 (H E = 0.79 ± 0.0221) than from 2003 (H E = 0.67 ± 0.0301). In P. falciparum, the level of genetic diversity in 2003 (H E = 0.51 ± 0.0378) was higher than in 2004 (H E = 0.39 ± 0.0641) and all mixed samples were found during 2003.

A) Number of alleles per loci and B) percentage of alleles in low frequency (<5%) in P. vivax. Expected heterozygosity (H E ) in C) P. falciparum and in D) P. vivax using all samples from Tumeremo.
Population structure
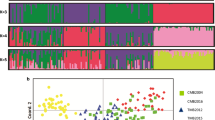
The population structures of P. falciparum and P. vivax were determined using the Structure 2.1 software. Single infections and mixed infections with only one polymorphic locus were included in the analysis with complete data or missing data in no more than three loci. The posterior probability for each cluster (K) was computed and the clustering patterns obtained with K = 4 was associated with the highest Delta K values (Delta K = mean (|L”(K)|)/sd(L(K))) for both species (Figure 2) [37]. Most parasites had their origin clearly assigned to a single cluster; few of them were fairly admixed, showed by bars partitioned into K coloured segments. Each fragment in the bar represents the estimated membership fractions, of each isolate or strain, in K clusters. However, the algorithm implemented in Structure might detect K-1 reasonable clusters, and subsume remaining low frequency variants in the Kth cluster. This might be indicated by higher genetic diversity in just one cluster. In each of the four P. falciparum clusters, similar numbers of samples were found (Table 3a); however, in P. vivax most of the samples were grouped either in the clusters labeled as “A” or “D” (Table 3b). In addition, when grouping Plasmodium haplotypes by year, P. falciparum samples from either 2003 or 2004 were also grouped in four clusters. Still, in 2003 haplotypes from falciparum cluster C were overrepresented while in 2004 haplotypes from falciparum cluster A were more abundant. Clusters B and D were present in almost the same frequency during both years. On the other hand, P. vivax samples from 2003 did group in four clusters whereas samples from 2004 were grouped in only three clusters; nevertheless, these results might be biased due to the lower number of 2004 samples included in the analysis. Due to the high prevalence of multi-clonal infections (13 out of 41) in the 2004 P. vivax samples it was difficult to determine the total number of haplotypes present in the sample so an estimate using the single infections was considered. From this description, it seems that the probability of sampling parasites from different clusters differs over time. The Kruskal-Wallis test was performed and showed to be significant for P. falciparum and P. vivax (p-values <0.01), indicating that the probabilities of sampling a parasite belonging to a given cluster changes over time. This is an indication of clonal expansions.

Population structure of A) P. falciparum (cluster A – pink, cluster B – yellow, cluster C – blue and cluster D – orange) and B) P. vivax (cluster A – orange, cluster B – yellow, cluster C – blue and cluster D – pink) inferred from microsatellite typing of Tumeremo haplotypes from 2003 and 2004 using the STRUCTURE program.
If parasite prevalence is due to events of clonal expansion, different clusters would be predominant at different time periods. Hence, the chronological sequence of clusters would not appear randomly. Since some samples were taken on the same day, the chronological order on the sample labels was followed. The Barton-David test was significant (p<10-11 for P. falciparum, p<10-7 for P. vivax) indicating that cluster-membership is not random over time, which is another indication for clonal expansion.
Furthermore, associations of time and cluster-membership were explored. The X2 and likelihood-X2-tests were both significant on an alpha level of 5% (all p-values <0.01). For P. vivax, ϕ = 0.1790, × C p = 0.5838, C p = 0.5838, and V = 0.4151, were obtained, indicating an association between time and cluster-membership. Likewise, for P. falciparum, ϕ = 0.8043, C p = 0.6267; and V = 0.4643 were obtained. However, it should be mentioned that the 85% of the cells in the contingency had expected counts less than 5, which renders the application of X2 tests and X2-based association measures problematic. It should be mentioned that. The sample size was too large to perform Fisher’s exact test, or rather the generalization of Freeman and Halton [44]. The statistics indicate that there is some association between time and cluster membership; however, this association is not very strong. Notably, the association appears stronger in P. falciparum than in P. vivax, which can be explained by the hypothesis of clonal expansion due to drug resistance. It may be possible that due to relapses in P. vivax the pattern of clonal expansion is distorted; however, data on the follow-up primaquine treatments is not available.
By using only six (Polya, TA60, ARA2, Pfg377, TA81 and 2490) of the 10 loci is enough to differentiate the four clusters with a 98% confidence interval. In addition, at least eight loci (TA1, Polya, TA60, Pfg377, PfPK2, TA109, TA42 and 2490) are needed to correctly identify the 41 unique haplotypes found in the 104 P. falciparum isolates determined with all ten loci. Thus, the number of loci used does not allow discriminating all individuals.
Plasmodium vivax samples were also reanalysed in order to obtain the four clusters and the different identifiable haplotypes from the 94 P. vivax samples (100 clones in total within single infections or mixed infections in one locus only). The set of loci used to identify P. vivax clusters (four representative loci from the population, which harbours average values of H E and percentage of alleles in low frequency, and two highly polymorphic loci to differentiate maximum number of individuals within cluster) were: locus 2.21, locus 12.335, locus MS1, locus MS6, locus MS8, and locus MS16, whereas the set of loci needed to differentiate the maximum number of haplotypes were: locus 6.34, locus 7.67, locus 8.332, locus MS2, locus MS8, locus MS9, locus MS10, locus MS15, locus MS16 and locus MS20. The Fst values between clusters in P. falciparum and P. vivax populations were also calculated to confirm if there was strong genetic differentiation within these subpopulations. The values were significant and ranged between 0.4 to 0.9 in P. falciparum and 0.3 to 0.8 in P. vivax. These results confirm that there is a strong genetic differentiation within these subpopulations (Table 4).
Linkage disequilibrium
In order to calculate linkage disequilibrium, the data was organized to follow the stepwise mutation model when possible. Significant multilocus linkage disequilibrium was found in P. falciparum and P. vivax isolates according to the standard index of association (ISA2003 = 0.1236, p<0.01; ISA2004 = 0.172, p<0.01 and ISA2003 = 0.3776, p<0.01; ISA2004 = 0.1448, p<0.01, respectively) and to pairwise comparisons using Arlequin 3.11. Linkage disequilibrium was also calculated considering the time of collection of the isolates, with unique haplotypes and excluding microsatellites in the same chromosome. In all analyses performed significant linkage disequilibrium was found. Further analysis to calculate R in P. falciparum (Figures 3, A and B) and P. vivax (Figure 4, A and B) conducted between pairs of microsatellites and using all information, including mixed infections, also revealed linkage disequilibrium in both species. Additionally, we tested for pairwise linkage disequilibrium using Freeman-Halton’s (“Fisher’s exact”) test (Figures 3, C and D and Figure 4, C and D). To rule out the possibility that association resulted from combining highly different subpopulations in P. vivax, linkage disequilibrium was also calculated using pairwise analysis within the genetic clusters present in both years and a significant result was found for cluster D (ISA = 0.1136, p-value <0.001). Furthermore, significant LD (ISA = 0.172, p-value <0.05) was also found in P. vivax cluster B, which was only present in 2003.

R values of linkage disequilibrium in P. falciparum samples from A) 2003 and B) 2004 and the respective outcomes of Freeman-Halton’s test for significant deviations from linkage equilibrium (C and D) at a significance level of 0.05.

R values of linkage disequilibrium in P. vivax samples from A) 2003 and B) 2004 the respective outcomes of Freeman-Halton’s test for significant deviations from linkage equilibrium (C and D) at a significance level of 0.05.
Discussion
Low-transmission areas are of great interest because they are potential targets for malaria elimination. Even further, they allow for testing how to deploy resources effectively at the final stages of the elimination process. Under such conditions, the precise monitoring of malaria infections is indispensable. Whereas molecular marker-based approaches could enrich malaria surveillance [5], it needs to be ascertained whether such methods - besides their general popularity - are cost-effective for detecting substructures in parasite populations that are valuable for elimination programmes, especially in these low-transmission settings. One step in the process of translating population-genetic concepts into epidemiology is to determine whether an available set of markers has the sensitivity to detect patterns that are epidemiologically informative, especially when performing cross-sectional studies in a narrow period of time that simply allow to see a predominant clonal expansion. Whereas, hypothetically, the number of markers could be increased up to levels that can rule out many confounding factors; in reality, malaria elimination programmes face limited funds, samples, and human resources that can be invested in molecular surveillance.
In general, highly polymorphic loci are suitable to identify individuals within populations whereas more conserved (less polymorphic) loci are useful to establish clusters or parasite populations within a given endemic area. For the first purpose, imperfect microsatellites would be more appropriate due to the complexity on their core sequence which results in greater allelic diversity, while dinucleotides and trinucleotides are more conserved and best represent the local population diversity. However, there are always exceptions in each of the categories established. Distinctions between loci have practical consequences. Highly polymorphic markers have many alleles in low frequency that are more likely to fluctuate by random events (e.g. random genetic drift but also inadequate sampling); thus, a local genotype could not be detected at a given time and then “appear” as new (e.g. been identified as an introduction) after deploying control interventions. Those cases could be considered “imported” rather than the result of residual transmission, misleading the programme to conclude that the control strategy was more efficacious than in reality. This problem is particularly important if the only baseline information is provided by a cross-sectional study that simply detected a predominant clonal expansion. On the contrary, the number of treatment “failures” (putative recrudescent cases) could be over-estimated and conclude that a drug treatment has reduced its efficacy by using low polymorphic marker that cannot properly differentiate genotypes within a cluster. Thus, in order to get a fine scale fingerprint of the epidemiologically relevant events targeted by a given molecular surveillance programme, the number and kind of microsatellites used should consider the characteristics of the population under study. Those characteristics include the replacement of clusters at least between two transmission seasons.
This investigation provides some basic information about how to design a pilot (baseline) investigation that could support prospective molecular population-based studies, especially in areas with low genetic diversity and low transmission as the one found in Tumeremo, Venezuela. Those steps are summarized in a flow chart (Figure 5). First, an extended set of loci should be evaluated in order to determine how many reliable work (e.g. can be consistently amplified or the pattern in the electropherogram is unambiguously interpretable) in that area. Then, based on that set of loci, the number of mixed infections, haplotypes and clusters circulating in a given geographic area during at least two transmission seasons can be evaluated. The assumption is that the information collected from the original set of loci (in this case, 22 loci in P. vivax) provides the best possible resolution given the available resources. Based on the identified structures using the extended set of loci, it is possible to estimate the minimum number of microsatellite loci needed to detect such structure and differentiate Plasmodium populations (clusters) in that specific geographic area. The same baseline data can be used to estimate the minimum number of microsatellites needed to differentiate individuals within these clusters. Hypothetically, since each of the samples analysed originated from a distinct individual, many confounding effects could be reduced by simply increasing the number of markers. However, the number of loci that can be used in a given study is finite, simply because they are tied to the available resources. Thus, pilot studies are needed in order to determine the best resolution that can be obtained using molecular markers in a given context.

Flow chart illustrating the steps to follow in a Plasmodium population genetic study. The goal is to identify appropriate microsatellite markers for haplotypic determination (to differentiate between reinfection and recrudescence cases) and to determine clusters or subpopulations (to evaluate clonal expansions and possible imported cases).
Considering all data analysed for P. falciparum and P. vivax, only 45% of the P. falciparum and 85% of the P. vivax samples at the individual level were correctly identified. In order to increase the percentage of individual identification for P. falciparum in a place like Tumeremo that underwent a strong drug selective pressure, a greater number of polymorphic loci are needed since the heterozygosity per locus is much lower compared with P. vivax. This represents a problem since the analysis of more than 10 loci per sample could be costly. It is also important to emphasize that there are clusters of individuals infected by highly related parasites (clonal expansions) and those simply cannot be easily separated. Thus, a manageable number of microsatellites will only identify haplotypes or clonal lineages per population, some of them stable through time [4, 47, 48]. Population-based studies should be designed considering such restrictions by targeting groups (e.g. arms in a drug efficacy study) or geographic areas where the turnover of such clones (or their persistence) could be informative at the population level. In the case of drug efficacy studies, the proportions of undifferentiated genotypes from prior and post treatment samples can be compared between groups providing a way to reduce the confounding effect of the local parasite demography. Unfortunately, such an approach may require high sample sizes.
Plasmodium vivax and P. falciparum circulating in Tumeremo differ according to the prevalence of mixed clone infections and the overall levels of genetic diversity. Since in P. falciparum drug resistant haplotypes are fixed [4, 28], it was expected that such early drug pressure will result in few clones present year-round [4]. However, both species exhibited significant linkage disequilibrium (LD), similar number of clusters/clones when considering haplotypes only, and a similar pattern of temporal replacement of clusters. In P. falciparum the four clusters found in 2003 were also found the following year. In P. vivax, however, three of the four clusters persisted through time. This could indicate a sporadic event in 2003 of a parasite lineage (cluster B) coming into the Tumeremo population from one of the shared borders with Guyana and Brazil. Moreover, significant LD was found in P. vivax within cluster B, which strengthens the idea of a group of minor frequency lineages that are not stable in time and that could be the result of imported cases. Detecting introductions is important especially in the context of control and elimination of the disease; those migration events could explain a local increase in incidence indicating that the deployed strategies/policies were effective in controlling the previously circulating parasites. Ideally, data from other populations should allow identifying the source of the migrant cases providing additional information about how malaria is maintained at a regional level.
A higher level of LD in P. vivax compared to P. falciparum was not expected since this could not be accounted by recent epidemic expansions of drug resistant lineages like in P. falciparum[4, 28]. Thus, the pattern observed in P. vivax is indicative of ongoing reduced levels of recombination due to the parasite demography. Overall, the local ecology appears to explain the turnover of clones/clusters in both parasites at this time scale. The temporal interval used in this study is appropriate in the context of surveillance when monitoring malaria prevalence from one malaria season to the next one. Plus, the effectiveness of local malaria control can be evaluated by observing changes (which could be the result of incoming gene flow) in the parasite population structure.
It is worth noting that in P. vivax, also cluster D had a significant level of LD among lineages within that cluster. When explored closely, this cluster D included different lineages found in low frequency that are stable through time. This cluster D then contrasts with cluster B; where the first seems to be stable in the population and composed by several minor haplotypes, the later (B) may indicate migration/introduction of haplotypes in lower frequency into the endemic area. Thus, a close observation of the linkage disequilibrium within clusters provides additional information that could explain the observed patterns of malaria transmission.
Conclusions
Molecular epidemiological studies should consider temporal heterogeneities in the parasite population structures. In the context of this investigation, clonal expansions in South America affect estimates of the local genetic diversity. Such dynamic will generate a distorted view if studies are based in a sample collected in a single time point. A second observation is that, with a reasonable number of loci, there is uncertainty in the separation of infections at individual level. Such uncertainties need to be incorporated whenever recrudescent cases need to be separated from new infections in the context of drug efficacy studies. Third, it is relatively easy to identify major clones or sub-populations with a small number of polymorphic microsatellites. Such information could be valuable to better understand gene flow and patterns of migration/re-introduction. It seems logical to contrast samples from at least two transmission seasons, even in gene flow studies, and use loci that allow detecting spatial connectivity so they are useful to track reintroductions at a local level or spatial movements at a regional level. Finally, it was observed that both P. falciparum and P. vivax exhibit similar patterns of clonal expansions. Whereas previous studies have found strong linkage disequilibrium in both parasites [1–5, 7], this is the first time that such dynamics are described in sympatric populations through time. Overall, it seems that such clonal temporal replacements take place even when drug resistant mutations could be fixed, as is the case for P. falciparum[4, 28].
The type of pilot study proposed in this article could be performed in any endemic area but it is particularly critical in those where clonal expansions are suspected, such is the case of seasonal and low transmission endemic areas. Even though only a limited number of loci could be considered for routine analysis, the use of well-chosen microsatellite loci represent a high sensitive method for cluster differentiation and provides a way to deeper analyse patterns of gene flow as well as parasite lineages maintained through time. Overall, this investigation highlights the need of locally evaluating the diversity of microsatellite loci during at least two transmission seasons before starting molecular epidemiologic investigations.
References
Imwong M, Nair S, Pukrittayakamee S, Sudimack D, Williams JT, Mayxay M, Newton PN, Kim JR, Nandy A, Osorio L, Carlton JM, White NJ, Day NP, Anderson TJ: Contrasting genetic structure in Plasmodium vivax populations from Asia and South America. Int J Parasitol. 2007, 37: 1013-1022. 10.1016/j.ijpara.2007.02.010.
Van den Eede P, Van der Auwera G, Delgado C, Huyse T, Soto-Calle VE, Gamboa D, Grande T, Rodriguez H, Llanos A, Anné J, Erhart A, D'Alessandro U: Multilocus genotyping reveals high heterogeneity and strong local population structure of the Plasmodium vivax population in the Peruvian Amazon. Malar J. 2010, 9: 151-10.1186/1475-2875-9-151.
Pumpaibool T, Arnathau C, Durand P, Kanchanakhan N, Siripoon N, Suegorn A, Sitthi-Amorn C, Renaud F, Harnyuttanakorn P: Genetic diversity and population structure of Plasmodium falciparum in Thailand, a low transmission country. Malar J. 2009, 8: 155-10.1186/1475-2875-8-155.
Griffing SM, Mixson-Hayden T, Sridaran S, Alam MT, McCollum AM, Cabezas C, Marquiño Quezada W, Barnwell JW, De Oliveira AM, Lucas C, Arrospide N, Escalante AA, Bacon DJ, Udhayakumar V: South American Plasmodium falciparum after the malaria eradication era: clonal population expansion and survival of the fittest hybrids. PLoS One. 2011, 6: e23486-10.1371/journal.pone.0023486.
Arnott A, Barry AE, Reeder JC: Understanding the population genetics of Plasmodium vivax is essential for malaria control and elimination. Malar J. 2012, 10: 11-
Chenet SM, Tapia LL, Escalante AA, Durand S, Lucas C, Bacon DJ: Genetic diversity and population structure of genes encoding vaccine candidate antigens of Plasmodium vivax. Malar J. 2012, 11: 68-10.1186/1475-2875-11-68.
Iwagami M, Fukumoto M, Hwang SY, Kim SH, Kho WG, Kano S: Population structure and transmission dynamics of Plasmodium vivax in the Republic of Korea based on microsatellite DNA analysis. PLoS Negl Trop Dis. 2012, 6: e1592-10.1371/journal.pntd.0001592.
Wright S: Isolation by distance. Genetics. 1943, 2: 114-38.
Meirmans PG: The trouble with isolation by distance. Mol Ecol. 2012, 21: 2839-46. 10.1111/j.1365-294X.2012.05578.x.
Gauthier C, Tibayrenc M: Population structure of malaria parasites: the driving epidemiological forces. Acta Trop. 2005, 3: 241-250.
Mu J, Awadalla P, Duan J, McGee KM, Joy DA, McVean GA, Su XZ: Recombination hotspots and population structure in Plasmodium falciparum. PLoS Biol. 2005, 3: e335-10.1371/journal.pbio.0030335.
Volkman SK, Sabeti PC, DeCaprio D, Neafsey DE, Schaffner SF, Milner DA, Daily JP, Sarr O, Ndiaye D, Ndir O, Mboup S, Duraisingh MT, Lukens A, Derr A, Stange-Thomann N, Waggoner S, Onofrio R, Ziaugra L, Mauceli E, Gnerre S, Jaffe DB, Zainoun J, Wiegand RC, Birren BW, Hartl DL, Galagan JE, Lander ES, Wirth DF: A genome-wide map of diversity in Plasmodium falciparum. Nat Genet. 2007, 39: 113-119. 10.1038/ng1930.
Neafsey DE, Schaffner SF, Volkman SK, Park D, Montgomery P, Milner DA, Lukens A, Rosen D, Daniels R, Houde N, Cortese JF, Tyndall E, Gates C, Stange-Thomann N, Sarr O, Ndiaye D, Ndir O, Mboup S, Ferreira MU, Moraes Sdo L, Dash AP, Chitnis CE, Wiegand RC, Hartl DL, Birren BW, Lander ES, Sabeti PC, Wirth DF: Genome-wide SNP genotyping highlights the role of natural selection in Plasmodium falciparum population divergence. Genome Biol. 2008, 9: R171-10.1186/gb-2008-9-12-r171.
Cheeseman IH, Miller BA, Nair S, Nkhoma S, Tan A, Tan JC, Al Saai S, Phyo AP, Moo CL, Lwin KM, McGready R, Ashley E, Imwong M, Stepniewska K, Yi P, Dondorp AM, Mayxay M, Newton PN, White NJ, Nosten F, Ferdig MT, Anderson TJ: A major genome region underlying artemisinin resistance in malaria. Science. 2012, 336: 79-82. 10.1126/science.1215966.
Orjuela-Sánchez P, Karunaweera ND, da Silva-Nunes M, da Silva NS, Scopel KK, Gonçalves RM, Amaratunga C, Sá JM, Socheat D, Fairhust RM, Gunawardena S, Thavakodirasah T, Galapaththy GL, Abeysinghe R, Kawamoto F, Wirth DF, Ferreira MU: Single-nucleotide polymorphism, linkage disequilibrium and geographic structure in the malaria parasite Plasmodium vivax: prospects for genome-wide association studies. BMC Genet. 2010, 11: 65-
Payseur BA, Jing P, Haasl RJ: A genomic portrait of human microsatellite variation. Mol Biol Evol. 2011, 28: 303-312. 10.1093/molbev/msq198.
Russell B, Suwanarusk R, Lek-Uthai U: Plasmodium vivax genetic diversity: microsatellite length matters. Trends Parasitol. 2006, 22: 399-401. 10.1016/j.pt.2006.06.013.
Wootton JC, Feng X, Ferdig MT, Cooper RA, Mu J, Baruch DI, Magill AJ, Su XZ: Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature. 2002, 418: 320-323. 10.1038/nature00813.
McCollum AM, Basco LK, Tahar R, Udhayakumar V, Escalante AA: Hitchhiking and selective sweeps of Plasmodium falciparum sulfadoxine and pyrimethamine resistance alleles in a population from central Africa. Antimicrob Agents Chemother. 2008, 52: 4089-4097. 10.1128/AAC.00623-08.
McCollum AM, Schneider KA, Griffing SM, Zhou Z, Kariuki S, Ter-Kuile F, Shi YP, Slutsker L, Lal AA, Udhayakumar V, Escalante AA: Differences in selective pressure on dhps and dhfr drug resistant mutations in western Kenya. Malar J. 2012, 11: 77-10.1186/1475-2875-11-77.
Nyachieo A, VAN Overmeir C, Laurent T, Dujardin JC, D'Alessandro U: Plasmodium falciparum genotyping by microsatellites as a method to distinguish between recrudescent and new infections. AmJTrop Med Hyg. 2005, 73: 210-3.
Greenhouse B, Myrick A, Dokomajilar C, Woo JM, Carlson EJ, Rosenthal PJ, Dorsey G: Validation of microsatellite markers for use in genotyping polyclonal Plasmodium falciparum infections. AmJTrop Med Hyg. 2006, 75: 836-842.
Orjuela-Sánchez P, da Silva NS, da Silva-Nunes M, Ferreira MU: Recurrent parasitemias and population dynamics of Plasmodium vivax polymorphisms in rural Amazonia. AmJTrop Med Hyg. 2009, 81: 961-968. 10.4269/ajtmh.2009.09-0337.
Restrepo E, Imwong M, Rojas W, Carmona-Fonseca J, Maestre A: High genetic polymorphism of relapsing P. vivax isolates in northwest Colombia. Acta Trop. 2011, 119: 23-29. 10.1016/j.actatropica.2011.03.012.
Sunnucks P: Efficient genetic markers for population biology. Trends Ecol Evol. 2000, 15: 199-203. 10.1016/S0169-5347(00)01825-5.
Mwangi JM, Omar SA, Ranford-Cartwright LC: Comparison of microsatellite and antigen-coding loci for differentiating recrudescing Plasmodium falciparum infections from reinfections in Kenya. Int J Parasitol. 2006, 36: 329-336. 10.1016/j.ijpara.2005.10.013.
Schneider KA, Kim Y: An analytical model for genetic hitchhiking in the evolution of antimalarial drug resistance. Theor Popul Biol. 2010, 78: 93-108. 10.1016/j.tpb.2010.06.005.
McCollum AM, Mueller K, Villegas L, Udhayakumar V, Escalante AA: Common origin and fixation of Plasmodium falciparum dhfr and dhps mutations associated with sulfadoxine-pyrimethamine resistance in a low-transmission area in South America. Antimicrob Agents Chemother. 2007, 51: 2085-2091. 10.1128/AAC.01228-06.
Schneider KA, Kim Y: Approximations for the hitchhiking effect caused by the evolution of antimalarial-drug resistance. J Math Biol. 2011, 62: 789-832. 10.1007/s00285-010-0353-9.
Moreno JE, Rubio-Palis Y, Páez E, Pérez E, Sánchez V: Abundance, biting behaviour and parous rate of anopheline mosquito species in relation to malaria incidence in gold-mining areas of southern Venezuela. Med Vet Entomol. 2007, 21: 339-349. 10.1111/j.1365-2915.2007.00704.x.
Imwong M, Sudimack D, Pukrittayakamee S, Osorio L, Carlton JM, Day NP, White NJ, Anderson TJ: Microsatellite variation, repeat array length, and population history of Plasmodium vivax. Mol Biol Evol. 2006, 23: 1016-1018. 10.1093/molbev/msj116.
Karunaweera ND, Ferreira MU, Munasinghe A, Barnwell JW, Collins WE, King CL, Kawamoto F, Hartl DL, Wirth DF: Extensive microsatellite diversity in the human malaria parasite Plasmodium vivax. Gene. 2008, 410: 105-112. 10.1016/j.gene.2007.11.022.
Anderson TJ, Su XZ, Bockarie M, Lagog M, Day KP: Twelve microsatellite markers for characterization of Plasmodium falciparum from finger-prick blood samples. Parasitology. 1999, 119: 113-125. 10.1017/S0031182099004552.
Park SDE: Trypanotolerance in West African Cattle and the Population Genetic Effects of Selection. PhD thesis. 2001, University of Dublin, Microsatellite tool kit version 3.1.1 available from: http://www.animalgenomics.ucd.ie/sdepark/ms-toolkit/
Nash D, Nair S, Mayxay M, Newton PN, Guthmann JP, Nosten F, Anderson TJ: Selection strength and hitchhiking around two anti-malarial resistance genes. Proc Biol Sci. 2005, 272: 1153-1161. 10.1098/rspb.2004.3026.
Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics. 2000, 155: 945-959. Structure 2.3.3 available from: http://pritch.bsd.uchicago.edu/structure.html
Dent Earl A, vonHoldt , Bridgett M: STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012, 4: 359-361. 10.1007/s12686-011-9548-7. Structure harvester available from: http://taylor0.biology.ucla.edu/struct_harvest/
Jakobsson M, Rosenberg NA: CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007, 23: 1801-1806. 10.1093/bioinformatics/btm233. CLUMPP 1.1.2 available from: http://www.stanford.edu/group/rosenberglab/clumpp.html
Rosenberg NA: Distruct: a program for the graphical display of population structure. Molecular Ecology Notes. 2004, 4: 137-138. Distruct 1.1 available at: http://www.stanford.edu/group/rosenberglab/distruct.html
Goudet J: FSTAT Version 1.2: a computer program to calculate F-statistics. J. Heredity. 1995, 86: 485-486. FSTAT 2.9.3.2 available from: http://www2.unil.ch/popgen/softwares/fstat.htm
Laval EG, Schneider S: Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform. 2005, 1: 47-50.
Haubold B, Hudson RR: LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics. 2000, 16: 847-848. 10.1093/bioinformatics/16.9.847. LIAN 3.5 available from: http://adenine.biz.fh-weihenstephan.de/cgi-bin/lian/lian.cgi.pl.
Maruyama T: Stochastic integrals and their application to population genetics. Molecular Evolution, Protein Polymorphism and the Neutral Theory. Edited by: Kimura M. 1982, Tokyo: Japan Scientific Societies Press, 151-166.
Freeman GH, Halton JH: Note on an Exact Treatment of Contingency, Goodness of Fit and Other Problems of Significance. Biometrika. 1951, 38: 141-149.
Barton DE, David F: Multiple runs. Biometrika. 1957, 44: 168-170.
Ferreira MU, Karunaweera ND, da Silva-Nunes M, da Silva NS, Wirth DF, Hartl DL: Population structure and transmission dynamics of Plasmodium vivax in rural Amazonia. J Infect Dis. 2007, 195: 1218-1226. 10.1086/512685.
Urdaneta L, Lal A, Barnabe C, Oury B, Goldman I, Ayala FJ, Tibayrenc M: Evidence for clonal propagation in natural isolates of Plasmodium falciparum from Venezuela. Proc Natl Acad Sci USA. 2001, 98: 6725-6729. 10.1073/pnas.111144998.
Razakandrainibe FG, Durand P, Koella JC, De Meeüs T, Rousset F, Ayala FJ, Renaud F: "Clonal" population structure of the malaria agent Plasmodium falciparum in high-infection regions. Proc Natl Acad Sci USA. 2005, 102: 17388-93. 10.1073/pnas.0508871102.
Acknowledgements
This study was supported by the grants NIH R01 GM084320 and the International Center of Excellence for Malaria Research: Latin American Center for Malaria Research and Control, project NIH 1U19AI089702. We thank Maria Pacheco for reading and commenting the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SC conducted the molecular genetic studies, performed the genetic diversity experiments, analysed the data and wrote the first draft of the manuscript. KS performed the statistical analysis and contributed to the write up of the final draft. LV provided the samples and contributed to the editing of the manuscript. AE designed the project, supervised and directed the research and contributed to the writing and editing of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12936_2012_2579_MOESM1_ESM.pdf
Additional file 1: Number of alleles per loci and expected heterozygosity per year in P. vivax (A and B) and in P. falciparum (C and D) using all samples from Tumeremo.(PDF 222 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chenet, S.M., Schneider, K.A., Villegas, L. et al. Local population structure of Plasmodium: impact on malaria control and elimination. Malar J 11, 412 (2012). https://doi.org/10.1186/1475-2875-11-412
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-11-412