
Abstract
Tumors are not merely masses of neoplastic cells but complex tissues composed of cellular and noncellular elements. This review provides recent data on the main components of a dynamic system, such as carcinoma associated fibroblasts that change the extracellular matrix (ECM) topology, induce stemness and promote metastasis-initiating cells. Altered production and characteristics of collagen, hyaluronan and other ECM proteins induce increased matrix stiffness. Stiffness along with tumor growth-induced solid stress and increased interstitial fluid pressure contribute to tumor progression and therapy resistance. Second, the role of immune cells, cytokines and chemokines is outlined. We discuss other noncellular characteristics of the tumor microenvironment such as hypoxia and extracellular pH in relation to neoangiogenesis. Overall, full understanding of the events driving the interactions between tumor cells and their environment is of crucial importance in overcoming treatment resistance and improving patient outcome.
Similar content being viewed by others


Background
Tumor progression is partly a result of evolving crosstalk between different cell types within the tumor and its surrounding supportive tissue or tumor stroma [1]. Invasive tumor cells interact with the microenvironment and remodel it into a milieu supportive of tumor growth and tumor progression. The altered environment is recognizable under light microscope as desmoplasia and this is used for assessing invasion [2]. The importance of the microenvironment in tumor progression is shown using model systems. It has been shown on animal xenografts that injection of purified malignant epithelial cells results in the formation of histologically complex tumors, with 80% of the cells being stromal [3]. Further, injection of non-transformed mammary epithelial cells into irradiated mammary stromal fat pads, resulted in increased tumor growth compared to those injected into contralateral, non-irradiated mammary fat pads. Irradiated stromal cells altered the microenvironment and this resulted in tumor promotion [4]. Genetic alterations that initiate carcinoma, occur in the epithelium but events that promote tumor progression involve the stroma. In some cases, the trigger for neoplastic progression is speculated to come from signals within the stromal microenvironment [5]. Cancer cells release stroma-modulating growth factors such as fibroblast growth factor, members of the VEGF family, PDGF, EGFR ligands, interleukins, colony-stimulating factors, transforming growth factor β and many others [6]. These factors act in a paracrine manner, disrupt normal tissue homeostasis, resulting in stromal reactions such as angiogenesis and the inflammatory response [7, 8].
Fibroblasts in tumor progression and therapy resistance
Carcinoma associated fibroblasts (CAF) are believed to influence tumor behavior and outcome and thus knowledge of their biology is of importance to an overall understanding of cancer. CAFs are large, spindle-shaped mesenchymal cells that share characteristics with smooth muscle cells and fibroblasts [6]. They constitute a significant component of the stroma and represent the cells responsible for the change of extracellular matrix composition into one with increased amounts of collagens (desmoplastic response) [3]. Currently, no precise definition of CAFs exists because of the different cellular origin and markers expressed: CAFs are likely to derive from resident fibroblasts and marrow-derived mesenchymal precursor cells, whereas their generation through epithelial-mesenchymal transition (EMT) of tumor cells is more controversial [6, 9, 10]. CAFs are not only phenotypically but also functionally distinct from their normal counterparts and are identified immunocyto-/histochemically based on different markers such as α-smooth muscle actin (α-SMA), vimentin, desmin, fibroblast specific protein −1, PDGFR α and β, and fibroblast activation protein or their combinations; CAFs generally lose caveolin 1, PTEN, p21, or have mutated TP53 [11, 12]. Some of these differences are reversible, whereas others persist when the fibroblasts are removed from the vicinity of the carcinoma cells. Their gene expression differences are due to epigenetic and genetic alterations [13, 14] and relate to the stage of their differentiation [15]. The evidence above shows that CAFs, like tumor cells, are heterogenous, not only between different but also within the same type of cancer.
CAFs promote tumor progression in several ways such as secretion of multiple factors and MMPs, inducing stemness, EMT, epigenetic changes, etc. [11, 16]. Recent breast cancer gene expression profiles of the stromal compartment, have revealed significantly different gene sets than normal mammary stroma, with increased cytokines, ECM molecules and proteases [17, 18]. They alter the three dimensional ECM scaffold and support tumor cells that eventually metastasize and activate immune cells to enhance the ECM-degrading capacity [19]. Secreted ECM components such as tenascin reveal pro-migratory activity [20]. TGF-β induces HGF expression by fibroblasts and also induces the transition of fibroblasts to myofibroblasts by increasing α-SMA and tenascin C expression [21]. Gene expression changes, reported by Rajski et al., [22], which were induced by IGF-I in human breast fibroblasts, contained several soluble factors, such as periostin which is involved in bone metastasis and angiogenesis [23–25], tenascin, which enhances tumor cell proliferation [26], as well as LOXL1, a member of the lysyl oxidase family. LOXL1 like LOXL2, may act in the vicinity of epithelial cells during tissue remodeling and collagen cross-linking. LOXL2 has been reported to be involved in invasiveness [27] and specifically expressed by fibroblasts in tumor tissue [28]. The presence of these factors indicates that the IGF-I activated stroma enhances proliferation and the metastatic potential of the cancer cells. In this sense, periostin and tenascin C also activate developmental pathways for the viability of metastasis-initiating cells in the lungs [29]. In the pulmonary parenchyma, TGF-β3 stimulates myofibroblasts to produce periostin which binds stromal Wnt factors Wnt1 and Wnt3a for presentation to stem-like metastasis-initiating cells [30]. Myofibroblasts and the cancer cells themselves also produce tenascin C which promotes the intracellular functioning of the Wnt and Notch pathways [29]. The Wnt pathway is known to control stem cell maintenance in a variety of tissues [31] and tumors [32, 30, 33, 34]. We previously identified another cancer associated extracellular matrix protein, asporin which may interact with the Wnt pathway [35–38]. Asporin was also identified as one of the most CAF-enriched molecules using gene ontology analysis and further suggested as a possible EMT marker due to colocalization with ZEB1 both in the stroma and epithelium of prostate cancer [39]. In breast and prostate carcinomas, mutations of critical tumor suppressor genes like PTEN and TP53 have been reported to occur in either epithelial or stromal cells in a mutually exclusive fashion [40, 41]. Such findings indicate establishment of a vicious circle, in which mutations in the carcinoma drive alterations in the stroma that again promote carcinoma progression [40, 41]. According to Dudley et al. [42], breast CAFs possess a nonmutated but functionally deficient form of p53 and TP53 mutation status may be a predictor of CAF-mediated chemoresistance [43]. The role of CAFs in chemo/endocrine and target resistance is well-reviewed in Mao et al. [11] using breast cancer as example. Besides the genetic mechanisms described above, there is also evidence supporting the involvement of epigenetic changes in the cancer stroma as a contributor to cancer progression. These include histone modifications and alterations in the expression of DNA methyltransferases, chromatin modifying factors and microRNAs [11]. Direct contact between stromal and tumor cells allows minor populations of the latter to evade chemotherapy. For example, adhesion of melanoma cells to fibroblast monolayers through N-cadherin activates AKT, which blocks BAD and significantly reduces the cytotoxic effects of the chemotherapy drug, cisplatin [44]. In summary, CAFs play a critical role in determining many aspects of tumor behavior and overall outcome.
Matrix topology, stiffness and solid stress
Cancer initiation and progression are largely dependent on the physical and chemical features of the adjacent environment particularly matrix topology (architecture) and stiffness. These features are determined by the size of biopolymer (proteins, proteoglycans and glycosaminoglycans) fibers and the density of the fiber network [45]. Connective tissue is characterized by different fiber arrangements ranging from loose or random to highly aligned structures. ECM topology can provide important regulation of cell motility through physical cues that geometrically constrain adhesion sites to guide directional migration [46]. Cancer cells display aligning behavior, called contact guidance through which they actively remodel the ECM fibers surrounding a tumor, using contractile force to align the fibers perpendicularly to the tumor [47, 45]. Dense fibrous collagen that is characteristic of breast cancer stroma forms radial patterns extending away from tumors [48]. On the other hand, the reticular orientation of the collagen matrix surrounding mammary glands may anchor and/or restrain cells. Thus, non-linear matrix reduces invasion while linear structure promotes it (reviewed in [46]).
Matrix concentration and post-translational modifications such as glycosylation and cross-linking (e.g. by LOXL1 and LOXL2) affect the mechanical properties, including visco-elasticity or stiffness (reviewed in [49]). Tumors are stiffer than their normal adjacent tissue. For example, healthy mammary gland is highly compliant (elastic modulus E = ~200 Pa), while the average tumor is over an order of magnitude stiffer (E = ~4,000 Pa). Both the tumor-surrounding stroma and vasculature exhibit increased stiffness (E = ~800–1,000 Pa and ~450 Pa, respectively) [47]. Increased matrix stiffness is also observed in fibrotic lungs, scar tissue and irradiated or aged tissue [50].
ECM topology and stiffness can influence mechanosensing and activate intracellular signaling to promote directional cell migration. Integrin receptors and the physical arrangement of adhesions could trigger orientation of the cytoskeleton, and matrix orientation can also stabilize leading edge protrusions to promote directionally persistent migration in which specific signaling pathways (via vinculin, talin, FAK, p130CAS and filamin A) are involved [45, 46]. Cancer cells recognize an increase in ECM stiffness and respond by generating increased traction forces on their surroundings by regulating focal adhesion formation and growth factor signaling. For this purpose, the cell has several options: it can either force the network fibers apart and remodel the shape until it can pass through the pore, or tumor cell degrades the fiber matrix with the help of proteolytic enzymes (reviewed in [45]). This in turn, enhances growth, survival, and invasion of tumor cells by promoting focal adhesion maturation and signaling through actomyosin contractility [51]. Increased tumor tissue stiffness has been linked to tumor progression, direct stem cell differentiation, cell–cell and cell–matrix adhesion, hyaluronan synthesis, and expression of genes that play important roles in invasion and metastasis [52–54].
Another important tumor characteristic is growth-induced solid stress (Figure 1). As tumor cells proliferate they sequentially create new solid material (i.e. cells and matrix components) which pushes against the surrounding tumor microenvironment. In normal tissue, the expansion of the tumor microenvironment is resisted by the enclosing microenvironment. However, cancer cells proliferate uncontrollably, ignoring contact inhibition and their expansion imposes elastic strain on the surrounding tumor microenvironment, storing stress through the deformation of compliant structures and collapsing more fragile structures, such as blood and lymphatic vessels (Figure 1). Interestingly, this solid stress is accumulated within the tumor and maintained even after the tumor is excised [55]. The known contribution of the ECM to solid stress includes both collagen and hyaluronan. Collagen resists tensile stress because it becomes stiffer as it is stretched. This finding is true for both capsular and interstitial collagen because the ECM in tumors is extensively cross-linked. Whereas hyaluronan resists compression, its negatively charged chains repel, owing to electrostatic repulsion and trap water, forming a poorly compressible matrix [55]. The compression of vessels by solid stress creates two potential barriers to drug delivery. First, the collapse of blood vessels hinders access of systemically administered drugs. This collapse might explain, in part, the fact that tumors with more ECM might be more resistant to treatment. For instance, pancreatic ductal adenocarcinomas, chondrosarcomas, and chordomas are rich in ECM and refractory to chemotherapy [56–58]. Second, the lack of lymphatic vessel function reduces drainage, leading to uniformly elevated interstitial fluid pressure (please see below). As a result, the transport of therapeutics, like antibodies and nanoparticles, is reduced because the dominant means of transport becomes diffusion which is a very slow process for large particles and macromolecules [59]. In this sense, decreasing solid stress by the inexpensive angiotensin inhibitor, losartan, enhances drug delivery and potentiates chemotherapy by decompressing tumor blood vessels [60].

Schematic of the biomechanical forces in the tumor microenvironment. As tumor cells proliferate they sequentially create new solid material (i.e. cells and matrix components) which generate radial and circumferential solid stresses. In the tumor center, circumferential and radial stresses are compressive while in the periphery, radial stress is compressive and circumferential stress is tensile (direction indicated with arrows). Compressive stresses in the tumor interior squeeze tumor components, including lymphatic and blood vessels (note compressed lumen of blood vessel and high density of cells and extracellular matrix in dark green). After the tumor is cut and the stresses are released, the tumor interior decompresses (note extended lumen of blood vessel, relaxed cells and extracellular matrix in light green). Reproduced and modified with permission from Prof. Rakesh K. Jain [55].
Immune cells in tumor microenvironment
Besides endothelial cells and fibroblasts, the tumor microenvironment also harbors innate and adaptive immune cells. It is a complex and highly dynamic system that should concomitantly work to eradicate a tumor. However, once a system is deformed, immunity becomes a benefit for the tumor and provides a very important cue to its development and progression [61]. The tumor-promoting effect of chronic inflammation has been reported many times [62, 63]. However, how tumors promote inflammation and engage inflammatory cells in this process, is still being intensively studied. Macrophage density in the tumor generally negatively correlates with relapse-free and overall survival but localization also seems to be important. The association between TAM (tumor associated macrophage)-density and survival of cancer patients depends on the intratumoral or peritumoral macrophages counted [15]. TAMs promote the metastatic capacity of cancer cells in stromal or perivascular areas, while around the necrosis, in a hypoxic state, they stimulate angiogenesis [64]. A fibroblast secreted protein-1 (FSP1), also called S100A4 and mts1, is secreted by both fibroblasts and cancer cells, and also possibly by macrophages [65], making the environment more favourable to tumor progression by regulating inflammation and angiogenesis and promoting metastasis [66, 67]. FSP1 is proangiogenic, possibly mediated either by the activation of plasminogen or through the transcriptional up-regulation of MMP13 [68]. Both of these proteinases play a role in endothelial cell invasion [5].
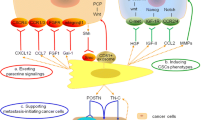
Immune cells, particularly macrophages and neutrophils are sources of chemokines, growth factors and proteases, as well as DNA-damaging reactive oxygen and nitrogen species. The gene expression profile of macrophages isolated from malignant tumors significantly differs from wound or resting peritoneal macrophage profile, with increased number of proliferation-associated genes [69]. Chemokines are produced not only by activated macrophages but by stromal and even cancer cells themselves. As an example, CXCL12 activates CXCR4 both on the surface of immune cells and on hematopoetic and endothelial precursors. The receptor is also expressed in some cancer cells. Accordingly, CXCL12 has several consequences: i) attraction of immune cells leading to tissue destruction, favoring invasion and metastasis; ii) promotion of growth and survival of cancer cells expressing the CXCR4 receptor; iii) recruitment of precursor cells for vasculogenesis; iv) activation of CXCR4 may also lead to greatly increased production of TNFα which itself exhibits other effects. Inflammatory cytokines, overexpressed by tumor cells recruit monocytes (macrophages), lymphocytes and neutrophils to tumor stroma, where they release VEGF, HGF, metalloproteinase 2 and interleukin 8 which affect endothelial cells and contribute to tumor progression [70]. Up-regulation of tumor-regulated interleukins 6 and1β is also associated with the inflammatory network, tumorigenesis, angiogenesis, and metastasis in breast, prostate, and pancreatic cancers [19]. Breast CAFs initiate and mediate tumorigenesis through a macrophage-recruitment inflammatory signature and is dependent on NF-kB signaling [71]. Recent studies demonstrate that colony stimulating factor (CSF)-1 which represents the main growth and differentiation factor for macrophages is overexpressed in breast, ovarian and prostate cancers [72]. Granulocyte CSF and granulocyte-macrophage CSF also contribute to progression of various cancers through recruitment of monocytes, macrophages and neutrophils into the tumor vicinity (reviewed in [6, 73, 74]).
Tumor vasculature, hypoxia and interstitial fluid pressure
Tumor angiogenesis is an important factor in proliferation, metastasis, and drug sensitivity. Primary tumors without vasculature are small and dormant, while the growth of the tumor mass creates hypoxic conditions in the center of the tumor that induce expression of VEGF and subsequent tumor vascularization [75]. CAFs are also suggested to be an important source for growth factors and cytokines recruiting endothelial cells. These are involved in the establishment of the cancer stem cell niche and metastatic spread of tumor cells into distant organs [76]. Angiogenesis in malignant tumors, as measured by microvessel density correlates with clinicopathological factors or poor survival in many cancer types (reviewed in [77]). Tumor-endothelial cell interactions are mediated mostly by cell surface adhesion molecules (i.e. integrins, cadherins, immunoglobulins and selectins) [78, 79]. CD34, CD31, and factor-VIII-related antigen are commonly used as tumor endothelial cell markers and microvessel density is determined by immunodetection of blood vessels with these markers. However, the markers identify not only neovessels but also pre-existing large ones [80]. Nestin has recently received attention as a marker of newly formed endothelial cells [81, 82] and seems promising in the evaluation of neoangiogenesis of different tumors.
Hypoxia is also characteristic of abnormal tumor microenvironment that is intrinsically linked to the formation of neovasculature and is clinically associated with metastasis and poor patient outcome [83, 84]. Diffusion-limited hypoxia is a consequence of tumor cells that are distant from the vascular supply. Such cells are exposed to prolonged or chronic hypoxia and tumor cells are viable in such environments for hours or a few days [85, 86]. Hypoxia induces oncogene expression, enhances DNA mutation rate, and selects for cells with increased apoptotic thresholds [83, 87]. Hypoxia drives tumor progression through increased matrix deposition, cross-linking and remodeling and, enhances collagen turnover and its fibril deposition [88]. Hypoxia-inducible factor (HIF)-1α plays an integral role in the body’s response to low oxygen. It is one of the primary genes involved in the homeostatic process which can increase vascularization in hypoxic areas. HIF-1α allows for survival and proliferation of cancer cells due to its angiogenic properties and its inhibition prevents the spread of cancer [89]. Many of the responses implicated in resistance to anti-angiogenic therapy may be mediated by HIF-1α activated genes [90]. The well-described inducers of EMT, Snail, Slug and Twist are themselves induced by hypoxia [91]. Hypoxia may also affect stem cells [92], and studies on this particular subpopulation of cells in tumors would be relevant to the metastatic process as cells surviving under hypoxic conditions become aggressive and pluripotent. Development of novel hypoxia-targeted therapies include bioreductive prodrugs, HIF-1 targeting, and genetic engineering of anaerobic bacteria [93].
Low extracellular pH is another consequence of the abnormal metabolism in the tumor and supportive factor for its progression. Products of anaerobic glycolysis - lactic acid and carbonic acid (produced by carbonic anhydrase from CO2 and H2O), are the known sources of H + ions in tumors [94, 95]. The imbalance between increased production of H + ions and their reduced removal, lowers the extracellular pH in tumors. The mean pH profiles also decrease in tumors with increasing distance from nearest blood vessels. Low extracellular pH causes stress-induced alteration of gene expression, including the upregulation of VEGF and IL-8 in tumor cells in vitro[96]. Coordinated study of pH, pO2, and VEGF expression in vivo[97] indicated that in low pH or oxygenated regions, tissue pH, but not pO2, regulates VEGF promoter activity. Conversely, in hypoxic or neutral pH regions, tissue pO2 and not pH regulates VEGF expression [97]. Tissue pO2 and pH appear to regulate VEGF transcription in tumors independently. These data suggest that these key microenvironmental parameters in solid tumors regulate angiogenic factors in a complementary manner.
Another feature of the pathophysiology of the tumor microenvironment, is elevated interstitial fluid pressure (IFP) ranging from 10 to 100 mmHg [98, 99] while IFP of normal tissue is around zero [100]. It is thought that the tumor vasculature is the driving force in increasing tumor IFP [101, 102]. In contrast to normal vasculature which is characterized by dichotomous branching, tumor vasculature is unorganized and has trifurcations and branches with uneven diameters. Large inter-endothelial junctions, increased numbers of fenestrations, vesicles and vesico-vacuolar channels and a lack of normal basement membrane are often found in tumor vessels [103]. Due to ultrastructural alterations in the tumor vessel wall, vascular permeability in solid tumors is generally higher than that in various normal tissues [100]. Tumors also lack lymphatic vessels or the intratumoral vessels are non-functional [104, 105] and as a result, excess fluid accumulates in the interstitium, extending the elastic ECM and elevating IFP. Using the model of IFP regulation Heldin et al. [106] showed that fibroblasts actively regulate the tension applied to the ECM through integrins which enable them to exert or modify tension on the collagen fibre network, thereby modulating the elasticity of the ECM in response to hyaluronan and proteoglycan expansion [106]. According to Oldberg et al. [107], inflammatory processes in carcinomas promote synthesis of collagen binding proteoglycan fibromodulin by stroma cells, leading to the formation of a dense and stiff collagen scaffold and a high IFP. Interstitial fluid pressure may have important clinical implications with regard to cancer therapy. Roh et al. [108] reported an inverse relationship between tumor IFP and tissue oxygenation and hypothesized that IFP may aid in predicting the efficacy of radiation therapy. Elevated tumor IFP can also act as a barrier to delivery of therapeutic agents, thereby reducing their efficacy. In this sense, multiple studies have demonstrated improved uptake of chemotherapeutic drugs following a reduction in tumor IFP [60, 59, 109, 110].
Conclusion
Interactions during which the tumor creates a microenvironment favourable for proliferation, for the recruitment of new blood vessels, and for the stimulation of the production of proteases that can degrade adjacent tissues, increase the likelihood of tumor development and invasion. Growing evidence points towards a key role of the multiple cellular and noncellular components of the tumor microenvironment such as cancer associated fibroblasts, immune and endothelial cells as well as matrix topology and stiffness, interstitial fluid pressure, growth induced solid stress, hypoxia and extracellular pH. Full understanding of all the events driving the interaction of tumor cells with their environment is of crucial importance in overcoming treatment resistance and in better patient outcome.
References
Liotta LA, Kohn EC: The microenvironment of the tumour-host interface. Nature. 2001, 411: 375-379. 10.1038/35077241.
Koperek O, Asari R, Niederle B, Kaserer K: Desmoplastic stromal reaction in papillary thyroid microcarcinoma. Histopathology. 2011, 58: 919-924. 10.1111/j.1365-2559.2011.03791.x.
Dudley AC, Shih SC, Cliffe AR, Hida K, Klagsbrun M: Attenuated p53 activation in tumour-associated stromal cells accompanies decreased sensitivity to etoposide and vincristine. Br J Cancer. 2008, 99: 118-125. 10.1038/sj.bjc.6604465.
Barcellos-Hoff MH, Ravani SA: Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 2000, 60: 1254-1260.
Dranoff G: Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004, 4: 11-22. 10.1038/nrc1252.
Mueller MM, Fusenig NE: Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004, 4: 839-849. 10.1038/nrc1477.
Baeriswyl V, Christofori G: The angiogenic switch in carcinogenesis. Semin Cancer Biol. 2009, 19: 329-337. 10.1016/j.semcancer.2009.05.003.
Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, van de Rijn M, Botstein D, Brown PO: Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004, 2: E7-10.1371/journal.pbio.0020007.
Kalluri R, Zeisberg M: Fibroblasts in cancer. Nat Rev Cancer. 2006, 6: 392-401. 10.1038/nrc1877.
Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA: Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007, 449: 557-563. 10.1038/nature06188.
Mao Y, Keller ET, Garfield DH, Shen K, Wang J: Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013, 32: 303-315. 10.1007/s10555-012-9415-3.
Ferrara N: VEGF as a therapeutic target in cancer. Oncology. 2005, 69 (Suppl 3): 11-16.
Herrera M, Herrera A, Dominguez G, Silva J, Garcia V, Garcia JM, Gomez I, Soldevilla B, Munoz C, Provencio M, Campos-Martin Y, Garcia de Herreros A, Casal I, Bonilla F, Pena C: Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci. 2013, 104: 437-444. 10.1111/cas.12096.
Patocs A, Zhang L, Xu Y, Weber F, Caldes T, Mutter GL, Platzer P, Eng C: Breast-cancer stromal cells with TP53 mutations and nodal metastases. N Engl J Med. 2007, 357: 2543-2551. 10.1056/NEJMoa071825.
Baronzio G, Freitas I, Kwaan HC: Tumor microenvironment and hemorheological abnormalities. Semin Thromb Hemost. 2003, 29: 489-497.
Elenbaas B, Weinberg RA: Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res. 2001, 264: 169-184. 10.1006/excr.2000.5133.
Singer CF, Gschwantler-Kaulich D, Fink-Retter A, Haas C, Hudelist G, Czerwenka K, Kubista E: Differential gene expression profile in breast cancer-derived stromal fibroblasts. Breast Cancer Res Treat. 2008, 110: 273-281. 10.1007/s10549-007-9725-2.
Casey T, Bond J, Tighe S, Hunter T, Lintault L, Patel O, Eneman J, Crocker A, White J, Tessitore J, Stanley M, Harlow S, Weaver D, Muss H, Plaut K: Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res Treat. 2009, 114: 47-62. 10.1007/s10549-008-9982-8.
Schulz WA, Burchardt M, Cronauer MV: Molecular biology of prostate cancer. Mol Hum Reprod. 2003, 9: 437-448. 10.1093/molehr/gag064.
Chauhan VP, Stylianopoulos T, Boucher Y, Jain RK: Delivery of molecular and nanoscale medicine to tumors: transport barriers and strategies. Annu Rev Chem Biomol Eng. 2011, 2: 281-298. 10.1146/annurev-chembioeng-061010-114300.
Untergasser G, Gander R, Lilg C, Lepperdinger G, Plas E, Berger P: Profiling molecular targets of TGF-beta1 in prostate fibroblast-to-myofibroblast transdifferentiation. Mech Ageing Dev. 2005, 126: 59-69. 10.1016/j.mad.2004.09.023.
Rajski M, Zanetti-Dallenbach R, Vogel B, Herrmann R, Rochlitz C, Buess M: IGF-I induced genes in stromal fibroblasts predict the clinical outcome of breast and lung cancer patients. BMC Med. 2010, 8: 1-10.1186/1741-7015-8-1.
Kharaishvili G, Cizkova M, Bouchalova K, Mgebrishvili G, Kolar Z, Bouchal J: Collagen triple helix repeat containing 1 protein, periostin and versican in primary and metastatic breast cancer: an immunohistochemical study. J Clin Pathol. 2011, 64: 977-982. 10.1136/jclinpath-2011-200106.
Sasaki H, Yu CY, Dai M, Tam C, Loda M, Auclair D, Chen LB, Elias A: Elevated serum periostin levels in patients with bone metastases from breast but not lung cancer. Breast Cancer Res Treat. 2003, 77: 245-252. 10.1023/A:1021899904332.
Schmidt-Hansen B, Ornas D, Grigorian M, Klingelhofer J, Tulchinsky E, Lukanidin E, Ambartsumian N: Extracellular S100A4(mts1) stimulates invasive growth of mouse endothelial cells and modulates MMP-13 matrix metalloproteinase activity. Oncogene. 2004, 23: 5487-5495. 10.1038/sj.onc.1207720.
Ruiz C, Huang W, Hegi ME, Lange K, Hamou MF, Fluri E, Oakeley EJ, Chiquet-Ehrismann R, Orend G: Growth promoting signaling by tenascin-C [corrected]. Cancer Res. 2004, 64: 7377-7385. 10.1158/0008-5472.CAN-04-1234.
Akiri G, Sabo E, Dafni H, Vadasz Z, Kartvelishvily Y, Gan N, Kessler O, Cohen T, Resnick M, Neeman M, Neufeld G: Lysyl oxidase-related protein-1 promotes tumor fibrosis and tumor progression in vivo. Cancer Res. 2003, 63: 1657-1666.
Hockel M, Vaupel P: Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001, 93: 266-276. 10.1093/jnci/93.4.266.
Oskarsson T, Massague J: Extracellular matrix players in metastatic niches. EMBO J. 2012, 31: 254-256. 10.1038/emboj.2011.469.
Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, Huelsken J: Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2012, 481: 85-89.
Zeng YA, Nusse R: Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell. 2010, 6: 568-577. 10.1016/j.stem.2010.03.020.
Reya T, Clevers H: Wnt signalling in stem cells and cancer. Nature. 2005, 434: 843-850. 10.1038/nature03319.
Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H: Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009, 457: 608-611. 10.1038/nature07602.
Kharaishvili G, Simkova D, Makharoblidze E, Trtkova K, Kolar Z, Bouchal J: Wnt signaling in prostate development and carcinogenesis. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2011, 155: 11-18. 10.5507/bp.2011.016.
Turashvili G, Bouchal J, Baumforth K, Wei W, Dziechciarkova M, Ehrmann J, Klein J, Fridman E, Skarda J, Srovnal J, Hajduch M, Murray P, Kolar Z: Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer. 2007, 7: 55-10.1186/1471-2407-7-55.
Turtoi A, Musmeci D, Wang Y, Dumont B, Somja J, Bevilacqua G, De Pauw E, Delvenne P, Castronovo V: Identification of novel accessible proteins bearing diagnostic and therapeutic potential in human pancreatic ductal adenocarcinoma. J Proteome Res. 2011, 10: 4302-4313. 10.1021/pr200527z.
Chouaib S, Kieda C, Benlalam H, Noman MZ, Mami-Chouaib F, Ruegg C: Endothelial cells as key determinants of the tumor microenvironment: interaction with tumor cells, extracellular matrix and immune killer cells. Crit Rev Immunol. 2010, 30: 529-545. 10.1615/CritRevImmunol.v30.i6.30.
Klee EW, Bondar OP, Goodmanson MK, Dyer RB, Erdogan S, Bergstralh EJ, Bergen HR, Sebo TJ, Klee GG: Candidate serum biomarkers for prostate adenocarcinoma identified by mRNA differences in prostate tissue and verified with protein measurements in tissue and blood. Clin Chem. 2012, 58: 599-609. 10.1373/clinchem.2011.171637.
Orr B, Riddick AC, Stewart GD, Anderson RA, Franco OE, Hayward SW, Thomson AA: Identification of stromally expressed molecules in the prostate by tag-profiling of cancer-associated fibroblasts, normal fibroblasts and fetal prostate. Oncogene. 2012, 31: 1130-1142. 10.1038/onc.2011.312.
Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C: Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet. 2002, 32: 355-357. 10.1038/ng1013.
Shao R, Bao S, Bai X, Blanchette C, Anderson RM, Dang T, Gishizky ML, Marks JR, Wang XF: Acquired expression of periostin by human breast cancers promotes tumor angiogenesis through up-regulation of vascular endothelial growth factor receptor 2 expression. Mol Cell Biol. 2004, 24: 3992-4003. 10.1128/MCB.24.9.3992-4003.2004.
D’Adamo DR: Appraising the current role of chemotherapy for the treatment of sarcoma. Semin Oncol. 2011, 38 (Suppl 3): S19-S29.
Sonnenberg M, van der Kuip H, Haubeis S, Fritz P, Schroth W, Friedel G, Simon W, Murdter TE, Aulitzky WE: Highly variable response to cytotoxic chemotherapy in carcinoma-associated fibroblasts (CAFs) from lung and breast. BMC Cancer. 2008, 8: 364-10.1186/1471-2407-8-364.
Li G, Satyamoorthy K, Herlyn M: N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001, 61: 3819-3825.
Brabek J, Mierke CT, Rosel D, Vesely P, Fabry B: The role of the tissue microenvironment in the regulation of cancer cell motility and invasion. Cell Commun Signal. 2010, 8: 22-10.1186/1478-811X-8-22.
Petrie RJ, Doyle AD, Yamada KM: Random versus directionally persistent cell migration. Nat Rev Mol Cell Biol. 2009, 10: 538-549. 10.1038/nrm2729.
Kraning-Rush CM, Reinhart-King CA: Controlling matrix stiffness and topography for the study of tumor cell migration. Cell Adh Migr. 2012, 6: 274-279. 10.4161/cam.21076.
Sidani M, Wyckoff J, Xue C, Segall JE, Condeelis J: Probing the microenvironment of mammary tumors using multiphoton microscopy. J Mammary Gland Biol Neoplasia. 2006, 11: 151-163. 10.1007/s10911-006-9021-5.
Durand RE, Raleigh JA: Identification of nonproliferating but viable hypoxic tumor cells in vivo. Cancer Res. 1998, 58: 3547-3550.
Mammoto A, Mammoto T, Kanapathipillai M, Wing Yung C, Jiang E, Jiang A, Lofgren K, Gee EP, Ingber DE: Control of lung vascular permeability and endotoxin-induced pulmonary oedema by changes in extracellular matrix mechanics. Nat Commun. 2013, 4: 1759-
Boghaert E, Gleghorn JP, Lee K, Gjorevski N, Radisky DC, Nelson CM: Host epithelial geometry regulates breast cancer cell invasiveness. Proc Natl Acad Sci U S A. 2012, 109: 19632-19637. 10.1073/pnas.1118872109.
Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM: Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009, 139: 891-906. 10.1016/j.cell.2009.10.027.
Folgueras AR, Pendas AM, Sanchez LM, Lopez-Otin C: Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int J Dev Biol. 2004, 48: 411-424. 10.1387/ijdb.041811af.
Reinhart-King CA: How matrix properties control the self-assembly and maintenance of tissues. Ann Biomed Eng. 2011, 39: 1849-1856. 10.1007/s10439-011-0310-9.
Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, Smith BL, Ferrone CR, Hornicek FJ, Boucher Y, Munn LL, Jain RK: Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci U S A. 2012, 109: 15101-15108. 10.1073/pnas.1213353109.
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR: Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012, 21: 418-429. 10.1016/j.ccr.2012.01.007.
Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M: Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009, 324: 1457-1461. 10.1126/science.1171362.
Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P, Popovic Z, Huang P, Bawendi MG, Boucher Y, Jain RK: Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013, 4: 2516-
Honn KV, Tang DG: Adhesion molecules and tumor cell interaction with endothelium and subendothelial matrix. Cancer Metastasis Rev. 1992, 11: 353-375. 10.1007/BF01307187.
Horino Y, Takahashi S, Miura T, Takahashi Y: Prolonged hypoxia accelerates the posttranscriptional process of collagen synthesis in cultured fibroblasts. Life Sci. 2002, 71: 3031-3045. 10.1016/S0024-3205(02)02142-2.
Hu M, Yao J, Cai L, Bachman KE, van den Brule F, Velculescu V, Polyak K: Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005, 37: 899-905. 10.1038/ng1596.
Castellsague X, Bosch FX, Munoz N: Environmental co-factors in HPV carcinogenesis. Virus Res. 2002, 89: 191-199. 10.1016/S0168-1702(02)00188-0.
Nelson WG, DeWeese TL, DeMarzo AM: The diet, prostate inflammation, and the development of prostate cancer. Cancer Metastasis Rev. 2002, 21: 3-16. 10.1023/A:1020110718701.
Lewis CE, Pollard JW: Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66: 605-612. 10.1158/0008-5472.CAN-05-4005.
Inoue T, Plieth D, Venkov CD, Xu C, Neilson EG: Antibodies against macrophages that overlap in specificity with fibroblasts. Kidney Int. 2005, 67: 2488-2493. 10.1111/j.1523-1755.2005.00358.x.
Goetz JG, Minguet S, Navarro-Lerida I, Lazcano JJ, Samaniego R, Calvo E, Tello M, Osteso-Ibanez T, Pellinen T, Echarri A, Cerezo A, Klein-Szanto AJ, Garcia R, Keely PJ, Sanchez-Mateos P, Cukierman E, Del Pozo MA: Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011, 146: 148-163. 10.1016/j.cell.2011.05.040.
Fukumura D, Jain RK: Tumor microenvironment abnormalities: causes, consequences, and strategies to normalize. J Cell Biochem. 2007, 101: 937-949. 10.1002/jcb.21187.
Semenza GL: Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003, 3: 721-732. 10.1038/nrc1187.
De Wever O, Nguyen QD, Van Hoorde L, Bracke M, Bruyneel E, Gespach C, Mareel M: Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18: 1016-1018.
Zumsteg A, Christofori G: Corrupt policemen: inflammatory cells promote tumor angiogenesis. Curr Opin Oncol. 2009, 21: 60-70. 10.1097/CCO.0b013e32831bed7e.
Kobayashi Y: The role of chemokines in neutrophil biology. Front Biosci. 2008, 13: 2400-2407. 10.2741/2853.
Lin CW, Shen SC, Ko CH, Lin HY, Chen YC: Reciprocal activation of macrophages and breast carcinoma cells by nitric oxide and colony-stimulating factor-1. Carcinogenesis. 2010, 31: 2039-2048. 10.1093/carcin/bgq172.
Coussens LM, Werb Z: Inflammation and cancer. Nature. 2002, 420: 860-867. 10.1038/nature01322.
Balkwill F: Cancer and the chemokine network. Nat Rev Cancer. 2004, 4: 540-550. 10.1038/nrc1388.
Egeblad M, Littlepage LE, Werb Z: The fibroblastic coconspirator in cancer progression. Cold Spring Harb Symp Quant Biol. 2005, 70: 383-388. 10.1101/sqb.2005.70.007.
Li L, Neaves WB: Normal stem cells and cancer stem cells: the niche matters. Cancer Res. 2006, 66: 4553-4557. 10.1158/0008-5472.CAN-05-3986.
Meert AP, Paesmans M, Martin B, Delmotte P, Berghmans T, Verdebout JM, Lafitte JJ, Mascaux C, Sculier JP: The role of microvessel density on the survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2002, 87: 694-701. 10.1038/sj.bjc.6600551.
Buess M, Rajski M, Vogel-Durrer BM, Herrmann R, Rochlitz C: Tumor-endothelial interaction links the CD44(+)/CD24(−) phenotype with poor prognosis in early-stage breast cancer. Neoplasia. 2009, 11: 987-1002.
Heldin CH, Rubin K, Pietras K, Ostman A: High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 2004, 4: 806-813. 10.1038/nrc1456.
Ishiwata T, Matsuda Y, Naito Z: Nestin in gastrointestinal and other cancers: effects on cells and tumor angiogenesis. World J Gastroenterol. 2011, 17: 409-418. 10.3748/wjg.v17.i4.409.
Teranishi N, Naito Z, Ishiwata T, Tanaka N, Furukawa K, Seya T, Shinji S, Tajiri T: Identification of neovasculature using nestin in colorectal cancer. Int J Oncol. 2007, 30: 593-603.
Kolar Z, Ehrmann J, Turashvili G, Bouchal J, Mokry J: A novel myoepithelial/progenitor cell marker in the breast?. Virchows Arch. 2007, 450: 607-609. 10.1007/s00428-007-0403-x.
Lunt SJ, Chaudary N, Hill RP: The tumor microenvironment and metastatic disease. Clin Exp Metastasis. 2009, 26: 19-34. 10.1007/s10585-008-9182-2.
Grum-Schwensen B, Klingelhofer J, Berg CH, El-Naaman C, Grigorian M, Lukanidin E, Ambartsumian N: Suppression of tumor development and metastasis formation in mice lacking the S100A4(mts1) gene. Cancer Res. 2005, 65: 3772-3780. 10.1158/0008-5472.CAN-04-4510.
Sutherland RM, Franko AJ: On the nature of the radiobiologically hypoxic fraction in tumors. Int J Radiat Oncol Biol Phys. 1980, 6: 117-120. 10.1016/0360-3016(80)90215-1.
Devarajan-Ketha H, Craig TA, Madden BJ, Robert Bergen H 3rd, Kumar R: The sclerostin-bone protein interactome. Biochem Biophys Res Commun. 2012, 417: 830-835. 10.1016/j.bbrc.2011.12.048.
Duff MD, Mestre J, Maddali S, Yan ZP, Stapleton P, Daly JM: Analysis of gene expression in the tumor-associated macrophage. J Surg Res. 2007, 142: 119-128. 10.1016/j.jss.2006.12.542.
Helmlinger G, Sckell A, Dellian M, Forbes NS, Jain RK: Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin Cancer Res. 2002, 8: 1284-1291.
Schauer IG, Sood AK, Mok S, Liu J: Cancer-associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia. 2011, 13: 393-405.
Bagley RG: Endosialin: from vascular target to biomarker for human sarcomas. Biomark Med. 2009, 3: 589-604. 10.2217/bmm.09.54.
Sun S, Ning X, Zhang Y, Lu Y, Nie Y, Han S, Liu L, Du R, Xia L, He L, Fan D: Hypoxia-inducible factor-1alpha induces Twist expression in tubular epithelial cells subjected to hypoxia, leading to epithelial-to-mesenchymal transition. Kidney Int. 2009, 75: 1278-1287. 10.1038/ki.2009.62.
Keith B, Simon MC: Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007, 129: 465-472. 10.1016/j.cell.2007.04.019.
Sun JD, Liu Q, Wang J, Ahluwalia D, Ferraro D, Wang Y, Duan JX, Ammons WS, Curd JG, Matteucci MD, Hart CP: Selective tumor hypoxia targeting by hypoxia-activated prodrug TH-302 inhibits tumor growth in preclinical models of cancer. Clin Cancer Res. 2012, 18: 758-770. 10.1158/1078-0432.CCR-11-1980.
Garin-Chesa P, Old LJ, Rettig WJ: Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc Natl Acad Sci U S A. 1990, 87: 7235-7239. 10.1073/pnas.87.18.7235.
Pouyssegur J, Dayan F, Mazure NM: Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006, 441: 437-443. 10.1038/nature04871.
Xu L, Fukumura D, Jain RK: Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: mechanism of low pH-induced VEGF. J Biol Chem. 2002, 277: 11368-11374. 10.1074/jbc.M108347200.
Erler JT, Weaver VM: Three-dimensional context regulation of metastasis. Clin Exp Metastasis. 2009, 26: 35-49. 10.1007/s10585-008-9209-8.
Nathanson SD, Nelson L: Interstitial fluid pressure in breast cancer, benign breast conditions, and breast parenchyma. Ann Surg Oncol. 1994, 1: 333-338. 10.1007/BF03187139.
Milosevic M, Fyles A, Hedley D, Pintilie M, Levin W, Manchul L, Hill R: Interstitial fluid pressure predicts survival in patients with cervix cancer independent of clinical prognostic factors and tumor oxygen measurements. Cancer Res. 2001, 61: 6400-6405.
Erler JT, Cawthorne CJ, Williams KJ, Koritzinsky M, Wouters BG, Wilson C, Miller C, Demonacos C, Stratford IJ, Dive C: Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol Cell Biol. 2004, 24: 2875-2889. 10.1128/MCB.24.7.2875-2889.2004.
Lunt SJ, Kalliomaki TM, Brown A, Yang VX, Milosevic M, Hill RP: Interstitial fluid pressure, vascularity and metastasis in ectopic, orthotopic and spontaneous tumours. BMC Cancer. 2008, 8: 2-10.1186/1471-2407-8-2.
Jain RK, Tong RT, Munn LL: Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: insights from a mathematical model. Cancer Res. 2007, 67: 2729-2735. 10.1158/0008-5472.CAN-06-4102.
Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, Xu L, Hicklin DJ, Fukumura D, di Tomaso E, Munn LL, Jain RK: Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004, 6: 553-563.
Padera TP, Kadambi A, di Tomaso E, Carreira CM, Brown EB, Boucher Y, Choi NC, Mathisen D, Wain J, Mark EJ, Munn LL, Jain RK: Lymphatic metastasis in the absence of functional intratumor lymphatics. Science. 2002, 296: 1883-1886. 10.1126/science.1071420.
Leu AJ, Berk DA, Lymboussaki A, Alitalo K, Jain RK: Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res. 2000, 60: 4324-4327.
Fukumura D, Xu L, Chen Y, Gohongi T, Seed B, Jain RK: Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001, 61: 6020-6024.
Oldberg A, Kalamajski S, Salnikov AV, Stuhr L, Morgelin M, Reed RK, Heldin NE, Rubin K: Collagen-binding proteoglycan fibromodulin can determine stroma matrix structure and fluid balance in experimental carcinoma. Proc Natl Acad Sci U S A. 2007, 104: 13966-13971. 10.1073/pnas.0702014104.
Roh HD, Boucher Y, Kalnicki S, Buchsbaum R, Bloomer WD, Jain RK: Interstitial hypertension in carcinoma of uterine cervix in patients: possible correlation with tumor oxygenation and radiation response. Cancer Res. 1991, 51: 6695-6698.
Vlahovic G, Ponce AM, Rabbani Z, Salahuddin FK, Zgonjanin L, Spasojevic I, Vujaskovic Z, Dewhirst MW: Treatment with imatinib improves drug delivery and efficacy in NSCLC xenografts. Br J Cancer. 2007, 97: 735-740. 10.1038/sj.bjc.6603941.
Pietras K, Rubin K, Sjoblom T, Buchdunger E, Sjoquist M, Heldin CH, Ostman A: Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002, 62: 5476-5484.
Acknowledgements
This work was supported in part by grants NT13573 from the Czech Ministry of Health and NPU I LO1304 from the Czech Ministry of Education. DS was also supported by LF_2014_003.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GK conceived the idea, did literature search and drafted the manuscript; DS did literature search and contributed in drafting the manuscript; MG, NN and KB were involved in discussion and drafting the manuscript; JB supervised the project, made substantial contributions to the concept and design of manuscript, and drafted the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Kharaishvili, G., Simkova, D., Bouchalova, K. et al. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer Cell Int 14, 41 (2014). https://doi.org/10.1186/1475-2867-14-41
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2867-14-41