
Abstract
Background
Oral Squamous Cell Carcinoma (OSCC) is a major cause of cancer death worldwide, which is mainly due to recurrence leading to treatment failure and patient death. Histological status of surgical margins is a currently available assessment for recurrence risk in OSCC; however histological status does not predict recurrence, even in patients with histologically negative margins. Therefore, molecular analysis of histologically normal resection margins and the corresponding OSCC may aid in identifying a gene signature predictive of recurrence.
Methods
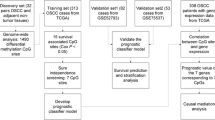
We used a meta-analysis of 199 samples (OSCCs and normal oral tissues) from five public microarray datasets, in addition to our microarray analysis of 96 OSCCs and histologically normal margins from 24 patients, to train a gene signature for recurrence. Validation was performed by quantitative real-time PCR using 136 samples from an independent cohort of 30 patients.
Results
We identified 138 significantly over-expressed genes (> 2-fold, false discovery rate of 0.01) in OSCC. By penalized likelihood Cox regression, we identified a 4-gene signature with prognostic value for recurrence in our training set. This signature comprised the invasion-related genes MMP1, COL4A1, P4HA2, and THBS2. Over-expression of this 4-gene signature in histologically normal margins was associated with recurrence in our training cohort (p = 0.0003, logrank test) and in our independent validation cohort (p = 0.04, HR = 6.8, logrank test).
Conclusion
Gene expression alterations occur in histologically normal margins in OSCC. Over-expression of the 4-gene signature in histologically normal surgical margins was validated and highly predictive of recurrence in an independent patient cohort. Our findings may be applied to develop a molecular test, which would be clinically useful to help predict which patients are at a higher risk of local recurrence.
Similar content being viewed by others


Background
Oral Squamous Cell Carcinoma (OSCC) accounts for 24% of all head and neck cancers [1]. Currently available protocols for treatment of OSCCs include surgery, radiotherapy and chemotherapy. Complete surgical resection is the most important prognostic factor [2], since failure to completely remove a primary tumor is the main cause of patient death. Accuracy of the resection is based on the histological status of the margins, as determined by microscopic evaluation of frozen sections. Presence of epithelial dysplasia or tumor cells in the surgical resection margins is associated with a significant risk (66%) of local recurrence [3]. However, even with histologically normal surgical margins, 10-30% of OSCC patients will still have local recurrence [4], which may lead to treatment failure and patient death.
Since histological status of surgical resection margins alone is not an independent predictor of local recurrence [5], histologically normal margins may harbor underlying genetic changes, which increase the risk of recurrence [6, 7]. To date, candidate-gene approach studies have identified genetic alterations in surgical resection margins in head and neck squamous cell carcinoma (HNSCC) from different disease sites, e.g., oral cavity, pharynx/hypopharynx, larynx [6–16]. Genetic alterations identified in HNSCC included over-expression of eIF4E [6, 9], TP53 [7, 11] and CDKN2A/P16 proteins [7]. Other alterations reported included promoter hypermethylation of CDKN2A/P16 [13] and TP53 mutations [12, 16]. In addition, promoter hypermethylation of CDKN2A, CCNAI and DCC was associated with decreased time to head and neck cancer recurrence [10].
Despite all these studies, genetic alterations identified to date have not been used clinically in the assessment of surgical margins, and no study has developed a gene signature that can accurately predict which patients with OSCC are at a higher risk of disease recurrence. This may be due to the lack of studies using high-throughput analysis of multiple surgical margins and matched OSCCs to identify deregulated genes with prognostic value for recurrence. One probable reason for this deficit is that the application of strict inclusion criteria (tumors of a single anatomical site, with all margins being histologically negative, and with multiple margins of adequate RNA quantity and quality for expression analyses), presents a challenge for obtaining adequate sample size to develop a prognostic signature that can predict recurrence in oral carcinoma. We addressed this challenge through a hypothesis-driven approach, based on the hypothesis that commonly over-expressed genes in OSCC and a subset of histologically normal margins are biomarkers of recurrence. We used meta-analysis of published microarray studies to help identify such genes with high confidence, along with global gene expression analysis of histologically normal margins and their corresponding OSCCs in our own study.
Methods
Patients
This work was performed with the approval of the University Health Network Research Ethics Board. All patients signed their informed consent before sample collection, and had surgery as their primary treatment. Tissue samples were prospectively collected at time of surgery from the Toronto General Hospital, Toronto, Ontario, Canada. The inclusion criteria of patients were based on primary disease, tumor site (oral cavity), histology of the primary tumor (squamous cell carcinoma), and histological status of surgical resection margins. All patients included in this study had a final pathology report of histologically normal resection margins, meaning none of the patients had a positive margin. An experienced head and neck pathologist (BP-O) performed histological evaluation of all surgical margins (frozen section analysis, as per standard of care) to ensure that they were histologically normal. A section from each tumor sample (OSCC) was stained with hematoxylin-eosin, to confirm the presence of at least 80% tumor cells. The primary tumor, histologically normal margins and a distant normal oral tissue were collected from each patient, in both training and validation cohorts. All tissue samples were snap-frozen in liquid nitrogen until RNA extraction and gene expression analysis. We collected a total of 232 tissue samples from 54 patients (divided into training and validation sets), as described below.
Samples used for microarrays (training set)
96 samples (histologically normal margins, OSCC and adjacent normal oral tissues) from 24 patients were used for oligonucleotide microarray analysis. Patient clinical data for the training set are shown in Table 1.
Samples used for quantitative real-time reverse-transcription PCR (RQ-PCR) (validation set)
136 samples (histologically normal margins, OSCC and adjacent normal oral tissues) from an independent cohort of 30 patients were used for RQ-PCR validation analysis. Patient clinical data for the validation set samples are shown in Table 2.
RNA Isolation, Microarrays and Validation Experiments
The detailed protocol for RNA isolation and the experimental details for microarrays using the Human Genome HG-U133A 2.0 plus oligonucleotide arrays (Affymetrix, Santa Clara, CA, USA), as well as validation experiments by RQ-PCR are described in Additional file 1, Methods S1.
Bioinformatic Analyses
All bioinformatic analyses of array data were performed in the R language and environment for statistical computing (version 2.10.0) implemented on CentOS 5.1 on an IBM HS21 Linux cluster. We pre-processed each public data set using GCRMA normalization [17] with updated Entrez Gene-based chip definition files [18], using the affy R package (version 1.24.2) [19]. Genes with evidence of tumor-normal differential expression across all public datasets, with a False Discovery Rate (FDR) of 0.01 and fold-change ≥ 2, were identified using the rankprod R package [20]. Gene Ontology enrichment analysis was performed with the GOstats R package (version 2.12.0) [21]. We also used GOEAST (Go Enrichment Analysis Software Toolkit) [22] for graphical representation of GO annotations. Analyses are reproducible using the code (available upon request); data have been deposited in NCBI's Gene Expression Omnibus [23] and are accessible through GEO series accession number GSE31056 http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE31056.
Meta-analysis of published microarray studies
Our goal was to identify a prognostic signature for recurrence in OSCC, based on the hypothesis that gene expression deregulation occurring in OSCC would be an early indicator of recurrence if gene expression changes are present in a subset of histologically normal surgical resection margins. We performed gene expression profiling of both resection margins and tumors with the purpose of (1) identifying over-expressed genes in tumors as potential markers of recurrence in histologically normal margins, and (2) finding a subset of those genes predictive of recurrence. In order to generate a very high-confidence gene set, we augmented the analysis of our data with a meta-analysis of five published microarray studies [24–28] to reliably identify a set of genes significantly deregulated in OSCC compared to normal oral tissues. These five public data sets were selected based on the availability of raw microarray data, as well as for the inclusion of both oral cavity tumors and either adjacent normal tissues or oral tissues from healthy individuals. Although data from Pyeon et al. [28] included HPV positive and HPV negative head and neck carcinomas from different anatomic sites, we selected oral carcinomas only, which are mainly negative for HPV infection, as shown in a recent study performed by our group [29]. This meta-analysis sample set was composed of a total of 199 samples (141 OSCCs, 38 adjacent normal tissues and 20 healthy normal tissues) from 141 oral cancer patients and 20 healthy individuals (without cancer) (Table 3). We pre-processed data from the different array platforms with updated chip definition files, as described above, to correct outdated probe mapping information from older platforms. We used a Rank Product analysis for the public studies, which considered only the ranking of genes by differential expression between pairs of samples within studies [20, 30], avoiding batch and platform-related effects which would occur from directly combining expression values from the different studies. Genes were selected with evidence of up-regulation in tumors with a False Discovery Rate (FDR) of 0.01 and fold-change ≥ 2. We chose to focus on over-expressed genes only, since histologically normal margins may contain only a fraction of genetically altered cells, and the presence of genetically normal cells would likely make down-regulated genes unreliable markers. By using the intersection of genes identified both by meta-analysis and the in-house array training set, we retained only genes that were reproducibly over-expressed compared to normal oral tissues from healthy patients and histologically normal margins. These strict selection criteria for gene signature candidates, based on prior hypothesis, helped to reduce the risk of over-fitting during Cox regression analysis.
Data analysis of in-house microarray study
Microarray results from our study were normalized as described above, along with normal oral tissue samples from healthy individuals also assayed by Affymetrix Human Genome U133 Plus 2.0 oligonucleotide arrays (downloaded from GEO accession number GSE6791). Probesets with low expression (75th percentile below log2(100)) or low variance (IQR on log2 scale < 0.5) were filtered [31] as well as the quality control probe sets. The treat function from LIMMA: Linear Models for Microarray Analysis (version 3.2.1) [32] was used to identify genes ≥ 2-fold up-regulated in tumors compared to margins from our study, with FDR = 0.01. We took the intersection of genes identified in both the meta-analysis and in-house microarray experiment, and this intersection analysis produced a list of 138 genes, which were used as the feature set for developing the gene signature for recurrence.
Penalized Cox regression
LASSO penalized Cox regression was used to train a predictive risk score from the maximum expression of the 138 genes in any margin of each patient (all these 138 genes being found to be over-expressed in OSCC). Normalized expression values were first converted to Z-scores for each gene, to ensure equal effect of the penalty on all coefficients, as is standard for penalized regression. We applied LASSO penalized Cox regression as implemented in the penalized R package (version 0.9-27) [33], to condition a linear risk score with time to recurrence as the event of interest. In keeping with the hypothesis that over-expression of these genes is predictive of recurrence, we constrained the regression coefficients to positive values, and selected the L1 penalty parameter by optimizing 10-fold cross-validated likelihood. Since the cross-validated likelihood depends on the randomized folding of the data, we repeated the regression 50 times and selected the model with the highest cross-validated likelihood.
Results
Patient characteristics: OSCC recurrence
As shown in Table 1 and Table 2, 9/24 patients (training set) and 7/30 patients (independent validation set) had disease recurrence. Median time to local recurrence (by Kaplan Meier estimate) of patients in the training set was 32 months (range 2-34 months). Similarly, patients from the validation set recurred within 2-36 months. All patients had local recurrence, and some patients also had regional and/or distant failure (detailed data in Tables 1 and 2). Median (by reverse Kaplan-Meier estimate) and range of follow-up times of patients were 20 months (1.4-57 months) in the training set and 23 months (1-81 months) in the validation set.
Differentially expressed genes in margins, OSCC and normal oral tissues
Meta-analysis of the five public data sets (Table 3) identified 667 up-regulated genes in OSCC; results are provided in Additional file 2, Table S1. Requiring two-fold up-regulation in tumors in both the meta-analysis of public datasets and our microarray experiment, with FDR < 0.01, identified 138 over-expressed genes (Additional file 3, Table S2). The expression patterns of these genes in tumors, margins, and normal oral tissue samples from healthy individuals are shown as a heatmap in Figure 1. As seen in the hierarchical clustering, the 138 genes accurately discriminate between the tumors and margins of the in-house study, as well as normal oral tissues from a publicly available study (as described in Methods). We performed LASSO penalized regression of these 138 genes and selected a total of six genes. Of these six genes, we retained four genes with the largest coefficients (MMP1, COL4A1, P4HA2 and THBS2), and eliminated two genes with very small coefficients (PXDN and PMEPA1), which together contributed less than 5% of the magnitude of the risk score. These four genes constituted a 4-gene signature of recurrence. In the heatmap, Gene cluster "B" contains three of the four genes in the signature (COL4A1, P4HA2 and THBS2), and it shows frequent up-regulation in the surgical margins compared to the normal oral tissues. Strikingly, MMP1, found in gene cluster "A", shows less frequent over-expression in the margins, but has extreme differential expression between margins and OSCCs (400-fold up-regulation as detected by microarrays, and 800-fold up-regulation, validated by RQ-PCR). The proteins encoded by the 138 genes are also shown in a protein interaction network that highlights the most highly inter-connected proteins (Figure 2 and Additional file 1, Methods S1). In the heatmap, the main features of clusters A and B are the large number of interacting MMP proteins in cluster A, which contains MMP1, and collagens plus TGFB1 in cluster B, which also contains P4HA2, THBS2 and COL4A1 genes of the signature. The large number of MMPs and collagen proteins are closely connected; in particular, MMP9 interacts with both THBS2 and COL4A1, and indirectly with MMP1.

Heatmap of 138 genes up-regulated in OSCC. Expression values for each row (gene) are scaled to z-scores for visualization. Margins and tumors annotated with darker colors above the heatmap are from patients who experienced recurrence.

Protein-protein interaction network of 138 genes. I2D version 1.72 was used to identify protein interactions for the 138 genes shown in the heatmap. The resulting network was visualized using NAViGaTOR 2.1.14 http://ophid.utoronto.ca/navigator. Color of nodes corresponds to Gene Ontology biological function, as described in the legend. Red-highlighted squares represent the four genes in the signature of OSCC recurrence.
Results of Gene Ontology (GO) enrichment analysis of all 138 genes are presented in Additional file 4, Table S3. Graphical representations of GO annotations are shown in Additional file 5, GO biological processes, Additional file 6, GO cellular component and Additional file 7, GO molecular function.
Over-expressed genes in surgical margins: prognostic value for recurrence
We examined these 138 genes (see above) for univariate association with recurrence in the microarray training data, which, since recurrence was not used in the selection of these genes, constitutes the first validation of our study hypothesis. We found significant enrichment for univariate association with recurrence in these 138 genes (see Additional file 8, Figure S1 for comparison of the distribution of nominal p-values with a null distribution empirically determined by permutation of the outcome labels).
Four-gene prognostic signature for OSCC recurrence
We performed quantitative PCR validation of the 4-gene signature (MMP1, COL4A1, P4HA2 and THBS2) in a separate patient cohort (Table 2). This validation analysis confirmed that all four genes were up-regulated in margins and OSCC samples from patients with disease recurrence compared to margins and OSCCs from patients who did not recur (Figure 3A). The risk score was calculated using the maximum expression in any margin of each patient, and was dichotomized at the median score. These risk groups were predictive of recurrence in an independent test cohort assayed by RQ-PCR (136 samples; N = 30 patients) (HR = 6.8, p = 0.04, logrank test) (Figure 3B). In multivariate Cox analysis, correction for T (tumor size) and N (nodal status) resulted only in a slight increase in significance level of the four-gene risk score (p = 0.06, likelihood ratio test). Clinical variables, alone or in combination, were not predictive of recurrence in either training or validation cohorts. The coefficients of the 4-gene risk score, for use with Z-score scaled expression values, are summarized in Table 4.

Heatmap of validation data and Kaplan-Meier plot of disease recurrence. (A) Unsupervised hierarchical clustering of the quantitative real-time PCR (validation data) showing the maximum expression levels of MMP1, P4HA2, THBS2 and COL4A1 in margins from patients with and without recurrence and with a follow-up time ≥ 12 months. Margins annotated with darker green above the heatmap are from patients who experienced recurrence. Margins from patients with locally recurrent tumors show increased expression levels of the four-gene signature compared to patients who did not recur. (B) Kaplan-Meier plot of quantitative real-time PCR data for patients in the validation set. Patients are assigned to high or low risk based on their four-gene signature risk score. As seen in the Kaplan-Meier plot, patients with over-expression of the 4-gene signature are at high risk for disease recurrence; all patients who experienced recurrence in the validation set are in the high risk group, suggesting that over-expression of this signature was highly predictive of recurrence in the validation set.
In addition, we performed a survival Receiver Operating Characteristic (ROC) analysis to demonstrate the sensitivity and specificity of the model in the test set. This analysis showed the effect of altering the cutoff threshold between high and low-risk patients. Our results showed that the use of a very high threshold to define high-risk patients predicts a majority of recurrences at a low false-positive rate (20%). The area under the ROC curve (AUC) for recurrence within 36 months was 0.73, an improvement over the expected AUC of 0.5 for unpredictive models (Additional file 9, Figure S2). While we maintained the standard median cutoff due to the limited sample size, a larger study in the future may be able to further fine-tune the cutoff threshold to optimize sensitivity and specificity in the context of the relative risks that treatment options informed by this prognostic score entail.
Effect of reducing the number of available margins
Simulating the selection of only a single margin from each patient, the 4-gene signature maintained a predictive effect in both the training and validation sets (median HR = 2.2 in the training set and 1.8 in the validation set, with 82% and 99% of bootstrapped hazard ratios greater than the no-effect value of HR = 1) (Additional file 10, Figure S3). Results from the bootstrap simulations showed smaller hazard ratios, compared with hazard ratios obtained when using the maximum expression value from several margins. This difference could not be explained by association between the number of margins and recurrence in the training set (HR = 1.17, p = 0.69 logrank test) or the validation set (HR = 1.30, p = 0.24 logrank test). However, a plausible explanation for this difference is that increasing the number of observed margins increases the probability of observing genetic alterations spreading asymmetrically from the tumor, since genetic alterations are not seen in all surgical margins.
Discussion
It is known that histologically normal margins may harbor genetic changes also found in the primary tumor, as shown by studies in HNSCC, including oral carcinomas [7]. In oral carcinoma, local recurrence may arise from cancer cells left behind after surgery, undetectable by histopathology (minimal residual cancer), or from fields of genetically altered cells with the potential to give rise to a new carcinoma [34]; such fields precede the tumor and can be detected in the surrounding mucosa (surgical resection margins). Molecular changes that are commonly detected in margins as well as the corresponding tumor could indicate that pre-malignant or malignant clones were able to migrate to the surrounding tissue, giving rise to a primary tumor recurrence [35].
Herein, we show that global gene expression analysis of histologically normal margins and OSCC is a valuable approach for the identification of deregulated genes and pathways associated with OSCC recurrence. We used a multi-step procedure including our in-house whole-genome expression profiling experiment and a meta-analysis of five published microarray datasets to develop a 4-gene signature (MMP1, COL4A1, THBS2 and P4HA2) for prediction of recurrence in OSCC. This signature is based on genes found to be consistently over-expressed in OSCC as compared to normal oral mucosa; these genes are also over-expressed in a subset of histologically normal surgical resection margins, and their over-expression in such margins provides an indication of the presence of genetic changes before histological alterations can be detected by histology. Notably, our initial analyses reveal that this 4-gene signature predicted recurrence in two of the patients (Pts. 17 and 20, Table 2, validation set) who had not recurred until the latest update of the clinical data for recurrence status. Both of these patients had local recurrence, 8 and 19 months after surgery, respectively.
Genes identified in the 4-gene signature (MMP1, COL4A1, THBS2 and P4HA2) play major roles in cell-cell and/or cell-matrix interaction, and invasion. The direct and indirect partners of these genes are illustrated in Figure 2. The functions of two genes (P4HA2 and THBS2) in our signature of OSCC recurrence and their roles in cancer are not well understood. P4HA2 encodes a key enzyme involved in collagen synthesis, and its over-expression has been previously reported in papillary thyroid cancer [36]. THBS2 is a matricellular protein that encodes an adhesive glycoprotein and interacts with other proteins to modulate cell-matrix interactions [37]. Interestingly, THBS2 is associated with tumor growth in adult mouse tissues [37]. The two other genes in our OSCC recurrence signature (COL4A1 and MMP1) are better characterized in cancer. COL4A1 encodes the major type IV alpha collagen chain and is one of the main components of basement membranes. Basement membranes have several important biological roles, and are essential for embryonic development, proper tissue architecture, and tissue remodeling [38]. COL4A1 binds other collagens (COL4A2, 3, 4, 5 and 6), as well as LAMC2 (laminin, gamma 2), TGFB1 (transforming growth factor, beta 1), among other proteins (Figure 2) http://www.ihop-net.org, playing a relevant role in extracellular matrix-receptor interaction and focal adhesion [39]. The extracellular matrix undergoes constant remodeling; during this process, proteins such as MMP1 can degrade the extracellular matrix proteins (e.g., collagen IV), and contribute to invasion and metastasis [40]. In cancer, combined over-expression of COL4A1 and LAMC2 can distinguish OSCC from clinically normal oral cavity/oropharynx tissues [41]; this latter study suggests that COL4A1 over-expression may be a useful biomarker for early detection of malignancy.
MMP1 belongs to the family of matrix metalloproteases, which are key proteases involved in extracellular matrix (ECM) degradation [42]. MMP1 encodes a collagenase, which is secreted by tumor cells as well as by stromal cells stimulated by the tumor; this secreted enzyme is responsible for breaking down interstitial collagens type I, II and III in normal physiological processes (e.g., tissue remodeling) as well as disease processes (e.g., cancer) [42]. It is believed that the mechanism of up-regulation of most of the MMPs is likely due to transcriptional changes, which may occur following alterations in oncogenes and/or tumor suppressor genes [42]. Indeed, MMP1 has been previously reported as consistently over-expressed in oral carcinoma compared to adjacent normal tissues [43–45] and suggested as a biomarker of malignant transformation in precursor lesions in oral cancer [44, 46, 47]. In HNSCC, over-expression of several genes with roles in invasion and metastasis, including MMPs, were previously associated with treatment failure of HNSCC [48]. In our study, MMP1 was over-expressed in a subset of margins exclusively from patients with recurrent OSCC, and showed the highest fold-change of up-regulation in OSCC compared to margins. Our results are in agreement with the literature findings of MMP1 over-expression associated with progression of pre-malignant oral lesions; in this context, our data showing MMP1 over-expression in histologically normal surgical margins from patients who developed local recurrence support the notion that MMP1 may be involved in initial steps of cellular transformation and tumorigenesis, as well as invasion of oral carcinoma cells. Indeed, matrix metalloproteinases play an important role not only in invasion and metastasis but also in early stages of cancer development/progression [42].
Our data suggest that histologically normal surgical resection margins that over-express MMP1, COL4A1, THBS2 and P4HA2 are indicative of an increased risk of recurrence in OSCC. Patients at higher risk of recurrence could potentially benefit from closer disease monitoring and/or adjuvant post-operative radiation treatment, even in the absence of other clinical and histopathological indicators, such as advanced disease stage and perineural invasion.
Conclusion
Considering that our 4-gene signature showed prognostic value for recurrence in two independent patient cohorts, over-expression of this signature may be used for molecular analysis of histologically negative margins in oral carcinoma.
References
Parkin DM, Pisani P, Ferlay J: Global cancer statistics. CA Cancer J Clin. 1999, 49 (1): 33-64. 10.3322/canjclin.49.1.33. 31
Sawair FA, Irwin CR, Gordon DJ, Leonard AG, Stephenson M, Napier SS: Invasive front grading: reliability and usefulness in the management of oral squamous cell carcinoma. J Oral Pathol Med. 2003, 32 (1): 1-9. 10.1034/j.1600-0714.2003.00060.x.
Jones AS, Bin Hanafi Z, Nadapalan V, Roland NJ, Kinsella A, Helliwell TR: Do positive resection margins after ablative surgery for head and neck cancer adversely affect prognosis? A study of 352 patients with recurrent carcinoma following radiotherapy treated by salvage surgery. Br J Cancer. 1996, 74 (1): 128-132. 10.1038/bjc.1996.327.
Leemans CR, Tiwari R, Nauta JJ, van der Waal I, Snow GB: Recurrence at the primary site in head and neck cancer and the significance of neck lymph node metastases as a prognostic factor. Cancer. 1994, 73 (1): 187-190. 10.1002/1097-0142(19940101)73:1<187::AID-CNCR2820730132>3.0.CO;2-J.
Brandwein-Gensler M, Teixeira MS, Lewis CM, Lee B, Rolnitzky L, Hille JJ, Genden E, Urken ML, Wang BY: Oral squamous cell carcinoma: histologic risk assessment, but not margin status, is strongly predictive of local disease-free and overall survival. Am J Surg Pathol. 2005, 29 (2): 167-178. 10.1097/01.pas.0000149687.90710.21.
Nathan CA, Amirghahri N, Rice C, Abreo FW, Shi R, Stucker FJ: Molecular analysis of surgical margins in head and neck squamous cell carcinoma patients. Laryngoscope. 2002, 112 (12): 2129-2140. 10.1097/00005537-200212000-00003.
Bilde A, von Buchwald C, Dabelsteen E, Therkildsen MH, Dabelsteen S: Molecular markers in the surgical margin of oral carcinomas. J Oral Pathol Med. 2009, 38 (1): 72-78.
Nathan CA, Liu L, Li BD, Abreo FW, Nandy I, De Benedetti A: Detection of the proto-oncogene eIF4E in surgical margins may predict recurrence in head and neck cancer. Oncogene. 1997, 15 (5): 579-584. 10.1038/sj.onc.1201216.
Nathan CA, Franklin S, Abreo FW, Nassar R, De Benedetti A, Glass J: Analysis of surgical margins with the molecular marker eIF4E: a prognostic factor in patients with head and neck cancer. J Clin Oncol. 1999, 17 (9): 2909-2914.
Tan HK, Saulnier P, Auperin A, Lacroix L, Casiraghi O, Janot F, Fouret P, Temam S: Quantitative methylation analyses of resection margins predict local recurrences and disease-specific deaths in patients with head and neck squamous cell carcinomas. Br J Cancer. 2008, 99 (2): 357-363. 10.1038/sj.bjc.6604478.
van der Toorn PP, Veltman JA, Bot FJ, de Jong JM, Manni JJ, Ramaekers FC, Hopman AH: Mapping of resection margins of oral cancer for p53 overexpression and chromosome instability to detect residual (pre)malignant cells. J Pathol. 2001, 193 (1): 66-72. 10.1002/1096-9896(2000)9999:9999<::AID-PATH785>3.0.CO;2-J.
van Houten VM, Leemans CR, Kummer JA, Dijkstra J, Kuik DJ, van den Brekel MW, Snow GB, Brakenhoff RH: Molecular diagnosis of surgical margins and local recurrence in head and neck cancer patients: a prospective study. Clin Cancer Res. 2004, 10 (11): 3614-3620. 10.1158/1078-0432.CCR-03-0631.
Goldenberg D, Harden S, Masayesva BG, Ha P, Benoit N, Westra WH, Koch WM, Sidransky D, Califano JA: Intraoperative molecular margin analysis in head and neck cancer. Arch Otolaryngol Head Neck Surg. 2004, 130 (1): 39-44.
Franklin S, Pho T, Abreo FW, Nassar R, De Benedetti A, Stucker FJ, Nathan CO: Detection of the proto-oncogene eIF4E in larynx and hypopharynx cancers. Arch Otolaryngol Head Neck Surg. 1999, 125 (2): 177-182.
Taioli E, Ragin C, Wang XH, Chen J, Langevin SM, Brown AR, Gollin SM, Garte S, Sobol RW: Recurrence in oral and pharyngeal cancer is associated with quantitative MGMT promoter methylation. BMC Cancer. 2009, 9: 354-10.1186/1471-2407-9-354.
van Houten VM, Tabor MP, van den Brekel MW, Kummer JA, Denkers F, Dijkstra J, Leemans R, van der Waal I, Snow GB, Brakenhoff RH: Mutated p53 as a molecular marker for the diagnosis of head and neck cancer. J Pathol. 2002, 198 (4): 476-486. 10.1002/path.1242.
Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F: A Model-Based Background Adjustment for Oligonucleotide Expression Arrays. Journal of the American Statistical Association. 2004, 99 (468): 909-917. 10.1198/016214504000000683.
Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, et al: Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33 (20): e175-10.1093/nar/gni179.
Gautier L, Cope L, Bolstad BM, Irizarry RA: affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004, 20 (3): 307-315. 10.1093/bioinformatics/btg405.
Hong F, Breitling R, McEntee CW, Wittner BS, Nemhauser JL, Chory J: RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics. 2006, 22 (22): 2825-2827. 10.1093/bioinformatics/btl476.
Falcon S, Gentleman R: Using GOstats to test gene lists for GO term association. Bioinformatics. 2007, 23 (2): 257-258. 10.1093/bioinformatics/btl567.
Zheng Q, Wang XJ: GOEAST: a web-based software toolkit for Gene Ontology enrichment analysis. Nucleic Acids Res. 2008, W358-363. 36 Web Server
Edgar R, Domrachev M, Lash AE: Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30 (1): 207-210. 10.1093/nar/30.1.207.
Toruner GA, Ulger C, Alkan M, Galante AT, Rinaggio J, Wilk R, Tian B, Soteropoulos P, Hameed MR, Schwalb MN, et al: Association between gene expression profile and tumor invasion in oral squamous cell carcinoma. Cancer Genet Cytogenet. 2004, 154 (1): 27-35. 10.1016/j.cancergencyto.2004.01.026.
Ye H, Yu T, Temam S, Ziober BL, Wang J, Schwartz JL, Mao L, Wong DT, Zhou X: Transcriptomic dissection of tongue squamous cell carcinoma. BMC Genomics. 2008, 9: 69-10.1186/1471-2164-9-69.
Kuriakose MA, Chen WT, He ZM, Sikora AG, Zhang P, Zhang ZY, Qiu WL, Hsu DF, McMunn-Coffran C, Brown SM, et al: Selection and validation of differentially expressed genes in head and neck cancer. Cell Mol Life Sci. 2004, 61 (11): 1372-1383. 10.1007/s00018-004-4069-0.
Sticht C, Freier K, Knopfle K, Flechtenmacher C, Pungs S, Hofele C, Hahn M, Joos S, Lichter P: Activation of MAP kinase signaling through ERK5 but not ERK1 expression is associated with lymph node metastases in oral squamous cell carcinoma (OSCC). Neoplasia. 2008, 10 (5): 462-470.
Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ, Woodworth CD, Connor JP, Haugen TH, Smith EM, et al: Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007, 67 (10): 4605-4619. 10.1158/0008-5472.CAN-06-3619.
Machado J, Reis PP, Zhang T, Simpson C, Xu W, Perez-Ordonez B, Goldstein DP, Brown DH, Gilbert RW, Gullane PJ, et al: Low prevalence of human papillomavirus in oral cavity carcinomas. Head Neck Oncol. 2010, 2: 6-10.1186/1758-3284-2-6.
Breitling R, Herzyk P: Rank-based methods as a non-parametric alternative of the T-statistic for the analysis of biological microarray data. J Bioinform Comput Biol. 2005, 3 (5): 1171-1189. 10.1142/S0219720005001442.
Bioinformatics and computational biology solutions using R and Bioconductor. [http://www.worldcat.org/isbn/0387251464]
McCarthy DJ, Smyth GK: Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics. 2009, 25 (6): 765-771. 10.1093/bioinformatics/btp053.
Goeman JJ: Penalized estimation in the Cox proportional hazards model. Biometrical journal Biometrische Zeitschrift. 2010, 52 (1): 70-84.
Tabor MP, Brakenhoff RH, van Houten VM, Kummer JA, Snel MH, Snijders PJ, Snow GB, Leemans CR, Braakhuis BJ: Persistence of genetically altered fields in head and neck cancer patients: biological and clinical implications. Clin Cancer Res. 2001, 7 (6): 1523-1532.
Ha PK, Califano JA: The molecular biology of mucosal field cancerization of the head and neck. Crit Rev Oral Biol Med. 2003, 14 (5): 363-369. 10.1177/154411130301400506.
Jarzab B, Wiench M, Fujarewicz K, Simek K, Jarzab M, Oczko-Wojciechowska M, Wloch J, Czarniecka A, Chmielik E, Lange D, et al: Gene expression profile of papillary thyroid cancer: sources of variability and diagnostic implications. Cancer Res. 2005, 65 (4): 1587-1597. 10.1158/0008-5472.CAN-04-3078.
Bornstein P, Kyriakides TR, Yang Z, Armstrong LC, Birk DE: Thrombospondin 2 modulates collagen fibrillogenesis and angiogenesis. J Investig Dermatol Symp Proc. 2000, 5 (1): 61-66. 10.1046/j.1087-0024.2000.00005.x.
Sado Y, Kagawa M, Naito I, Ueki Y, Seki T, Momota R, Oohashi T, Ninomiya Y: Organization and expression of basement membrane collagen IV genes and their roles in human disorders. J Biochem. 1998, 123 (5): 767-776.
Hoffmann R, Valencia A: A gene network for navigating the literature. Nat Genet. 2004, 36 (7): 664-10.1038/ng0704-664.
Tanzer ML: Current concepts of extracellular matrix. J Orthop Sci. 2006, 11 (3): 326-331. 10.1007/s00776-006-1012-2.
Chen C, Mendez E, Houck J, Fan W, Lohavanichbutr P, Doody D, Yueh B, Futran ND, Upton M, Farwell DG, et al: Gene expression profiling identifies genes predictive of oral squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2008, 17 (8): 2152-2162. 10.1158/1055-9965.EPI-07-2893.
Egeblad M, Werb Z: New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002, 2 (3): 161-174. 10.1038/nrc745.
Suhr ML, Dysvik B, Bruland O, Warnakulasuriya S, Amaratunga AN, Jonassen I, Vasstrand EN, Ibrahim SO: Gene expression profile of oral squamous cell carcinomas from Sri Lankan betel quid users. Oncol Rep. 2007, 18 (5): 1061-1075.
Yen CY, Chen CH, Chang CH, Tseng HF, Liu SY, Chuang LY, Wen CH, Chang HW: Matrix metalloproteinases (MMP) 1 and MMP10 but not MMP12 are potential oral cancer markers. Biomarkers. 2009, 14 (4): 244-249. 10.1080/13547500902829375.
Saleh A, Zain RB, Hussaini H, Ng F, Tanavde V, Hamid S, Chow AT, Lim GS, Abraham MT, Teo SH, et al: Transcriptional profiling of oral squamous cell carcinoma using formalin-fixed paraffin-embedded samples. Oral Oncol. 2010, 46 (5): 379-386. 10.1016/j.oraloncology.2010.02.022.
Jordan RC, Macabeo-Ong M, Shiboski CH, Dekker N, Ginzinger DG, Wong DT, Schmidt BL: Overexpression of matrix metalloproteinase-1 and -9 mRNA is associated with progression of oral dysplasia to cancer. Clin Cancer Res. 2004, 10 (19): 6460-6465. 10.1158/1078-0432.CCR-04-0656.
Estilo CL, P Oc, Talbot S, Socci ND, Carlson DL, Ghossein R, Williams T, Yonekawa Y, Ramanathan Y, Boyle JO, et al: Oral tongue cancer gene expression profiling: Identification of novel potential prognosticators by oligonucleotide microarray analysis. BMC Cancer. 2009, 9: 11-10.1186/1471-2407-9-11.
Ginos MA, Page GP, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE, Ondrey FG, Adams GL, Gaffney PM: Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004, 64 (1): 55-63. 10.1158/0008-5472.CAN-03-2144.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/11/437/prepub
Acknowledgements and funding
The authors acknowledge Dr. Jerry Machado and Dr. Mahadeo Sukhai for constructive criticism of this manuscript. This work was supported by the Ontario Institute for Cancer Research (OICR) (SKR, JI, PG, IJ, and BPO) and the Galloway Fund (RG, SKR), the Canada Research Chair Program (IJ) and the CIHR Catalyst Grant #202370 (IJ). Computational facilities were supported by the Canada Research Chair Program, Canada Foundation for Innovation (Grant #12301 and #203383), Ontario Research Fund (GL2-01-030), and IBM. This research was funded in part by the Ontario Ministry of Health and Long Term Care (MOHLTC). The views expressed do not necessarily reflect those of the MOHLTC.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PPR, BPO, IJ and SKR designed the study. LW, IJ and MP performed bioinformatics and statistical data analyses. PPR, LW, IJ and SKR drafted the manuscript. YX and NKC helped with quantitative PCR experiments and data interpretation. NNG, GCW and AAM helped with study design, identified patients and collected samples. NNG and AAM helped draft the manuscript. NNG, CS, DG, DB, RG, PG and JI helped identify patients, collected samples and provided detailed clinical data of patients. All authors read and approved the final manuscript.
Patricia P Reis, Levi Waldron contributed equally to this work.
Electronic supplementary material
12885_2011_2912_MOESM1_ESM.DOC
Additional file 1: Methods S1. Description of methods used for RNA isolation, oligonucleotide array experiments, quantitative real-time reverse-transcription PCR validation and protein-protein interaction network analysis. (DOC 42 KB)
12885_2011_2912_MOESM2_ESM.XLS
Additional file 2: Table S1. Results of meta-analysis of the five public data sets identified 667 up-regulated genes in OSCC. (XLS 1 MB)
12885_2011_2912_MOESM3_ESM.XLS
Additional file 3: Table S2. List of 138 over-expressed genes, with FDR < 0.01, identified in both the meta-analysis of public datasets and our microarray experiment. (XLS 234 KB)
12885_2011_2912_MOESM4_ESM.XLS
Additional file 4: Table S3. Results of Gene Ontology enrichment analysis of the 138 genes identified as over-expressed in both the public datasets and our microarray experiment. (XLS 502 KB)
12885_2011_2912_MOESM5_ESM.PDF
Additional file 5: GO biological function. Graphical representation of GO annotation (biological function). (PDF 377 KB)
12885_2011_2912_MOESM8_ESM.PNG
{kind=link}
Additional file 8: Figure S1. Distribution of nominal p-values for univariate association of the 138 genes identified as over-expressed in OSCC with recurrence. P-values were determined by Cox regression using the maximum expression in any margin of each patient. The empirical null distribution was determined from association of these same genes with 1,000 permutations of the outcome labels. The observed nominal p-values are significantly enriched for small values (p = 0.001, Kolmogorov-Smirnov test). It is worth noting that recurrence was not used at any stage in the selection of these genes. (PNG 6 KB)
12885_2011_2912_MOESM9_ESM.PNG
{kind=link}
Additional file 9: Figure S2. Survival Receiver Operating Characteristic curve for recurrence at 36 months in the test set. Using a high threshold to define high-risk patients predicts a majority of recurrences (true positives) at a low false-positive rate (20%). While we maintained the standard median cutoff for this study due to the limited sample size, a larger study in the future may be able to further tune the cutoff threshold to optimize sensitivity and specificity in the context of the relative risks that treatment options informed by this prognostic score entail. The area under the ROC curve (AUC) for recurrence within 36 months is 0.73, which is an improvement over the expected AUC of 0.5 for non-predictive risk scores. (PNG 9 KB)
12885_2011_2912_MOESM10_ESM.PNG
{kind=link}
Additional file 10: Figure S3. Bootstrap validation of four-gene signature risk score in training and validation sets. Density lines represent the distribution of hazard ratios observed in 1,000 re-samplings of a single margin, randomly chosen, from each patient. (PNG 83 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Reis, P.P., Waldron, L., Perez-Ordonez, B. et al. A gene signature in histologically normal surgical margins is predictive of oral carcinoma recurrence. BMC Cancer 11, 437 (2011). https://doi.org/10.1186/1471-2407-11-437
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-11-437