
Abstract
Background
Biomarkers that predict clinical response, tumor recurrence or patient survival are severely lacking for most cancers, particularly for oral and pharyngeal cancer. This study examines whether gene-promoter methylation of tumor DNA correlates with survival and recurrence rates in a population of patients with oral or pharyngeal cancer.
Methods
The promoter methylation status of the DNA repair gene MGMT and the tumor suppressor genes CDKN2A and RASSF1 were evaluated by methylation-specific PCR in 88 primary oral and pharyngeal tumors and correlated with survival and tumor recurrence. Quantitative MGMT methylation was also assessed.
Results
29.6% of the tumors presented with MGMT methylation, 11.5% with CDKN2A methylation and 12.1% with RASSF1 methylation. MGMT promoter methylation was significantly associated with poorer overall and disease-free survival. No differences in methylation status of MGMT and RASSF1 with HPV infection, smoking or drinking habits were observed. A significant inverse trend with the amount of MGMT methylation and overall and disease-free survival was observed (ptrend = 0.002 and 0.001 respectively).
Conclusion
These results implicate MGMT promoter methylation as a possible biomarker for oral and pharyngeal cancer prognosis. The critical role of MGMT in DNA repair suggests that defective DNA repair may be correlative in the observed association between MGMT promoter methylation and tumor recurrence. Follow-up studies should include further quantitative MSP-PCR measurement, global methylation profiling and detailed analysis of downstream DNA repair genes regulated by promoter methylation.
Similar content being viewed by others


Background
Oral and pharyngeal squamous cell carcinoma is the most common malignant neoplasm of the head and neck region [1]. An estimated 34,360 new patients were diagnosed with squamous cell carcinoma of the head and neck and 7,550 deaths occurred in the U.S. in 2007, accounting for 2-3% of all malignancies [2]. About 30-50% of oral and pharyngeal cancer patients will develop a recurrence and/or a second primary tumor within two decades of the initial diagnosis. The five-year survival rate for oral cavity cancer has changed little between 1975 and 2002, approximately 48% [2]. Despite the progress made in early detection and therapy, early predictors of cancer recurrence at time of diagnosis are still missing for oral and pharyngeal cancer.
Epigenetic changes in cancer have traditionally been evaluated by measuring the status of CpG island cytosine methylation of a particular gene, such as MGMT [3]. It has been well-documented in cell lines, xenografts and in clinical trials, that methylation at discrete regions within CpG islands of a given gene promoter results in gene silencing and therefore prevents expression of the corresponding gene [3]. Several genes (oncogenes, tumor suppressor genes, DNA repair genes and growth control genes) have been shown to have an aberrant methylation profile (promoter hypermethylation) in tumors as compared to normal tissue or blood cells or cells in bodily fluids [4–6]. The altered DNA methylation landscape of the cancer epigenome is not limited to promoter hypermethylation of select genes, but also includes global hypomethylation as a prelude to oncogene activation and genome instability [7–9].
It has been suggested that aberrant methylation patterns could therefore act as a selective factor on neoplastic cells, influencing patients' survival and prognosis, particularly if methylation affects expression of a tumor suppressor gene such as CDKN2A, RASSF1 or a DNA repair gene such as MGMT. For example, aberrant gene hypermethylation of CDKN2A, p14ARF [10], RASSF1 [11–13] and the DNA repair gene MGMT [3] have been reported in tumor tissue of oral cavity cancer patients (see Table 1 for a more comprehensive list of genes methylated in oral cancers). In addition, promoter methylation of CDKN2A, MGMT, DAPK1, and CDH1 has been studied in relation to head and neck cancer survival [14–17]. However, the role of hypermethylation on outcome in oral and pharyngeal cancer patients, a special subset of head and neck cancers, has not yet been addressed.
Oral and pharyngeal cancers are for the large part squamous cell histological types, and are often anatomically grouped with head and neck cancers. Although the head and neck tumors have been historically grouped together due to similar etiology, the oral cavity, pharynx and larynx are unique structures with different functions and possibly different sensitivities to carcinogens, especially alcohol and tobacco. Human Papillomavirus is another etiologic agent involved in oral and pharyngeal tumors [18, 19]. In the study described herein, we tested the association between promoter methylation and survival in a cohort of 88 oral and pharyngeal cancer patients, focusing on three target genes: the DNA repair gene MGMT and the tumor suppressor genes CDKN2A and RASSF1.
Methods
Study population
A database of head and neck cancer cases was established in June 2004 at the University of Pittsburgh Medical Center [19] for patients undergoing surgical resection of their tumors between November 1992 and February 1997. Demographics, smoking and alcohol use, family history, tumor site, clinical characteristics of the initial primary tumor and subsequent tumors, follow-up data (such as disease outcome and time to next disease occurrence) through June 2007 were obtained from the University of Pittsburgh Tumor Registry and from the review of clinical charts. Patients' clinical and demographic information was re-verified from each patient's original de-identified and coded questionnaires, pathology and surgical reports. This database contains clinical, treatment and follow-up information for the first surgical resection at the time of enrollment and all subsequent resections for all patients enrolled in the study. Tumor site, histology, stage and grade were classified according to the American Joint Committee on Cancer (AJCC) ICD9 codes, ICD-morphology, stage and grade classification, respectively. Samples were collected under IRB approval of the University of Pittsburgh Head and Neck SPORE tissue bank. All tumors samples were consented by the SPORE tissue bank and IRB approval is for data analysis as described herein.
Information on 88 subjects undergoing surgical resection of their first primary tumors was selected from the original database; HPV status was determined for all the tumors by PCR as previously described [19]. Sites of the oral cavity included cheek, retromolar area, alveolar ridge, oral tongue, palate, floor of mouth and overlapping lesions of other and unspecified parts of the mouth. The oropharynx included sites involving the base of tongue, soft palate, tonsil and overlapping lesions of the oral cavity and pharynx.
Methylation-specific PCR
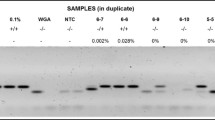
Previously extracted DNA from tumors was utilized in this study. DNA was isolated from primary fresh-frozen tumor tissue by guanidine thiocyanate extraction using the commercially available IsoQuick kit (MicroProbe, Garden Grove, CA) as we have described previously [20]. Methylation-specific PCR was used for the analysis of MGMT, RASSF1 and CDKN2A promoter methylation as described [3, 21–24] using the Zymo Research EZ DNA Methylation-Gold reagents (ZYMO Research) according to the manufacturer's instructions. The primers used to assess MGMT promoter methylation status were: Methylated MGMT allele - tttcgacgttcgtaggttttcgc and gcactcttccgaaaacgaaacg; annealing temperature = 58°C; expected amplicon size = 81 bp. Un-methylated MGMT allele - tttgtgttttgatgtttgtaggtttttgt and aactccacactcttccaaaaacaaaaca; annealing temperature = 58°C; expected amplicon size = 93 bp [3]. The primer sequences to assess RASSF1 and CDKN2A methylation status were previously described [17, 25]. A DNA methylated control using either Methylated or Unmethylated DNA (Chemicon; Millipore) is included in all the DNA modification and PCR reactions. PCR reaction products were then separated on 4% agarose gels containing ethidium bromide with 100 bp DNA Ladder Markers (Bioline) and examined under ultraviolet illumination to identify the distinct bands. Each PCR reaction was run in duplicate.
Quantitative Methylation Analysis
To determine the degree of methylation among subjects found to have MGMT hypermethylation by MSP, quantitative methylation analysis was performed using a pyrosequencing methylation assay [26, 27]. DNA samples that yielded a positive result in the MSP assay were then subjected to a quantitative methylation test using the PyroMark MGMT ID system, as per the manufacturer's instructions (Biotage, Inc.). Briefly, DNA was bisulfite treated using the Zymo Research EZ DNA Methylation-Gold reagents as indicated above. The treated DNA was amplified by PCR with MGMT-specific primers and twenty microliters of the amplicon was then subjected to pyrosequencing using the Biotage PyroMark Q24 System. CpG site quantification was performed with the Biotage methylation Software PyroQ-CpG™. The average percent methylation for MGMT was calculated from direct measurement of the percentage of methylation at five individual CpG sites.
Statistical analyses
Follow-up, demographic, clinical and laboratory data for the study population was extracted from the head and neck database and imported to a statistical software package for analysis. Statistical analyses were performed using the Intercooled STATA (version 8.2) software (StataCorp. LP, College Station TX). Vital status and recurrence were the primary statistical endpoints for survival. Overall survival was defined as the time period between the surgical resection of the initial primary tumor and death. Disease-free survival was defined as the time period between the surgical resection of the initial primary and tumor recurrence. All patients lost to follow-up were censored. Kaplan-Meier survivor functions for overall and disease-free survival were generated using STATA. The log-rank statistic was used to test the equality of survivor functions. Multivariable Cox proportional hazards models were created for overall and disease-free survival for both MSP data for CDKN2A, RASSF1A, and MGMT; and quantitative MGMT methylation data generated from the pyrosequencing assay. For the quantitative data models, degree of MGMT methylation status was classified as unmethylated, methylation index ≤ 6.9%, and methylation index > 6.9%, based on the median of the whole population, and was analyzed as a categorical variable. Age, race, gender, smoking status, alcohol consumption, treatment and stage at diagnosis were considered for inclusion in the model. All covariates with P < 0.25 were retained in the final model.
Results
Patient demographics
The description of the population under study is reported in Table 2. As expected from the epidemiology of oral and pharyngeal cancer, there was a 2:1 male:female ratio. The majority of the subjects were ever smokers (87.5%) and ever drinkers (81.8%). The average follow up was 65.8 ± 48.9 months. During the follow-up, 42.0% of the patients developed a recurrence, and 53.4% died. More than half of the patients (59%) were in stage III or IV at diagnosis; half of the patients were treated with surgery alone, half with a combination of surgery and radiotherapy and/or chemotherapy.
MGMT, CDKN2A and RASSF1 methylation status and outcome
Promoter methylation at MGMT was seen in 29.6% (26/88) of the tumors, 11.5% (7/61) showed CDKN2A methylation and 12.1% (8/66) RASSF1 methylation. The distribution of methylation status in the three genes studied according to several personal and clinical characteristics is presented in Table 3. Methylation of CDKN2A was significantly more frequent in never drinkers, and in less severe stages at diagnosis. Conversely, no association was observed between methylation status of MGMT, or RASSF1 and any of the variables analyzed, including smoking or drinking habits, stage at diagnosis or family history of cancer. MGMT promoter methylation was significantly inversely associated with overall survival (p = 0.049; Figure 1A) and disease-free survival (p = 0.013; Figure 1B). A non-significant association of promoter hypermethylation and both recurrence and survival was observed for CDKN2A (data not shown).

MGMT promoter methylation status. Methylation at the MGMT promoter was determined by methylation specific PCR, followed by gel electrophoresis. Shown are the plots of (A) overall patient survival time (months) and (B) disease-free survival time (months), according to MGMT methylation status (unmethylated or methylated).
In the multivariate Cox models, MGMT hypermethylation was significantly inversely associated with overall survival (HR = 2.17, 95% CI: 1.11-4.23), after adjustment for age and stage at diagnosis, smoking, alcohol consumption; and disease-free survival (HR = 3.49, 95% CI: 1.62-7.52), adjusting for age and stage at diagnosis, smoking, alcohol consumption, treatment. There was no association between hypermethylation of CDKN2A or RASSF1 and overall or disease-free survival (Table 4).
For a subset of the population (n = 61) information on methylation status of all three genes was available (Table 5). Only 1.6% of the samples showed simultaneous hypermethylation of MGMT, CDKN2A, and RASSF1, while 8.2% of the samples reported methylation in two of the investigated genes. These frequencies do not diverge significantly from expected frequencies derived from a random combination of methylated genes.
All of the HPV-positive tumors carried non-methylated CDKN2A promoters, while no differences in methylation status of MGMT and RASSF1 with HPV infection was observed.
Methylation status in peripheral blood
No RASSF1 or CDKN2A methylation was observed in DNA extracted from peripheral blood lymphocytes, while one sample (1.3%), a T4N0 M0 (stage IVa) squamous cancer of the oral cavity, showed MGMT methylation in the DNA sample obtained from blood cells.
Quantitative methylation analysis of MGMT
We were able to obtain pyrosequencing results for 20/26 samples that tested positive for MGMT promoter methylation by MSP (3 samples had no remaining DNA; and 3 resulted as "not determined"). The median MtI value was 6.9% methylation, which ranged from 0.6% to 52.6%. Patients with high levels of MGMT promoter methylation (> 6.9%; n = 10) showed an overall significant decreased survival relative to patients with no MGMT hypermethylation (HR = 4.38, 95% CI: 1.78-3.91), after adjustment for age, gender, alcohol consumption, and stage at diagnosis. There was no association between the presence of less than or equal to 6.9% methylation (n = 10) and overall survival (HR = 1.52, 95% CI: 0.59-3.91). A significant inverse trend in survival with the amount of MGMT methylation was observed (ptrend = 0.002; Table 4). Similar differences were observed in the Kaplan-Meier survival function (p = 0.025; Figure 2A).

Degree of MGMT promoter methylation. The degree of methylation at the MGMT promoter was determined by pyrosequencing (Pyromark MGMT ID system). Shown are the plots of (A) overall patient survival time (months) and (B) disease-free survival time (months), according to the degree of MGMT methylation (no detectable methylation or a level of methylation either greater than or less than 6.9%).
The degree of MGMT hypermethylation was also associated with poorer disease-free survival (ptrend = 0.001). Those with the highest degree of MGMT hypermethylation experienced poorer disease-free survival (HR = 5.46, 95% CI: 1.75-17.00) than patients with ≤ 6.9% MGMT methylation (HR = 3.03, 95% CI: 1.24-7.44) relative to subjects with no MGMT hypermethylation. A positive inverse trend was observed between the degree of MGMT methylation and disease-free survival (ptrend = 0.001). These results were observed in the unadjusted Kaplan-Meier survival function as well (p = 0.046; Figure 2B).
Discussion
Current research has shown that a number of tumor suppressor genes may be inactivated not only by genetic mechanisms such as deletions or point mutations, but also by hypermethylation or other similar epigenetic mechanisms [28] (see Table 1). While it is unclear whether environmental factors are responsible for gene hypermethylation, it has become clear that the presence of hypermethylation could be one of the predictors of prognosis [14–17, 29, 30]. We have analyzed here promoter methylation in three genes that have been involved in head and neck cancer prognosis, but focused our attention on the predictive values of these epigenetic events in cancer of the oral cavity and pharynx. Our results suggest that MGMT hypermethylation is one of the prognostic factors in oral and pharyngeal cancer patients' survival; similar findings were reported for head and neck patients in general by some authors [21, 23] but not by others [24].
Among the strengths of our study is the well characterized population of oral and pharyngeal cancers, which has detailed baseline epidemiological data on risk factors including HPV testing and complete therapy information, as well as complete follow-up data. In addition, a comparison between tissue and peripheral blood methylation from the same patient was possible because of the availability of both biological samples. A possible weakness of this study is that although this is the largest oral and pharyngeal cancer study using follow up data in relation to promoter methylation, the sample size is still relatively small. Additionally, the tumor samples used were not microdissected, leaving open the possibility of contamination from surrounding normal tissue and stroma, although previous studies have demonstrated that MGMT is not generally methylated in normal head and neck tissue [31, 32].
In our cohort of patients, a methylation frequency similar to that reported in the literature was observed for MGMT (25-52%) and for RASSF1 (0-8%), while a lower value for CDKN2A (23-67%) was observed [11]. It should be noted that the studies recently reviewed [11] were conducted on small populations from different ethnic backgrounds. It is possible that environmental exposures as well as distinct genetic pathways to cancer development may explain the wide range of frequencies reported in the literature [33]. We also analyzed the tumor methylation pattern of the combination of the three genes under study. We observed that in roughly half of the tumors, none of the three genes were methylated, while in a small proportion of the tissues (roughly 10%), at least two of the genes under study were hypermethylated. This suggests that different carcinogenetic pathways may be present in tumors that are otherwise similar for histology and location. Unfortunately, the small size of the study does not allow any further speculation on this issue. CDKN2A was not methylated in any of the HPV-positive samples. This observation is consistent with the hypothesis that HPV-associated head and neck cancers have a distinct etiology, since HPV-positive tumors are more likely to over-express p16 [34].
No direct association between promoter hypermethylation in MGMT or RASSF1 and age, sex, smoking, alcohol drinking, or tumor stage was observed, in agreement with recently reported data [16, 17, 35]. A possible association between drinking and stage with methylation of CDKN2A was observed, although small sample size requires confirmation of this result. The comparison of the findings in the tumor tissue and in the peripheral blood from the same patient showed no RASSF1 or CDKN2A methylation in any of the peripheral blood samples, and only one sample positive for MGMT methylation; a T4N0 M0 (stage IVa) squamous cancer of the oral cavity. Although no metastasis was identified at diagnosis, it is conceivable that the positive blood samples were due to circulating tumor cells from the advanced stage tumor. These results indicate that hypermethylation is tumor specific and is not a general characteristic of other tissues or cell types of the individual at risk for tumor recurrence.
Epigenetic changes in cancer, more recently referred to as the cancer epigenome, have traditionally been evaluated by measuring the status of CpG island cytosine methylation of a particular gene such as MGMT [3] using Methylation-Specific PCR (MSP-PCR). MSP-PCR is a well-established, straightforward and rapid PCR-based method for analysis of promoter methylation and gene silencing. However, MSP-PCR is not quantitative and therefore it was of interest to determine whether variable amounts of MGMT promoter methylation could be detected in these tumors as a function of biological characteristics of the tumor. To assess this, a quantitative pyrosequencing methylation assay was employed. The tumors found to be positively methylated by methylation-specific PCR indeed showed variable methylation levels ranging from 0.6% to 46.6% with a median level of 6.9%. This is in agreement with the findings of Shaw and colleagues [29], who report MGMT percent methylation ranging from 0.0%-45% among 37 head and neck tumor samples. The association between degree of MGMT hypermethylation and oral and pharyngeal cancer survival has not been reported before to our knowledge. Indirect evidence in support of our findings comes from previous reports on a negative correlation between MGMT mRNA levels and percent promoter methylation in head and neck tumors [26, 36].
Conclusion
We evaluated 88 primary oral and pharyngeal tumors for methylation of the promoter for the DNA repair gene MGMT and the tumor suppressor genes CDKN2A and RASSF1 using methylation-specific PCR. Further, quantitative MGMT methylation was assessed and these results were then correlated with survival and tumor recurrence. MGMT promoter methylation was significantly associated with poorer overall and disease-free survival and a significant trend with the amount of MGMT methylation and survival and recurrence was observed (ptrend = 0.002 and 0.001 respectively). These results implicate MGMT promoter methylation as a possible biomarker for oral and pharyngeal cancer prognosis. It is therefore conceivable that as the degree of methylation in head and neck cancers increases, genomic stability declines as a result of decreasing MGMT expression, thus leading to poorer prognosis and tumor recurrence. Further, the altered DNA methylation landscape of the cancer epigenome is not limited to promoter hypermethylation of select genes but also includes global hypomethylation as a prelude to oncogene activation and genome instability [7–9]. The critical role of MGMT in DNA repair suggests that defective DNA repair may be correlative in the observed association between MGMT promoter methylation and tumor recurrence. We therefore suggest that follow-up studies include (i) further quantitative methylation analysis, such as the MethyLight [37–42] or pyrosequencing assay [26, 27], (ii) a measurement of global methylation status in promoters and non-promoter regions [43] and (iii) a detailed analysis of downstream DNA repair genes regulated by methylation such as MLH1, MSH2, ATM etc. in an effort to identify a DNA repair gene methylation signature that may be responsive to patient outcome.
Abbreviations
- MGMT:
-
O6-methylguanine-DNA methyltransferase
- MSP-PCR:
-
Methylation-Specific PCR
- HPV:
-
human papillomavirus.
References
Funk GF, Karnell LH, Robinson RA, Zhen WK, Trask DK, Hoffman HT: Presentation, treatment, and outcome of oral cavity cancer: a National Cancer Data Base report. Head Neck. 2002, 24 (2): 165-180. 10.1002/hed.10004.
Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ: Cancer statistics, 2007. CA Cancer J Clin. 2007, 57 (1): 43-66. 10.3322/canjclin.57.1.43.
Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG: Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999, 59 (4): 793-797.
Reed AL, Califano J, Cairns P, Westra WH, Jones RM, Koch W, Ahrendt S, Eby Y, Sewell D, Nawroz H, et al: High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res. 1996, 56 (16): 3630-3633.
Maruya S, Issa JP, Weber RS, Rosenthal DI, Haviland JC, Lotan R, El-Naggar AK: Differential methylation status of tumor-associated genes in head and neck squamous carcinoma: incidence and potential implications. Clin Cancer Res. 2004, 10 (11): 3825-3830. 10.1158/1078-0432.CCR-03-0370.
Sanchez-Cespedes M, Esteller M, Wu L, Nawroz-Danish H, Yoo GH, Koch WM, Jen J, Herman JG, Sidransky D: Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res. 2000, 60 (4): 892-895.
Feinberg AP, Ohlsson R, Henikoff S: The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006, 7 (1): 21-33. 10.1038/nrg1748.
Wilson IM, Davies JJ, Weber M, Brown CJ, Alvarez CE, MacAulay C, Schubeler D, Lam WL: Epigenomics: mapping the methylome. Cell Cycle. 2006, 5 (2): 155-158. 10.4161/cc.5.2.2367.
Ting AH, McGarvey KM, Baylin SB: The cancer epigenome--components and functional correlates. Genes Dev. 2006, 20 (23): 3215-3231. 10.1101/gad.1464906.
Shintani S, Nakahara Y, Mihara M, Ueyama Y, Matsumura T: Inactivation of the p14(ARF), p15(INK4B) and p16(INK4A) genes is a frequent event in human oral squamous cell carcinomas. Oral Oncol. 2001, 37 (6): 498-504. 10.1016/S1368-8375(00)00142-1.
Ha PK, Califano JA: Promoter methylation and inactivation of tumour-suppressor genes in oral squamous-cell carcinoma. Lancet Oncol. 2006, 7 (1): 77-82. 10.1016/S1470-2045(05)70540-4.
Huang KH, Huang SF, Chen IH, Liao CT, Wang HM, Hsieh LL: Methylation of RASSF1A, RASSF2A, and HIN-1 is associated with poor outcome after radiotherapy, but not surgery, in oral squamous cell carcinoma. Clin Cancer Res. 2009, 15 (12): 4174-4180. 10.1158/1078-0432.CCR-08-2929.
Tran TN, Liu Y, Takagi M, Yamaguchi A, Fujii H: Frequent promoter hypermethylation of RASSF1A and p16INK4a and infrequent allelic loss other than 9p21 in betel-associated oral carcinoma in a Vietnamese non-smoking/non-drinking female population. J Oral Pathol Med. 2005, 34 (3): 150-156. 10.1111/j.1600-0714.2004.00292.x.
Wang J, Qin Y, Li B, Sun Z, Yang B: Detection of aberrant promoter methylation of GSTP1 in the tumor and serum of Chinese human primary hepatocellular carcinoma patients. Clin Biochem. 2006, 39 (4): 344-348. 10.1016/j.clinbiochem.2006.01.008.
Liu K, Zuo C, Luo QK, Suen JY, Hanna E, Fan CY: Promoter hypermethylation and inactivation of hMLH1, a DNA mismatch repair gene, in head and neck squamous cell carcinoma. Diagn Mol Pathol. 2003, 12 (1): 50-56. 10.1097/00019606-200303000-00007.
Qian XC, Brent TP: Methylation hot spots in the 5' flanking region denote silencing of the O6-methylguanine-DNA methyltransferase gene. Cancer Res. 1997, 57 (17): 3672-3677.
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB: Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996, 93 (18): 9821-9826. 10.1073/pnas.93.18.9821.
Hobbs CG, Sterne JA, Bailey M, Heyderman RS, Birchall MA, Thomas SJ: Human papillomavirus and head and neck cancer: a systematic review and meta-analysis. Clin Otolaryngol. 2006, 31 (4): 259-266. 10.1111/j.1749-4486.2006.01246.x.
Ragin CC, Taioli E, Weissfeld JL, White JS, Rossie KM, Modugno F, Gollin SM: 11q13 amplification status and human papillomavirus in relation to p16 expression defines two distinct etiologies of head and neck tumours. Br J Cancer. 2006, 95 (10): 1432-1438. 10.1038/sj.bjc.6603394.
Law JC, Whiteside TL, Gollin SM, Weissfeld J, El-Ashmawy L, Srivastava S, Landreneau RJ, Johnson JT, Ferrell RE: Variation of p53 mutational spectra between carcinoma of the upper and lower respiratory tract. Clin Cancer Res. 1995, 1 (7): 763-768.
Zuo C, Ai L, Ratliff P, Suen JY, Hanna E, Brent TP, Fan CY: O6-methylguanine-DNA methyltransferase gene: epigenetic silencing and prognostic value in head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2004, 13 (6): 967-975.
Ai L, Stephenson KK, Ling W, Zuo C, Mukunyadzi P, Suen JY, Hanna E, Fan CY: The p16 (CDKN2a/INK4a) tumor-suppressor gene in head and neck squamous cell carcinoma: a promoter methylation and protein expression study in 100 cases. Mod Pathol. 2003, 16 (9): 944-950. 10.1097/01.MP.0000085760.74313.DD.
Puri SK, Si L, Fan CY, Hanna E: Aberrant promoter hypermethylation of multiple genes in head and neck squamous cell carcinoma. Am J Otolaryngol. 2005, 26 (1): 12-17. 10.1016/j.amjoto.2004.06.007.
Dikshit RP, Gillio-Tos A, Brennan P, De Marco L, Fiano V, Martinez-Penuela JM, Boffetta P, Merletti F: Hypermethylation, risk factors, clinical characteristics, and survival in 235 patients with laryngeal and hypopharyngeal cancers. Cancer. 2007, 110 (8): 1745-1751. 10.1002/cncr.22975.
Burbee DG, Forgacs E, Zochbauer-Muller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, et al: Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst. 2001, 93 (9): 691-699. 10.1093/jnci/93.9.691.
Shaw RJ, Hall GL, Lowe D, Liloglou T, Field JK, Sloan P, Risk JM: The role of pyrosequencing in head and neck cancer epigenetics: correlation of quantitative methylation data with gene expression. Archives of otolaryngology--head & neck surgery. 2008, 134 (3): 251-256.
Tost J, Gut IG: DNA methylation analysis by pyrosequencing. Nature protocols. 2007, 2 (9): 2265-2275. 10.1038/nprot.2007.314.
Esteller M: Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007, 16 (Spec No 1): R50-59. 10.1093/hmg/ddm018.
Viswanathan M, Tsuchida N, Shanmugam G: Promoter hypermethylation profile of tumor-associated genes p16, p15, hMLH1, MGMT and E-cadherin in oral squamous cell carcinoma. Int J Cancer. 2003, 105 (1): 41-46. 10.1002/ijc.11028.
Li J, El-Naggar A, Mao L: Promoter methylation of p16INK4a, RASSF1A, and DAPK is frequent in salivary adenoid cystic carcinoma. Cancer. 2005, 104 (4): 771-776. 10.1002/cncr.21215.
Kato K, Hara A, Kuno T, Mori H, Yamashita T, Toida M, Shibata T: Aberrant promoter hypermethylation of p16 and MGMT genes in oral squamous cell carcinomas and the surrounding normal mucosa. Journal of cancer research and clinical oncology. 2006, 132 (11): 735-743. 10.1007/s00432-006-0122-8.
Righini CA, de Fraipont F, Timsit JF, Faure C, Brambilla E, Reyt E, Favrot MC: Tumor-specific methylation in saliva: a promising biomarker for early detection of head and neck cancer recurrence. Clin Cancer Res. 2007, 13 (4): 1179-1185. 10.1158/1078-0432.CCR-06-2027.
Moore LE, Huang WY, Chung J, Hayes RB: Epidemiologic considerations to assess altered DNA methylation from environmental exposures in cancer. Ann N Y Acad Sci. 2003, 983: 181-196. 10.1111/j.1749-6632.2003.tb05973.x.
Vang R, Gown AM, Farinola M, Barry TS, Wheeler DT, Yemelyanova A, Seidman JD, Judson K, Ronnett BM: p16 expression in primary ovarian mucinous and endometrioid tumors and metastatic adenocarcinomas in the ovary: utility for identification of metastatic HPV-related endocervical adenocarcinomas. Am J Surg Pathol. 2007, 31 (5): 653-663. 10.1097/01.pas.0000213369.71676.25.
Shaw RJ, Liloglou T, Rogers SN, Brown JS, Vaughan ED, Lowe D, Field JK, Risk JM: Promoter methylation of P16, RARbeta, E-cadherin, cyclin A1 and cytoglobin in oral cancer: quantitative evaluation using pyrosequencing. Br J Cancer. 2006, 94 (4): 561-568. 10.1038/sj.bjc.6602972.
Murakami J, Asaumi J, Maki Y, Tsujigiwa H, Nagatsuka H, Kokeguchi S, Inoue T, Kawasaki S, Tanaka N, MacPhee D, et al: Influence of CpG island methylation status in O6-methylguanine-DNA methyltransferase expression of oral cancer cell lines. Oncol Rep. 2004, 12 (2): 339-345.
Ogino S, Kawasaki T, Brahmandam M, Cantor M, Kirkner GJ, Spiegelman D, Makrigiorgos GM, Weisenberger DJ, Laird PW, Loda M, et al: Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn. 2006, 8 (2): 209-217. 10.2353/jmoldx.2006.050135.
Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW: MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28 (8): E32-10.1093/nar/28.8.e32.
Zeschnigk M, Bohringer S, Price EA, Onadim Z, Masshofer L, Lohmann DR: A novel real-time PCR assay for quantitative analysis of methylated alleles (QAMA): analysis of the retinoblastoma locus. Nucleic Acids Res. 2004, 32 (16): e125-10.1093/nar/gnh122.
Bastian PJ, Ellinger J, Heukamp LC, Kahl P, Muller SC, von Rucker A: Prognostic value of CpG island hypermethylation at PTGS2, RAR-beta, EDNRB, and other gene loci in patients undergoing radical prostatectomy. Eur Urol. 2007, 51 (3): 665-674. 10.1016/j.eururo.2006.08.008. discussion 674
Grote HJ, Schmiemann V, Geddert H, Rohr UP, Kappes R, Gabbert HE, Bocking A: Aberrant promoter methylation of p16(INK4a), RARB2 and SEMA3B in bronchial aspirates from patients with suspected lung cancer. Int J Cancer. 2005, 116 (5): 720-725. 10.1002/ijc.21090.
Trinh BN, Long TI, Laird PW: DNA methylation analysis by MethyLight technology. Methods. 2001, 25 (4): 456-462. 10.1006/meth.2001.1268.
Yuan E, Haghighi F, White S, Costa R, McMinn J, Chun K, Minden M, Tycko B: A single nucleotide polymorphism chip-based method for combined genetic and epigenetic profiling: validation in decitabine therapy and tumor/normal comparisons. Cancer Res. 2006, 66 (7): 3443-3451. 10.1158/0008-5472.CAN-05-3739.
Chakraborty S, Mohiyuddin SM, Gopinath KS, Kumar A: Involvement of TSC genes and differential expression of other members of the mTOR signaling pathway in oral squamous cell carcinoma. BMC Cancer. 2008, 8: 163-10.1186/1471-2407-8-163.
Imai T, Toyota M, Suzuki H, Akino K, Ogi K, Sogabe Y, Kashima L, Maruyama R, Nojima M, Mita H, et al: Epigenetic inactivation of RASSF2 in oral squamous cell carcinoma. Cancer Sci. 2008, 99 (5): 958-966. 10.1111/j.1349-7006.2008.00769.x.
Long NK, Kato K, Yamashita T, Makita H, Toida M, Hatakeyama D, Hara A, Mori H, Shibata T: Hypermethylation of the RECK gene predicts poor prognosis in oral squamous cell carcinomas. Oral Oncol. 2008, 44 (11): 1052-1058. 10.1016/j.oraloncology.2008.02.004.
Ai L, Vo QN, Zuo C, Li L, Ling W, Suen JY, Hanna E, Brown KD, Fan CY: Ataxia-telangiectasia-mutated (ATM) gene in head and neck squamous cell carcinoma: promoter hypermethylation with clinical correlation in 100 cases. Cancer Epidemiol Biomarkers Prev. 2004, 13 (1): 150-156. 10.1158/1055-9965.EPI-082-3.
Shaw RJ, Omar MM, Rokadiya S, Kogera FA, Lowe D, Hall GL, Woolgar JA, Homer J, Liloglou T, Field JK, et al: Cytoglobin is upregulated by tumour hypoxia and silenced by promoter hypermethylation in head and neck cancer. Br J Cancer. 2009, 101 (1): 139-144. 10.1038/sj.bjc.6605121.
Chang HW, Chan A, Kwong DL, Wei WI, Sham JS, Yuen AP: Detection of hypermethylated RIZ1 gene in primary tumor, mouth, and throat rinsing fluid, nasopharyngeal swab, and peripheral blood of nasopharyngeal carcinoma patient. Clin Cancer Res. 2003, 9 (3): 1033-1038.
Wu CL, Roz L, McKown S, Sloan P, Read AP, Holland S, Porter S, Scully C, Paterson I, Tavassoli M, et al: DNA studies underestimate the major role of CDKN2A inactivation in oral and oropharyngeal squamous cell carcinomas. Genes Chromosomes Cancer. 1999, 25 (1): 16-25. 10.1002/(SICI)1098-2264(199905)25:1<16::AID-GCC3>3.0.CO;2-2.
Ishida E, Nakamura M, Ikuta M, Shimada K, Matsuyoshi S, Kirita T, Konishi N: Promotor hypermethylation of p14ARF is a key alteration for progression of oral squamous cell carcinoma. Oral Oncol. 2005, 41 (6): 614-622. 10.1016/j.oraloncology.2005.02.003.
Uesugi H, Uzawa K, Kawasaki K, Shimada K, Moriya T, Tada A, Shiiba M, Tanzawa H: Status of reduced expression and hypermethylation of the APC tumor suppressor gene in human oral squamous cell carcinoma. Int J Mol Med. 2005, 15 (4): 597-602.
Marsit CJ, Posner MR, McClean MD, Kelsey KT: Hypermethylation of E-cadherin is an independent predictor of improved survival in head and neck squamous cell carcinoma. Cancer. 2008, 113 (7): 1566-1571. 10.1002/cncr.23770.
Hasegawa M, Nelson HH, Peters E, Ringstrom E, Posner M, Kelsey KT: Patterns of gene promoter methylation in squamous cell cancer of the head and neck. Oncogene. 2002, 21 (27): 4231-4236. 10.1038/sj.onc.1205528.
De Schutter H, Geeraerts H, Verbeken E, Nuyts S: Promoter methylation of TIMP3 and CDH1 predicts better outcome in head and neck squamous cell carcinoma treated by radiotherapy only. Oncol Rep. 2009, 21 (2): 507-513.
Hall GL, Shaw RJ, Field EA, Rogers SN, Sutton DN, Woolgar JA, Lowe D, Liloglou T, Field JK, Risk JM: p16 Promoter methylation is a potential predictor of malignant transformation in oral epithelial dysplasia. Cancer Epidemiol Biomarkers Prev. 2008, 17 (8): 2174-2179. 10.1158/1055-9965.EPI-07-2867.
Lee ES, Issa JP, Roberts DB, Williams MD, Weber RS, Kies MS, El-Naggar AK: Quantitative promoter hypermethylation analysis of cancer-related genes in salivary gland carcinomas: comparison with methylation-specific PCR technique and clinical significance. Clin Cancer Res. 2008, 14 (9): 2664-2672. 10.1158/1078-0432.CCR-07-1232.
Czerninski R, Krichevsky S, Ashhab Y, Gazit D, Patel V, Ben-Yehuda D: Promoter hypermethylation of mismatch repair genes, hMLH1 and hMSH2 in oral squamous cell carcinoma. Oral diseases. 2009, 15 (3): 206-213. 10.1111/j.1601-0825.2008.01510.x.
Gao F, Huang C, Lin M, Wang Z, Shen J, Zhang H, Jiang L, Chen Q: Frequent inactivation of RUNX3 by promoter hypermethylation and protein mislocalization in oral squamous cell carcinomas. Journal of cancer research and clinical oncology. 2009, 135 (5): 739-747. 10.1007/s00432-008-0508-x.
Subbalekha K, Pimkhaokham A, Pavasant P, Chindavijak S, Phokaew C, Shuangshoti S, Matangkasombut O, Mutirangura A: Detection of LINE-1s hypomethylation in oral rinses of oral squamous cell carcinoma patients. Oral Oncol. 2009, 45 (2): 184-191. 10.1016/j.oraloncology.2008.05.002.
Sogabe Y, Suzuki H, Toyota M, Ogi K, Imai T, Nojima M, Sasaki Y, Hiratsuka H, Tokino T: Epigenetic inactivation of SFRP genes in oral squamous cell carcinoma. Int J Oncol. 2008, 32 (6): 1253-1261.
Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J: Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008, 68 (7): 2094-2105. 10.1158/0008-5472.CAN-07-5194.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/9/354/prepub
Acknowledgements
We would like to thank Dr. Lin Zhang and members of his lab for helpful discussions on optimizing the MGMT MSP-PCR assay. This research was supported by a Research Scholar grant (RSG-05-246-01-GMC) from the American Cancer Society to RWS; P50CA097190 (Head and neck SPORE: Project 1) and the ECNIS project, Grant number: EU Contract 513943; 5P50CA097190 (Head and neck SPORE: minority supplement) to ET and SG and grants from NIH (R01DE10513 & R01DE14729) to SMG. This project is also funded, in part, under a grant with the Pennsylvania Department of Health. The Department of Health specifically disclaims responsibility for any analyses, interpretations or conclusions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
XHW carried out the MGMT MSP-PCR reactions and analysis. XHW and ARB carried out the MGMT pyrosequencing analysis. CR carried out the RASSF1 and CDKN2A MSP-PCR reactions and analysis, participated in the preparation of the manuscript and performed the data and statistical analysis. JC and SL made substantial contributions in data acquisition, statistical analysis and helped in manuscript and figure preparation. SMG provided the purified DNA samples. ET, SG and RWS wrote the manuscript and participated in study and experimental design. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Taioli, E., Ragin, C., Wang, Xh. et al. Recurrence in oral and pharyngeal cancer is associated with quantitative MGMT promoter methylation. BMC Cancer 9, 354 (2009). https://doi.org/10.1186/1471-2407-9-354
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-9-354