
Abstract
Background
Considering that a portion of the heterogeneity amongst previous replication studies may be due to a variable proportion of obese subjects in case-control designs, we assessed the association of genetic variants with type 2 diabetes (T2D) in large groups of obese and non-obese subjects.
Methods
We genotyped RETN, KCNJ11, HNF4A, HNF1A, GCK, SLC30A8, ENPP1, ADIPOQ, PPARG, and TCF7L2 polymorphisms in 1,283 normoglycemic (NG) and 1,581 T2D obese individuals as well as in 3,189 NG and 1,244 T2D non-obese subjects of European descent, allowing us to examine T2D risk over a wide range of BMI.
Results
Amongst non-obese individuals, we observed significant T2D associations with HNF1A I27L [odds ratio (OR) = 1.14, P = 0.04], GCK -30G>A (OR = 1.23, P = 0.01), SLC30A8 R325W (OR = 0.87, P = 0.04), and TCF7L2 rs7903146 (OR = 1.89, P = 4.5 × 10-23), and non-significant associations with PPARG Pro12Ala (OR = 0.85, P = 0.14), ADIPOQ -11,377C>G (OR = 1.00, P = 0.97) and ENPP1 K121Q (OR = 0.99, P = 0.94). In obese subjects, associations with T2D were detected with PPARG Pro12Ala (OR = 0.73, P = 0.004), ADIPOQ -11,377C>G (OR = 1.26, P = 0.02), ENPP1 K121Q (OR = 1.30, P = 0.003) and TCF7L2 rs7903146 (OR = 1.30, P = 1.1 × 10-4), and non-significant associations with HNF1A I27L (OR = 0.96, P = 0.53), GCK -30G>A (OR = 1.15, P = 0.12) and SLC30A8 R325W (OR = 0.95, P = 0.44). However, a genotypic heterogeneity was only found for TCF7L2 rs7903146 (P = 3.2 × 10-5) and ENPP1 K121Q (P = 0.02). No association with T2D was found for KCNJ11, RETN, and HNF4A polymorphisms in non-obese or in obese individuals.
Conclusion
Genetic variants modulating insulin action may have an increased effect on T2D susceptibility in the presence of obesity, whereas genetic variants acting on insulin secretion may have a greater impact on T2D susceptibility in non-obese individuals.
Similar content being viewed by others

Background
Type 2 diabetes (T2D) is characterized by defects in both insulin sensitivity and beta-cell dysfunction [1]. This disease involves a complex interaction between genetic variants and environment with obesity established as a primary risk factor [2]. In this regard, correlations between increased body fat and insulin resistance have been reliably demonstrated [3]. Recently, associations between polymorphisms and T2D were shown to be modulated according to obesity status.
ENPP1, ADIPOQ, PPARG and TCF7L2 single nucleotide polymorphisms (SNPs) have previously been associated with T2D [4–7] and have shown variable significance amongst different classes of obesity. It has been suggested that the ENPP1 K121Q and ADIPOQ -11,391G>A and -11,377C>G variants may confer greater T2D susceptibility in obese populations [8–10]. PPARG Pro12Ala, though controversial, was more associated with T2D amongst obese subgroups in our previous study [11–13]. Conversely, the TCF7L2 rs7903146 T allele has been shown to be more prevalent in T2D non-obese individuals compared to T2D obese subjects [7]. Interestingly, the variants in PPARG, ADIPOQ, and ENPP1, which elevate risk in obese populations, have been categorized as acting on insulin resistance while the TCF7L2 variant elevating risk in lean populations has been categorized as acting on insulin secretion [14–17]. These preliminary results suggest that different genetic architectures could increase T2D susceptibility according to the presence or absence of obesity.
Based on these observations, we have chosen to confirm previous findings and evaluate additional genetic variants which have a reported effect on either insulin action (RETN -420C>G) [18] or insulin secretion (KCNJ11 E23K, HNF4A rs1884614 and rs2144908, HNF1A I27L, GCK -30G>A, and SLC30A8 R325W) [19–25] and which have previously been associated with T2D in large scale studies or meta-analysis [21, 26–30]. The evaluated variants were tested for their differential association with T2D in obese [1,283 normoglycemic (NG) and 1,581 T2D] and non-obese subjects (3,189 NG and 1,244 T2D) of European descent.
Methods
Study population
All NG and T2D subjects were recruited in three different centers, two in France and one in Switzerland. The clinical characteristics of the studied groups are presented in Table 1.
Unrelated NG and T2D French white adults were recruited using a multimedia campaign run by the "Centre National de la Recherche Scientifique" (CNRS), the Department of Nutrition of the Paris Hôtel-Dieu Hospital and the Pasteur Institute of Lille.
Individuals both non-obese and NG (n = 2,122) of the French general population, aged between 30 and 65 years, participated in the cohort for the Data From an Epidemiological Study on the Insulin Resistance syndrome (D.E.S.I.R.), a 9-year follow-up study that aims to clarify the development of the insulin resistance syndrome and described previously [31]. Ethnic origin cannot be legally documented in France. We estimated the proportion of subjects having non-European ancestry from a subgroup of 654 subjects selected in the D.E.S.I.R. cohort, as previously described [32]. We genotyped 328 SNPs which were spaced by at least 5 Mb and highly differentiated among individuals from different continents (Fst > 0.2 based on the Perlegen dataset) [33]. Analysis using the STRUCTURE software identified only two individuals of non-european ancestry in a total of 654 individuals. From this analysis, the proportion of subjects having non-European ancestry was estimated to be 0.30% in the D.E.S.I.R. cohort. Additionally, all individuals born outside France were excluded from the study.
All NG and T2D Swiss white obese individuals were from Bern and Zurich as described previously [34]. They were added to the French samples of the same corpulence category. No heterogeneities were found between these two population groups for all SNPs [see Additional file 1].
In all centers, two classes of glycemic status were defined according to the 1997 American Diabetes Association criteria: NG, defined as fasting glucose < 6.1 mmol/l and T2D, defined as fasting plasma glucose ≥ 7.0 mmol/l. Two separate BMI categories were analyzed in this study, according to WHO international classification: non-obese (BMI < 30 kg/m2), and obese (BMI ≥ 30 kg/m2).
This genetic study was approved by the Ethical Committee of Hotel-Dieu in Corbeil-Essonnes and CHRU in Lille and informed consent was obtained from all participants.
SNP genotyping
High-throughput genotyping of all genetic variants was performed using the TaqMan® SNP Genotyping Assays (Applied Biosystems, Foster City, Calif. USA). The PCR primers and TaqMan probes were designed by Primer Express and optimized according to the manufacturer's protocol. There was a 96–99% genotyping success rate for all SNPs and the genotyping error rate was assessed by randomly genotyping 384 control and 384 T2D individuals. No difference was found from the first genotyping results, thus the genotyping error rate was 0% for each SNP. The genotypic distributions for all SNPs were in Hardy-Weinberg equilibrium (P ≥ 0.01) [see Additional file 2].
Statistical analysis
The association with T2D was calculated using a logistic regression model adjusted for age, gender, and BMI. The minimum detectable effect size with a statistical power of 80% was assessed using Quanto [see Additional file 3]. The Woolf test was applied to asses the genotypic heterogeneity between obese and non-obese groups. No Bonferroni correction was applied as, in the case of replication studies, it is unlikely to detect effects due to statistical fluctuation only. All P values are two-sided. SPSS (version 14.0.2) and R statistics (version 2.5.0) software were used for general statistics.
Results
All SNPs were evaluated for their contribution to T2D in the complete sample population (4,472 NG and 2,825 T2D), then in obese individuals (1,283 NG and 1,581 T2D) and non-obese subjects (3,189 NG and 1,244 T2D). Clinical characteristics and allelic distributions of the study population are summarized in Table 1 and Table 2, respectively. The best fitting genetic models were selected from previous studies [5–7, 13, 26–30, 35] and were in agreement with what we found in our analyses (data not shown).
All NG and T2D Individuals
We first evaluated the genetic variants amongst all NG and T2D individuals regardless of obesity status (Table 3). As previously reported in a more modestly-sized French set, T2D associations were confirmed in PPARG Pro12Ala [odds ratio (OR) = 0.81, P = 0.004], TCF7L2 rs7903146 (OR = 1.59, P = 7.9 × 10-27), and ENPP1 K121Q (OR = 1.15, P = 0.01) [7–9, 11]. Additionally, GCK -30G>A (OR 1.20, P = 0.001) and SLC30A8 R325W (OR = 0.91, P = 0.03) were also found to increase T2D risk.
In contrast, in our whole sample analysis, no evidence for association with T2D was found for the ADIPOQ variants -11,391G>A (OR = 0.99, P = 0.91) and -11,377C>G (OR = 1.08, P = 0.18), HNF1A I27L (OR = 1.01, P = 0.71), HNF4A variants rs1884614 (OR = 1.03, P = 0.58) and rs2144908 (OR = 1.04, P = 0.45), KCNJ11 E23K (OR = 0.9, P = 0.09), or RETN -420C>G (OR = 1.00, P = 0.99).
Non-Obese T2D Association
We then assessed the genetic variants amongst the non-obese population (BMI < 30 kg/m2) (Table 3). The risk alleles of HNF1A I27L (OR = 1.14, P = 0.04), GCK -30G>A (OR = 1.23, P = 0.01), SLC30A8 R325W (OR = 0.87, P = 0.04), and TCF7L2 rs7903146 (OR = 1.89, P = 4.5 × 10-23) conferred a significantly increased risk of T2D.
The ADIPOQ variants -11,391G>A (OR = 0.96, P = 0.71) and -11,377C>G (OR = 1.00, P = 0.97), ENPP1 K121Q (OR = 0.99, P = 0.94), PPARG Pro12Ala (OR = 0.85, P = 0.14), RETN -420C>G (OR = 0.41, P = 0.14), KCNJ11 E23K (OR = 0.96, P = 0.68), and HNF4A variants rs1884614 (OR = 1.05, P = 0.54) and rs2144908 (OR = 1.08, P = 0.33) were not associated with T2D amongst the non-obese group.
Obese T2D Association
In the obese group (BMI ≥ 30 kg/m2), associations with T2D were detected in PPARG Pro12Ala (OR 0.73, P = 0.004), ADIPOQ -11,377C>G (OR = 1.25, P = 0.015), ENPP1 K121Q (OR = 1.30, P = 0.003) and TCF7L2 rs7903146 (OR = 1.30, P = 1.1 × 10-4) (Table 3).
We did not find evidence for increased T2D risk for GCK -30G>A (OR = 1.15, P = 0.12), ADIPOQ -11,391G>A (OR = 1.07, P = 0.58), HNF1A I27L (OR = 0.96, P = 0.53), HNF4A variants rs1884614 (OR = 1.05, P = 0.54) and rs2144908 (OR = 1.04, P = 0.63), KCNJ11 E23K (OR = 0.96, P = 0.68), or SLC30A8 R325W (OR = 0.95, P = 0.44) amongst obese subjects.
Genotypic heterogeneity
Discrepancies in genotypic distribution were assessed by Woolf test between the obese and non-obese categories (Table 3). A genotypic heterogeneity was found for TCF7L2 rs7903146 (P = 3.2 × 10-5) and ENPP1 K121Q (P = 0.02). Their association with BMI change was then assessed in all studied subgroups. Significant effects on BMI were only detected in the T2D group for the TCF7L2 genetic variant [see Additional file 4]. Trends towards heterogeneity were found for ADIPOQ 11377C>G (P = 0.07) and HNF1A I27L (P = 0.06).
Discussion
This large case-control study further supports the hypothesis that the T2D risk contribution of ENPP1, ADIPOQ, PPARG and TCF7L2 SNPs may be modulated by obesity status [6–8, 11]. Notably, we made the novel finding that T2D risk, as conferred by HNF1A, GCK, and SLC30A8 genetic variants, may also be modified in the presence/absence of obesity. Despite the stringent standards exacted by the Woolf test, genotypic heterogeneity between obese and non-obese individuals was detected for TCF7L2 rs7903146 and ENPP1 K121Q, and suggested for ADIPOQ 11377C>G and HNF1A I27L SNPs. Future study designs containing more individuals will be needed to confirm all apparent genotypic heterogeneities.
The analysis of HNF4A, KCNJ11 and RETN polymorphisms did not support their role as T2D risk factors despite sufficient statistical power [see Additional file 3] to detect the effect sizes (1.08, 1.09 and 1.16, respectively) previously established by meta-analysis [21, 26, 27]. The inability of the current study to replicate previous findings is likely the result of true but modest contributions to T2D risk in the European population.
While it is known that defects in insulin action and insulin secretion are critical in T2D pathogenesis, their interaction with genetic and environmental factors is less clear. Interestingly, SLC30A8, GCK, and HNF1A genetic variants, all of which have been associated with insulin secretion [22–25], were only associated with T2D in non-obese individuals. The TCF7L2 genetic variant, another SNP modulating beta cell function [17, 36], was more associated with T2D in non-obese subjects than in obese individuals. Conversely, insulin resistance associated genetic variants of PPARG, ADIPOQ and ENPP1 [14–16, 37, 38] were found to be only associated in obese subjects. While obesity has been strongly associated with increased insulin resistance [3], our data suggests that insulin secretory variants confer a greater T2D risk in non-obese individuals while insulin sensitivity variants more significantly modulate T2D risk in obese subjects.
Currently, some dissension exists regarding the BMI modulation of the Pro12Ala SNP in T2D risk. Contrary to what was reported by Ghoussaini and colleagues [11], other studies of the PPARG Pro12Ala SNP suggest that the protective effect of the Ala allele may be greater when control BMI is lower [12, 13]. The present analysis was specifically designed to detect interactions by analyzing a large number of European subjects with no genotypic heterogeneity and characterized by a wide range of BMI. In the "Diabetes Prevention Program", 55 percent of participants were Caucasian, 45 percent were minorities, most were obese, and most had a family history of T2D [12]. Similarly, Ludivico and colleagues performed a combined analysis of Asian, North American, and European populations. Interestingly, the genetic effect on T2D was found to be ~30% stronger in Asians than in the two other more corpulent populations [13]. We therefore suggest that population heterogeneity may be the primary contributor to the overall observed BMI effect as supported by the ability of BMI to statistically explain the heterogeneity between these populations but not within Europeans [13]. Large ethnically-matched studies would be necessary to know if such interaction is found in non-European subjects.
These results illustrate the importance of BMI in population design as obesity mediated T2D associations may account for part of the difficulty encountered in replication studies. The ENPP1 K121Q variant may be illustrative of this point. Studies exploring the T2D risk contribution of the K121Q SNP amongst obese individuals [8, 10] have found an enhanced association in this subgroup. In the present study, we supported these results in finding no significant effect on T2D in non-obese individuals (OR = 0.99, P = 0.94), an association with T2D in obese subjects (OR = 1.30, P = 0.003) and a subsequent modest effect when obese and non-obese samples were analyzed together (OR = 1.15, P = 0.01). A portion of the heterogeneity amongst previous replication studies may be due to a variable fraction of obese subjects in study designs [5, 8, 39, 40]. Considering these results, future association studies should take into account obesity status when interpreting their data.
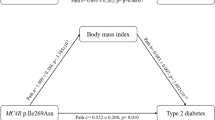
A representation of the possible relationship between T2D risk variants and obesity status is modeled in Figure 1. This study suggests that the magnitude of a variant's effect on T2D susceptibility is modulated by both obesity status and SNP function. Specifically, variants effecting insulin action more significantly increase T2D susceptibility in obese individuals while variants effecting insulin secretion confer greater T2D risk in non-obese individuals. In our study, data on waist/hip ratio were not available in most T2D cases. However, further analyses of the issue should consider this anthropometric parameter when analyzing interactions between obesity and T2D.

Functional relevance of genetic variants may affect their association with T2D by obesity status. The present figure does not take into account variants associated with T2D through their effect on BMI.
These data are concordant with results from previous epidemiological and physiological studies. Prospective data indicates that impaired beta cell function, not insulin resistance, predicts future T2D in non-obese subjects [41]. Furthermore, it was recently reported that interactions between TCF7L2 genetic variants and adiposity may lead to reduced beta-cell compensation in leaner individuals but facilitate improved compensation in more obese subjects [42]. Increased beta-cell mass has indeed been observed in the pancreata of obese compared to lean non-diabetic subjects [43]. These results support a primary role of insulin secretory modulating variants in the absence of obesity. Conversely, the hyperbolic relationship of insulin resistance and insulin secretion enables the beta-cell to adequately maintain glucose homeostasis for a large range of insulin resistance levels [44]. In non-obese subjects not affected by adiposity-induced insulin resistance, this mechanism may sufficiently compensate the effects of genetic variants increasing insulin resistance.
Obesity-induced insulin resistance is characterized by a series of events: i) impaired regulation of lipolysis in adipocytes, ii) increased circulating levels of free fatty acids [45], iii) ectopic fat storage in muscle and liver, iv) impaired ability of insulin to inhibit hepatic glucose production and muscular glucose uptake [46], v) increased beta cell death by apoptosis (glucolipotoxicity) [43, 47]. In obesity-prone populations, insulin resistance more accurately predicts development of T2D than does insulin secretory dysfunction [48]. Previous studies on lipodistrophy in both human and murine models have shown that adipose tissue plays a key role in insulin resistance [49]. Interestingly, PPARG, ADIPOQ, and ENPP1 SNPs only associated with T2D in obese subjects, and have a pivotal role in adipocyte differentiation, maturation, and action [50–52]. The effects of genetic polymorphisms leading to insulin resistance may therefore be worsened by obesity.
In the present study, we suggested that the genetic architecture of T2D may be different in obese and non-obese individuals. In a given ethnic group, the lack of replication in association studies may be, in part, due to different fractions of obesity in case-control designs. Novel SNPs recently identified in genome-wide association studies will prove important in the confirmation of these findings once their functions have been reliably demonstrated.
Abbreviations
- T2D:
-
Type 2 Diabetes
- NG:
-
Normoglycemic
- BMI:
-
Body Mass Index
- OR:
-
Odds ratio
- SNP:
-
Single Nucleotide Polymorphism.
References
Stumvoll M, Goldstein BJ, van Haeften TW: Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005, 365 (9467): 1333-1346. 10.1016/S0140-6736(05)61032-X.
Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, Marks JS: Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. Jama. 2003, 289 (1): 76-79. 10.1001/jama.289.1.76.
Kahn SE, Hull RL, Utzschneider KM: Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006, 444 (7121): 840-846. 10.1038/nature05482.
McAteer JB, Prudente S, Bacci S, Lyon HN, Hirschhorn JN, Trischitta V, Florez JC: The ENPP1 K121Q polymorphism is associated with type 2 diabetes in European populations: evidence from an updated meta-analysis in 42,042 subjects. Diabetes. 2008, 57 (4): 1125-1130. 10.2337/db07-1336.
Grarup N, Urhammer SA, Ek J, Albrechtsen A, Glumer C, Borch-Johnsen K, Jorgensen T, Hansen T, Pedersen O: Studies of the relationship between the ENPP1 K121Q polymorphism and type 2 diabetes, insulin resistance and obesity in 7,333 Danish white subjects. Diabetologia. 2006, 49 (9): 2097-2104. 10.1007/s00125-006-0353-x.
Vasseur F, Helbecque N, Dina C, Lobbens S, Delannoy V, Gaget S, Boutin P, Vaxillaire M, Lepretre F, Dupont S, Hara K, Clement K, Bihain B, Kadowaki T, Froguel P: Single-nucleotide polymorphism haplotypes in the both proximal promoter and exon 3 of the APM1 gene modulate adipocyte-secreted adiponectin hormone levels and contribute to the genetic risk for type 2 diabetes in French Caucasians. Hum Mol Genet. 2002, 11 (21): 2607-2614. 10.1093/hmg/11.21.2607.
Cauchi S, Choquet H, Gutierrez-Aguilar R, Capel F, Grau K, Proenca C, Dina C, Duval A, Balkau B, Marre M, Potoczna N, Langin D, Horber F, Sorensen TI, Charpentier G, Meyre D, Froguel P: Effects of TCF7L2 Polymorphisms on Obesity in European Populations. Obesity (Silver Spring). 2008, 16 (2): 476-482. 10.1038/oby.2007.77.
Meyre D, Bouatia-Naji N, Tounian A, Samson C, Lecoeur C, Vatin V, Ghoussaini M, Wachter C, Hercberg S, Charpentier G, Patsch W, Pattou F, Charles MA, Tounian P, Clement K, Jouret B, Weill J, Maddux BA, Goldfine ID, Walley A, Boutin P, Dina C, Froguel P: Variants of ENPP1 are associated with childhood and adult obesity and increase the risk of glucose intolerance and type 2 diabetes. Nat Genet. 2005, 37 (8): 863-867. 10.1038/ng1604.
Vasseur F, Helbecque N, Lobbens S, Vasseur-Delannoy V, Dina C, Clement K, Boutin P, Kadowaki T, Scherer PE, Froguel P: Hypoadiponectinaemia and high risk of type 2 diabetes are associated with adiponectin-encoding (ACDC) gene promoter variants in morbid obesity: evidence for a role of ACDC in diabesity. Diabetologia. 2005, 48 (5): 892-899. 10.1007/s00125-005-1729-z.
Bochenski J, Placha G, Wanic K, Malecki M, Sieradzki J, Warram JH, Krolewski AS: New polymorphism of ENPP1 (PC-1) is associated with increased risk of type 2 diabetes among obese individuals. Diabetes. 2006, 55 (9): 2626-2630. 10.2337/db06-0191.
Ghoussaini M, Meyre D, Lobbens S, Charpentier G, Clement K, Charles MA, Tauber M, Weill J, Froguel P: Implication of the Pro12Ala polymorphism of the PPAR-gamma 2 gene in type 2 diabetes and obesity in the French population. BMC Med Genet. 2005, 6: 11-10.1186/1471-2350-6-11.
Florez JC, Jablonski KA, Sun MW, Bayley N, Kahn SE, Shamoon H, Hamman RF, Knowler WC, Nathan DM, Altshuler D: Effects of the type 2 diabetes-associated PPARG P12A polymorphism on progression to diabetes and response to troglitazone. J Clin Endocrinol Metab. 2007, 92 (4): 1502-1509. 10.1210/jc.2006-2275.
Ludovico O, Pellegrini F, Di Paola R, Minenna A, Mastroianno S, Cardellini M, Marini MA, Andreozzi F, Vaccaro O, Sesti G, Trischitta V: Heterogeneous effect of peroxisome proliferator-activated receptor gamma2 Ala12 variant on type 2 diabetes risk. Obesity (Silver Spring). 2007, 15 (5): 1076-1081.
Deeb SS, Fajas L, Nemoto M, Pihlajamaki J, Mykkanen L, Kuusisto J, Laakso M, Fujimoto W, Auwerx J: A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat Genet. 1998, 20 (3): 284-287. 10.1038/3099.
Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T: The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001, 7 (8): 941-946. 10.1038/90984.
Maddux BA, Goldfine ID: Membrane glycoprotein PC-1 inhibition of insulin receptor function occurs via direct interaction with the receptor alpha-subunit. Diabetes. 2000, 49 (1): 13-19. 10.2337/diabetes.49.1.13.
Lyssenko V, Lupi R, Marchetti P, Del Guerra S, Orho-Melander M, Almgren P, Sjogren M, Ling C, Eriksson KF, Lethagen AL, Mancarella R, Berglund G, Tuomi T, Nilsson P, Del Prato S, Groop L: Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J Clin Invest. 2007, 117 (8): 2155-2163. 10.1172/JCI30706.
Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA: The hormone resistin links obesity to diabetes. Nature. 2001, 409 (6818): 307-312. 10.1038/35053000.
Nielsen EM, Hansen L, Carstensen B, Echwald SM, Drivsholm T, Glumer C, Thorsteinsson B, Borch-Johnsen K, Hansen T, Pedersen O: The E23K variant of Kir6.2 associates with impaired post-OGTT serum insulin response and increased risk of type 2 diabetes. Diabetes. 2003, 52 (2): 573-577. 10.2337/diabetes.52.2.573.
Lehto M, Bitzen PO, Isomaa B, Wipemo C, Wessman Y, Forsblom C, Tuomi T, Taskinen MR, Groop L: Mutation in the HNF-4alpha gene affects insulin secretion and triglyceride metabolism. Diabetes. 1999, 48 (2): 423-425. 10.2337/diabetes.48.2.423.
Johansson S, Raeder H, Eide SA, Midthjell K, Hveem K, Sovik O, Molven A, Njolstad PR: Studies in 3,523 Norwegians and meta-analysis in 11,571 subjects indicate that variants in the hepatocyte nuclear factor 4 alpha (HNF4A) P2 region are associated with type 2 diabetes in Scandinavians. Diabetes. 2007, 56 (12): 3112-3117. 10.2337/db07-0513.
Holmkvist J, Cervin C, Lyssenko V, Winckler W, Anevski D, Cilio C, Almgren P, Berglund G, Nilsson P, Tuomi T, Lindgren CM, Altshuler D, Groop L: Common variants in HNF-1 alpha and risk of type 2 diabetes. Diabetologia. 2006, 49 (12): 2882-2891. 10.1007/s00125-006-0450-x.
Terauchi Y, Takamoto I, Kubota N, Matsui J, Suzuki R, Komeda K, Hara A, Toyoda Y, Miwa I, Aizawa S, Tsutsumi S, Tsubamoto Y, Hashimoto S, Eto K, Nakamura A, Noda M, Tobe K, Aburatani H, Nagai R, Kadowaki T: Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance. J Clin Invest. 2007, 117 (1): 246-257. 10.1172/JCI17645.
Velho G, Froguel P, Clement K, Pueyo ME, Rakotoambinina B, Zouali H, Passa P, Cohen D, Robert JJ: Primary pancreatic beta-cell secretory defect caused by mutations in glucokinase gene in kindreds of maturity onset diabetes of the young. Lancet. 1992, 340 (8817): 444-448. 10.1016/0140-6736(92)91768-4.
Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, Styrkarsdottir U, Gretarsdottir S, Emilsson V, Ghosh S, Baker A, Snorradottir S, Bjarnason H, Ng MC, Hansen T, Bagger Y, Wilensky RL, Reilly MP, Adeyemo A, Chen Y, Zhou J, Gudnason V, Chen G, Huang H, Lashley K, Doumatey A, So WY, Ma RC, Andersen G, Borch-Johnsen K, et al: A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet. 2007, 39 (6): 770-775. 10.1038/ng2043.
Osawa H, Yamada K, Onuma H, Murakami A, Ochi M, Kawata H, Nishimiya T, Niiya T, Shimizu I, Nishida W, Hashiramoto M, Kanatsuka A, Fujii Y, Ohashi J, Makino H: The G/G genotype of a resistin single-nucleotide polymorphism at -420 increases type 2 diabetes mellitus susceptibility by inducing promoter activity through specific binding of Sp1/3. Am J Hum Genet. 2004, 75 (4): 678-686. 10.1086/424761.
van Dam RM, Hoebee B, Seidell JC, Schaap MM, de Bruin TW, Feskens EJ: Common variants in the ATP-sensitive K+ channel genes KCNJ11 (Kir6.2) and ABCC8 (SUR1) in relation to glucose intolerance: population-based studies and meta-analyses. Diabet Med. 2005, 22 (5): 590-598. 10.1111/j.1464-5491.2005.01465.x.
Weedon MN, Owen KR, Shields B, Hitman G, Walker M, McCarthy MI, Love-Gregory LD, Permutt MA, Hattersley AT, Frayling TM: Common variants of the hepatocyte nuclear factor-4alpha P2 promoter are associated with type 2 diabetes in the U.K. population. Diabetes. 2004, 53 (11): 3002-3006. 10.2337/diabetes.53.11.3002.
Winckler W, Burtt NP, Holmkvist J, Cervin C, de Bakker PI, Sun M, Almgren P, Tuomi T, Gaudet D, Hudson TJ, Ardlie KG, Daly MJ, Hirschhorn JN, Altshuler D, Groop L: Association of common variation in the HNF1alpha gene region with risk of type 2 diabetes. Diabetes. 2005, 54 (8): 2336-2342. 10.2337/diabetes.54.8.2336.
Winckler W, Weedon MN, Graham RR, McCarroll SA, Purcell S, Almgren P, Tuomi T, Gaudet D, Bostrom KB, Walker M, Hitman G, Hattersley AT, McCarthy MI, Ardlie KG, Hirschhorn JN, Daly MJ, Frayling TM, Groop L, Altshuler D: Evaluation of common variants in the six known maturity-onset diabetes of the young (MODY) genes for association with type 2 diabetes. Diabetes. 2007, 56 (3): 685-693. 10.2337/db06-0202.
Balkau B: [An epidemiologic survey from a network of French Health Examination Centres, (D.E.S.I.R.): epidemiologic data on the insulin resistance syndrome]. Rev Epidemiol Sante Publique. 1996, 44 (4): 373-375.
Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D, Polychronakos C, Froguel P: A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007, 445 (7130): 881-885. 10.1038/nature05616.
Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, Ballinger DG, Frazer KA, Cox DR: Whole-genome patterns of common DNA variation in three human populations. Science. 2005, 307 (5712): 1072-1079. 10.1126/science.1105436.
Branson R, Potoczna N, Kral JG, Lentes KU, Hoehe MR, Horber FF: Binge eating as a major phenotype of melanocortin 4 receptor gene mutations. N Engl J Med. 2003, 348 (12): 1096-1103. 10.1056/NEJMoa021971.
Silander K, Mohlke KL, Scott LJ, Peck EC, Hollstein P, Skol AD, Jackson AU, Deloukas P, Hunt S, Stavrides G, Chines PS, Erdos MR, Narisu N, Conneely KN, Li C, Fingerlin TE, Dhanjal SK, Valle TT, Bergman RN, Tuomilehto J, Watanabe RM, Boehnke M, Collins FS: Genetic variation near the hepatocyte nuclear factor-4 alpha gene predicts susceptibility to type 2 diabetes. Diabetes. 2004, 53 (4): 1141-1149. 10.2337/diabetes.53.4.1141.
Dahlgren A, Zethelius B, Jensevik K, Syvanen AC, Berne C: Variants of the TCF7L2 gene are associated with beta cell dysfunction and confer an increased risk of type 2 diabetes mellitus in the ULSAM cohort of Swedish elderly men. Diabetologia. 2007
Stefan N, Vozarova B, Funahashi T, Matsuzawa Y, Weyer C, Lindsay RS, Youngren JF, Havel PJ, Pratley RE, Bogardus C, Tataranni PA: Plasma adiponectin concentration is associated with skeletal muscle insulin receptor tyrosine phosphorylation, and low plasma concentration precedes a decrease in whole-body insulin sensitivity in humans. Diabetes. 2002, 51 (6): 1884-1888. 10.2337/diabetes.51.6.1884.
Kubaszek A, Pihlajamaki J, Karhapaa P, Vauhkonen I, Laakso M: The K121Q polymorphism of the PC-1 gene is associated with insulin resistance but not with dyslipidemia. Diabetes Care. 2003, 26 (2): 464-467. 10.2337/diacare.26.2.464.
Bacci S, Ludovico O, Prudente S, Zhang YY, Di Paola R, Mangiacotti D, Rauseo A, Nolan D, Duffy J, Fini G, Salvemini L, Amico C, Vigna C, Pellegrini F, Menzaghi C, Doria A, Trischitta V: The K121Q polymorphism of the ENPP1/PC-1 gene is associated with insulin resistance/atherogenic phenotypes, including earlier onset of type 2 diabetes and myocardial infarction. Diabetes. 2005, 54 (10): 3021-3025. 10.2337/diabetes.54.10.3021.
Weedon MN, Shields B, Hitman G, Walker M, McCarthy MI, Hattersley AT, Frayling TM: No evidence of association of ENPP1 variants with type 2 diabetes or obesity in a study of 8,089 U.K. Caucasians. Diabetes. 2006, 55 (11): 3175-3179. 10.2337/db06-0410.
Alvarsson M, Wajngot A, Cerasi E, Efendic S: K-value and low insulin secretion in a non-obese white population: predicted glucose tolerance after 25 years. Diabetologia. 2005, 48 (11): 2262-2268. 10.1007/s00125-005-1929-6.
Watanabe RM, Allayee H, Xiang AH, Trigo E, Hartiala J, Lawrence JM, Buchanan TA: Transcription factor 7-like 2 (TCF7L2) is associated with gestational diabetes mellitus and interacts with adiposity to alter insulin secretion in Mexican Americans. Diabetes. 2007, 56 (5): 1481-1485. 10.2337/db06-1682.
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC: Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003, 52 (1): 102-110. 10.2337/diabetes.52.1.102.
Larsson H, Ahren B: Islet dysfunction in insulin resistance involves impaired insulin secretion and increased glucagon secretion in postmenopausal women with impaired glucose tolerance. Diabetes Care. 2000, 23 (5): 650-657. 10.2337/diacare.23.5.650.
Boden G: Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes. 1997, 46 (1): 3-10. 10.2337/diabetes.46.1.3.
Saltiel AR, Kahn CR: Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001, 414 (6865): 799-806. 10.1038/414799a.
Prentki M, Nolan CJ: Islet beta cell failure in type 2 diabetes. J Clin Invest. 2006, 116 (7): 1802-1812. 10.1172/JCI29103.
Lillioja S, Mott DM, Spraul M, Ferraro R, Foley JE, Ravussin E, Knowler WC, Bennett PH, Bogardus C: Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. Prospective studies of Pima Indians. N Engl J Med. 1993, 329 (27): 1988-1992. 10.1056/NEJM199312303292703.
Garg A: Lipodystrophies. Am J Med. 2000, 108 (2): 143-152. 10.1016/S0002-9343(99)00414-3.
Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, Chatterjee VK, O'Rahilly S: Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999, 402 (6764): 880-883.
Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T: Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002, 8 (11): 1288-1295. 10.1038/nm788.
Liang J, Fu M, Ciociola E, Chandalia M, Abate N: Role of ENPP1 on adipocyte maturation. PLoS ONE. 2007, 2 (9): e882-10.1371/journal.pone.0000882.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/9/45/prepub
Acknowledgements
This work was partly supported by the French Governmental "Agence Nationale de la Recherche", and the charities: "Association Française des Diabétiques" and "Programme national de recherche sur le diabète". We thank Marianne Deweider and Frederic Allegaert for the DNA bank management and Stefan Gaget for his help on phenotype databases. We are indebted to all subjects who participated to this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SC managed the study, carried out the genetic analyses and drafted the manuscript. KTN carried out the genetic analyses, drafted the manuscript and carried out the genotyping experiments. HC carried out the genotyping experiments. FH participated in the design of the study. NP participated in the design of the database. BB participated in the design of the study. MM participated in the design of the study. GC participated in the design of the study. PF coordinated the study. DM conceived the study, and participated in its design and coordination. All authors read and approved the final manuscript.
Stéphane Cauchi, Kevin T Nead contributed equally to this work.
Electronic supplementary material
12881_2008_339_MOESM1_ESM.doc
Additional file 1: Supplementary table 1. Homogeneity in genotypic distributions between Swiss and French subjects (DOC 40 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Cauchi, S., Nead, K.T., Choquet, H. et al. The genetic susceptibility to type 2 diabetes may be modulated by obesity status: implications for association studies. BMC Med Genet 9, 45 (2008). https://doi.org/10.1186/1471-2350-9-45
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-9-45