
Abstract
Background
Terminal deletions of chromosome 4q are associated with a broad spectrum of phenotypes including cardiac, craniofacial, digital, and cognitive impairment. The rarity of this syndrome renders genotype-phenotype correlation difficult, which is further complicated by the widely different phenotypes observed in patients sharing similar deletion intervals.
Case presentation
Herein, we describe a boy with congenital hearing impairment and a variety of moderate syndromic features that prompted SNP array analysis disclosing a heterozygous 6.9 Mb deletion in the 4q35.1q35.2 region, which emerged de novo in the maternal germ line.
Conclusion
In addition to the index patient, we review 35 cases from the literature and DECIPHER database to attempt genotype-phenotype correlations for a syndrome with great phenotypic variability. We delineate intervals with recurrent phenotypic overlap, particularly for cleft palate, congenital heart defect, intellectual disability, and autism spectrum disorder. Broad phenotypic presentation of the terminal 4q deletion syndrome is consistent with incomplete penetrance of the individual symptoms.
Similar content being viewed by others
Background
Terminal deletions of chromosome 4q are a rare event with an approximate incidence of 1 in 100,000 [1, 2]. While the majority are de novo cases, an estimated 10-20% are the unbalanced product of a parental reciprocal translocation. Furthermore, some pediatric cases with classical phenotypes have inherited their 4q deletion from a parent described as either normal or only mildly affected [3–6]. Although there is a high degree of phenotypic variation in those presenting overlapping deletion intervals, there is a general consensus that chromosome 4q deletion syndrome is characterized by intellectual disability (ID), craniofacial dysmorphism, rotated or low-set ears, cleft palate (CP), micrognathia, congenital heart defects (CHD), craniofacial, skeletal and digital abnormalities, and occasionally autism spectrum disorder (ASD), behavioural disorders, and developmental delay [7–9]. Chromosome 4q deletions are divided in two different subgroups depending on the region of 4q that is deleted: interstitial, spanning the centromere through 4q28.3 and terminal, from 4q31.1 to 4qter. Although both deletion types each have highly variable phenotypic associations, terminal deletion cases present a broader phenotypic range including CHD, craniofacial and skeletal abnormalities. The 4q33 region has been proposed as critical for ulnar deficiency, cleft lip and palate, and brain development [10].
Herein, we report on an eight year-old boy with moderate dysmorphic features and a de novo deletion in the 4q35.1q35.2 region. By analyzing the considerable phenotypic variability of terminal 4q deletion cases from the literature and DECIPHER database, we attempt to delineate critical intervals for common phenotypic features.
Case presentation
Clinical report
The proband is the only child of two healthy unrelated parents of German ethnicity, born at a gestational age of 38.3 weeks, after an uncomplicated pregnancy and normal spontaneous delivery. Birth weight was 3,125 g (25th centile), APGAR scores of nine and ten at one and five minutes, respectively, cord blood pH was 7.3, and an unremarkable otoacoustic emissions newborn hearing screening test was recorded. At four months of age, he had bilateral hearing impairment in the 60 dB range and was fitted with hearing aids. We sequenced genes commonly screened for hearing loss, including GJB2 (MIM: 121011), GJB3 (MIM: 603324), and GJB6 (MIM: 604418). Sequencing disclosed a heterozygous mutation in GJB3 c.94C > T, p.Arg32Trp (rs1805063; minor allele frequency T = 0.015), which is a well-described autosomal recessive deafness gene requiring a second heterozygous mutation either in trans or in compound heterozygous configuration to convey hearing loss. A targeted deafness gene next generation sequencing panel was negative for other pathogenic mutations.
In the first year of life, he was diagnosed with aortic isthmus stenosis, corrected via balloon angioplasty, and a patent foramen ovale. He demonstrated shortened PQ intervals on an electrocardiogram indicative of an atrioventricular node irregularity. Regular pediatric cardiology follow-up was recommended. He also presented with chronic Eustachian tube dysfunction that was treated several times with myringotomy tubes, as well as a bifid uvula. In the fifth year of life, a submucous CP was detected. During the same year, he underwent corrective surgery for the CP and velopharyngeal insufficiency. Additionally, he presented with bilateral cryptorchidism that required testicular orchiopexie. An abdominal sonogram could not rule out the possibility of a left duplex kidney; urine analysis was within normal limits. Despite a small thyroid, he had normal thyroid function on lab testing. His blood profile was unremarkable apart from mild concurrent deficiencies of blood coagulation factors IX (56%), XI (48%) and XII (38%). He had an elevated prothrombin time of 46.5 s (normal: 25–39 s) and an elevated lupus anticoagulant confirmatory test of 1.26 (normal: 0.91-1.07). Further coagulation testing was negative for von Willebrand disease.
Psychological developmental evaluation at the age of three to four years showed mild general developmental delay (six months). Subsequent evaluations showed normal development. Neurological evaluation at the age five showed a lack of age-appropriate coordination. He also had delayed speech and language development, likely secondary, at least in part, to his hearing impairment and extensive hospitalization history. Currently, he attends regular school and does not require remedial classroom instruction.
Methods
Classical cytogenetic and fluorescence in situhybridization (FISH) analyses
Chromosomes of the proband and his parents were prepared from peripheral blood lymphocyte cultures and analyzed by GTG-banding at the 500 band resolution. FISH was carried out using selected BAC probes from the deleted region. BAC DNA was labelled by nick translocation with fluorescein-12-dUTP (Roche Diagnostics, Mannheim, Germany) or tetramethyl-rhodamine-5-dUTP (Roche), and hybridized overnight to denatured chromosomes. Image acquisition and analysis were performed using FISHView 2.0 software (Applied Spectral Imaging, Edingen-Neckarhausen, Germany).
Copy number variation and genotype analyses
Genomic DNA (gDNA) of the proband and his parents was prepared from peripheral blood by standard salt extraction method. The Illumina Omni1-Quad v1.0 SNP array (Illumina, San Diego, CA, USA), with >1.1 million SNP markers, was used for whole genome genotyping and copy number variation (CNV) detection. 200 ng gDNA were utilized in an Illumina Infinium HD Ultra Assay according to the manufacturer’s specifications. Data were analyzed using GenomeStudio (v2011.1) software with both cnvPartition 3.2.0 (Illumina) and QuantiSNP 2.2 copy number algorithm [11]. Genotypes of father, mother and proband were obtained from the SNP array for parent-of-origin determination. HaploPainter [12] was used in combination with manual intervention to illustrate the absence of maternal genotypes in the deletion patient. The terminal 4q monosomy was validated by real-time quantitative polymerase chain reaction (qPCR) of FRG1 exons 1, 8, and DUX4L6 using the SensiMix SYBR Green kit (Bioline, Luckenwalde, Germany).
Mapping critical intervals for terminal 4q deletion syndrome phenotypes
This study makes use of data generated by the DECIPHER consortium, which is funded by the Wellcome Trust (http://decipher.sanger.ac.uk). With the combined DECIPHER cases (nos. 278055, 248967, 249192, 249458, 249476, 249536, 249541, 249655, 251175, 253743, 254882, 256186, 257358, 264122, 264942, 267783, 269176, and 276704) and review of the literature [6–10, 13–22], phenotypic and deletion overlaps among individuals with monosomies spanning different sizes were delineated. We used the UCSC Genome Browser Custom Track (http://genome-euro.ucsc.edu/cgi-bin/hgCustom) to map these cases and targeted the narrowest critical interval for CP, CHD, ID, and ASD.
Results
Classical and molecular cytogenetic analyses
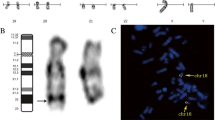
Conventional chromosome banding analysis of the proband revealed a 46,XY karyotype without gross abnormalities. However, the distal G-band negative region in the long arm of chromosome 4 corresponding to q35.1q35.2 appeared to be somewhat smaller in one of the homologs, suggestive of a loss of chromosome material (Figure 1A). Both parents had normal karyotypes without evidence of deletion on chromosome 4q.

Molecular karyotyping of the patient and his parents. (A) GTG-banding and FISH analysis of homologous chromosomes 4 in proband, mother and father. The chromosomes are counterstained with DAPI (blue). The proximal flanking BAC RP11-188P17 is labeled with fluorescein-dUTP (green) and the deleted BAC RP11-775P18 with rhodamine-dUTP (red). An arrowhead indicates the critical band q35.1q35.2 on the patient’s derivative chromosome. (B) Illumina SNP array analysis (B allele frequency and log R ratio) of the 4q35.1q35.2 region in the boy with terminal 4q deletion syndrome. (C) Selected genotypes in the deletion interval from the Illumina array are depicted for proband (left), mother (middle), and father (right). Mendelian transmission errors (absence of maternal genotypes) in the proband are indicated in gray.
To validate the deletion in the proband, SNP array analysis was performed which disclosed a 6.9 Mb heterozygous deletion on chromosome 4q35.1q35.2 (184,046,156-190,901,117 bp from rs17074417 to rs10005101, hg19) (Figure 1B). qPCR analysis of FRG1 exons 1, 8, and DUX4L6 confirmed that the distal deletion breakpoint extends beyond 190,939,252 bp (data not shown), encompassing a total of 42 annotated genes (18 OMIM genes). Based on these results, the proband’s karyotype could be assigned as 46,XY,del(4)(q35.1q35.2). SNP array analyses of maternal and paternal DNA did not indicate CNV for chromosome 4q in the parental karyotypes, consistent with a de novo deletion in the child. Informative SNPs from the terminal 4q region for which the mother and father have divergent genotypes revealed a loss of maternal genotypes in the child (Figure 1C), compatible with maternal origin of the deleted chromosome.
FISH analysis was performed with BACs from the proximal flanking region 4q35.1 (RP11-188P17) and the deleted region 4q35.1q35.2 (RP11-775P18, RP11-118M15, and RP11-652J12). As expected, the flanking BAC probe hybridized to both chromosomes in the proband and parental metaphase spreads. Probes from the deleted region recognized only one chromosome 4 homolog of the patient, but were present on both chromosome 4q35.1q35.2 copies of father and mother (Figure 1A). Obviously, the de novo deletion is not due to a cytogenetically cryptic subtelomeric translocation in a parental karyotype.
Genotype-phenotype correlation of terminal 4q deletion syndrome
We created a map of terminal 4q deletion syndrome cases through reviewing the literature and DECIPHER database. Figure 2 (upper section) presents 36 deletion cases (including our own) meeting our interval criteria and after controlling for normal CNV from the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home). Although we had to estimate best fit intervals for cases describing deletions using various low-resolution methods, we were able to roughly map out five critical regions for four common 4q deletion syndrome phenotypes: CP, CHD, ID, and ASD. Figure 2 (middle section) and Additional file 1: Table S1 show the gene content of the deletion region, with an emphasis on genes implicated in the various associated phenotypes. Additional file 2: Table S2 details case summaries including approximate deletion sizes, inheritance and phenotype information.

Genotype-phenotype correlation of terminal 4q deletions. The upper part of the figure presents the mapped deletion intervals in chromosome 4q31.1qter including the present case, marked with a box, and 35 additional cases from DECIPHER and the literature. The red bars delineate the deletion region for each case. Highlighted intervals indicate critical regions for common phenotypes among the cases. Depicted, from left to right, are the intervals for cleft palate (purple), congenital heart defect region 1 (light green), intellectual disability (dark green), congenital heart defect region 2 (red), and autism spectrum disorder (turquoise). Orange indicates the mapping interval of the DFNA24 locus. The middle section shows the gene content of the 4q31.1qter region. Likely disease-relevant genes overlapping with critical deletion intervals are boxed. The bottom diagram shows the deCODE recombination map, highlighting male and female recombination hotspots in the terminal 4q deletion syndrome region.
A locus (chr. 4: 155,600,001-158,373,133 bp) for CP (Figure 2, purple) was mapped with two out of three cases spanning this region having CP [7, 10]. The gene PDGFC is important for development of the palate with implication in non-syndromic orofacial clefting [23]. Furthermore, Pdgfc -/- knockout mice display clefting [24]. Thirteen cases under evaluation indicate CP, suggesting an additional critical interval involved in palate formation.
Congenital heart defects were mapped to two separate regions. The first region (Figure 2, light green) spans a large interval (chr. 4: 160,717,000-178,579,037 bp) unable to be further subdivided based on the cases presented. There are 17 cases with various cardiac phenotypes, 13 of which overlap with the proposed interval, with three individuals unique to this first CHD locus [10, 18, 21]. This interval contains two genes of interest (Additional file 1: Table S1). TLL1 is important for mammalian heart septation [25]. Mice with abnormalities in this gene die from blood circulation failure [26]. From mouse and zebrafish experiments, HAND2 is also involved in cardiac morphogenesis, angiogenesis, and formation of the right ventricle and aortic arch arteries and, interestingly, plays a role in limb formation [27, 28]. Although many individuals presented digital and forearm deficiencies, we were not able to clearly map these phenotypes to this region as well.
The second CHD locus (chr. 4: 184,046,156-186,997,806 bp) maps in a region containing 12 out of 17 overlapping cases with cardiac phenotypes (Figure 2, red), two of whom uniquely overlap with this region. The critical interval contains two adjacent genes, PDLIM3 and SORBS2, implicated in cardiac development. PDLIM3 is essential for right ventricular development and thought to enhance mechanical strength stability of cardiac muscle during mouse development [29]. SORBS2 is highly expressed in the intercalated disk in normal cardiac tissue [30]. Additionally, SORBS2 could have implication in CP formation, since case #20 [9] with CP has a small deletion (chr. 4: 186,533,075-186,997,806 bp) exclusively affecting SORBS2 and TLR3 (Figure 2, upper and middle section). Ten out of 13 total individuals with CP overlapped with this region, but the proximal border was too large to map an informative locus.
A smaller region (chr. 4: 171,144,641-175,897,427 bp) within the first CHD interval may account for ID, with eight of 15 individuals having ID (Figure 2, dark green). While no gene is presently linked to ID in this region, the gene SCRG1 is highly expressed in the brain and has differential regulation in schizophrenia and bipolar disorder [31] (Additional file 1: Table S1). Lack of genomic variation among healthy individuals in the Database of Genomic Variants and strong evolutionary conservation (data not shown) further emphasize the importance of normal copy number of this ID region.
A number of reports implicate chromosome 4q35.2 in ASD [20, 22]. While only four cases reviewed here have ASD (Additional file 2: Table S2), all four overlap one narrow interval (chr. 4: 187,234,067-188,424,639 bp) (Figure 2, turquoise) with only three genes (MTNR1A, FAT1 and F11), that was first reported in a boy with ASD [22]. FAT1 has been associated with bipolar affective disorder [32] and ASD [33], and is essential for controlling developmental cell proliferation [34].
Mild factor XI deficiency and elevated prothrombin time in our proband are presumably explained through deletion of F11[35, 36] and an adjacent coagulation gene, KLKB1[37]. Surprisingly, the mild bleeding tendencies that can be associated with F11 and KLKB1 deletions have not been discussed in great detail yet, although many children with terminal 4q deletion syndrome require multiple surgeries.
The first clinical symptom of our patient with a 6.9 Mb deletion (chr. 4: 184,046,156-190,901,117 bp) was mild to severe bilateral hearing loss. Two additional cases with larger deletions, DECIPHER case 256186 and the case from Calabrese et al., 1997 [16], also reported hearing impairment. In a Swiss-German kindred with autosomal dominant non-syndromic hearing loss, an autosomal dominant deafness locus, DFNA24 (MIM: 606282) was mapped to an 8.1 Mb region (chr. 4: 183,200,000-191,154,276 bp) (Figure 2, orange) on chromosome 4q35qter [38, 39]. However, in this context it is important to emphasize that 11 normal hearing terminal 4q deletion cases overlap completely and nine normal hearing cases overlap partially with the DFNA24 interval. Thus, loss of one locus copy is not sufficient to cause DFNA24. A cumulative effect of rare, pathogenic variants in different deafness genes scattered across the genome (i.e. haploinsufficiency for DFNA24) could contribute to hearing impairment [40]. Mouse knockout experiments suggest that Casp3, which is contained in the critical region, is required for proper functioning of the cochlea [41–43]. Casp3 -/- mice indicated sensorineural hearing loss, whereas Casp3 +/- mice displayed intermediate vestibular dysfunction, as well as marginally increased hair cell counts.
Discussion
Since its first description [44], the genotype-phenotype delineation of chromosome 4q deletion syndrome has been complicated by extensive inconsistencies reported among individuals with similar deletion intervals. With >170 genes residing in the terminal 4q region, delineation of the phenotypes associated with such deletions presents a tremendous task toward understanding the complete spectral presentation of a syndrome with excessive phenotypic variability. The patient we present was analyzed with a high resolution SNP array to delineate the deletion interval and the parental origin of the de novo rearrangement. We found it especially challenging to finely map disease-relevant intervals with the various low-resolution techniques that used GTG banding [7, 10, 13–15], FISH [16], and the different resolution arrays, including BAC aCGH [18], 1 Mb aCGH [19], 44 K aCGH [6, 17, 20], 105 K aCGH [9, 22], and 300 K SNP array [21]. Another limitation includes possible variations in the depth of clinical descriptions listed, especially those from the DECIPHER database, which were not as detailed as the published cases. Collectively, case-supported critical regions for several distinct phenotypes such as CP, CHD, ID, and ASD were defined. In this context, it is important to emphasize that most phenotypic features that are associated with terminal 4q deletion syndrome show incomplete penetrance and/or are rather unspecific, which renders genotype-phenotype correlations difficult.
The overwhelming majority of cases are de novo possibly due to errors during meiotic recombination leading to a loss of chromosomal material from one parental allele. Meiotic crossovers preferentially occur at non-random hotspots which have been mapped according to frequency and spatial distribution in both males and females [45]. The deCODE recombination map [46] of the 4q31.1qter region illustrates an enrichment of both male and female hotspots along the major part of this interval (Figure 2, bottom section). However, it is also possible that deletions arise in mitotically dividing spermatogonial and oogonial stem cells, respectively. The resulting germ-cell mosaicism would increase the likelihood of having another child with the same deletion.
In summary, the case presented here is the first to use a SNP array to determine the parent-of-origin of the large deletion. Assuming that the same gene(s) is underlying hearing impairment in terminal 4q deletion and DFNA24 patients, it may help further narrow the DFNA24 locus. Our case, in combination with the cases described in the literature and DECIPHER, accommodate a proposal of critical phenotypic intervals with possible genes of interest. This review is not intended as a holistic description of terminal chromosome 4q deletion syndrome. However, the ongoing reporting of precisely defined deletion intervals with higher resolution technologies will support eventual refinement and possible clarification of the genes and pathways responsible for the broad phenotypic presentation of deletions in this interval of chromosome 4q.
Consent
The study was approved by the Ethics Committee of the University of Würzburg. Full informed parental consent was obtained prior to initiating our investigation.
Abbreviations
- ASD:
-
Autism spectrum disorder
- BAC:
-
Bacterial artificial chromosome
- CHD:
-
Congenital heart defect
- CNV:
-
Copy number variation
- CP:
-
Cleft palate
- FISH:
-
Fluorescence in situ hybridization
- ID:
-
Intellectual disability
- PCR:
-
Polymerase chain reaction.
References
Strehle EM, Ahmed OA, Hameed M, Russell A: The 4q- syndrome. Genet Couns. 2001, 12: 327-339.
Strehle EM, Bantock HM: The phenotype of patients with 4q-syndrome. Genet Couns. 2003, 14: 195-205.
Descartes M, Keppler-Noreuil K, Knops J, Longshore JW, Finley WH, Carroll AJ: Terminal deletion of the long arm of chromosome 4 in a mother and two sons. Clin Genet. 1996, 50: 538-540.
Ravnan JB, Tepperberg JH, Papenhausen P, Lamb AN, Hedrick J, Eash D, Ledbetter DH, Martin CL: Subtelomere FISH analysis of 11 688 cases: an evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J Med Genet. 2006, 43: 478-489. 10.1136/jmg.2005.036350.
Balikova I, Menten B, de Ravel T, Le Caignec C, Thienpont B, Urbina M, Doco-Fenzy M, de Rademaeker M, Mortier G, Kooy F, van den Ende J, Devriendt K, Fryns JP, Speleman F, Vermeesch JR: Subtelomeric imbalances in phenotypically normal individuals. Hum Mutat. 2007, 28: 958-967. 10.1002/humu.20537.
Rossi MR, DiMaio MS, Xiang B, Lu K, Kaymakcalan H, Seashore M, Mahoney MJ, Li P: Clinical and genomic characterization of distal duplications and deletions of chromosome 4q: study of two cases and review of the literature. Am J Med Genet A. 2009, 149A: 2788-2794. 10.1002/ajmg.a.33088.
Giuffrè M, La Placa S, Carta M, Cataliotti A, Marino M, Piccione M, Pusateri F, Meli F, Corsello G: Hypercalciuria and kidney calcifications in terminal 4q deletion syndrome: further evidence for a putative gene on 4q. Am J Med Genet A. 2004, 126A: 186-190. 10.1002/ajmg.a.20561.
Strehle EM, Gruszfeld D, Schenk D, Mehta SG, Simonic I, Huang T: The spectrum of 4q- syndrome illustrated by a case series. Gene. 2012, 506: 387-391. 10.1016/j.gene.2012.06.087.
Strehle EM, Yu L, Rosenfeld JA, Donkervoort S, Zhou Y, Chen TJ, Martinez JE, Fan YS, Barbouth D, Zhu H, Vaglio A, Smith R, Stevens CA, Curry CJ, Ladda RL, Fan ZJ, Fox JE, Martin JA, Abdel-Hamid HZ, McCracken EA, McGillivray BC, Masser-Frye D, Huang T: Genotype-phenotype analysis of 4q deletion syndrome: proposal of a critical region. Am J Med Genet A. 2012, 158A: 2139-2151. 10.1002/ajmg.a.35502.
Keeling SL, Lee-Jones L, Thompson P: Interstitial deletion 4q32-34 with ulnar deficiency: 4q33 may be the critical region in 4q terminal deletion syndrome. Am J Med Genet. 2001, 99: 94-98. 10.1002/1096-8628(2000)9999:999<00::AID-AJMG1134>3.0.CO;2-D.
Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, Bassett AS, Seller A, Holmes CC, Ragoussis J: QuantiSNP: an objective Bayes hidden-Markov model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007, 35: 2013-2025. 10.1093/nar/gkm076.
Thiele H, Nürnberg P: HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005, 21: 1730-1732. 10.1093/bioinformatics/bth488.
Jefferson RD, Burn J, Gaunt KL, Hunter S, Davison EV: A terminal deletion of the long arm of chromosome 4 [46, XX, del(4)(q33)] in an infant with phenotypic features of Williams syndrome. J Med Genet. 1986, 23: 474-477. 10.1136/jmg.23.5.474.
Menko FH, Madan K, Baart JA, Beukenhorst HL: Robin sequence and a deficiency of the left forearm in a girl with a deletion of chromosome 4q33-qter. Am J Med Genet. 1992, 44: 696-698. 10.1002/ajmg.1320440532.
Borochowitz Z, Shalev SA, Yehudai I, Bar-el H, Dar H, Tirosh E: Deletion (4)(q33- > qter): a case report and review of the literature. J Child Neurol. 1997, 12: 335-337. 10.1177/088307389701200510.
Calabrese G, Giannotti A, Mingarelli R, Di Gilio MC, Piemontese MR, Palka G: Two newborns with chromosome 4 imbalances: deletion 4q33→q35 and ring r(4)(pterq35.2-qter). Clin Genet. 1997, 51: 264-267.
Quadrelli R, Strehle EM, Vaglio A, Larrandaburu M, Mechoso B, Quadrelli A, Fan YS, Huang T: A girl with del(4)(q33) and occipital encephalocele: clinical description and molecular genetic characterization of a rare patient. Genet Test. 2007, 11: 4-10. 10.1089/gte.2006.9995.
Kaalund SS, Møller RS, Teszas A, Miranda M, Kosztolanyi G, Ullmann R, Tommerup N, Tümer Z: Investigation of 4q-deletion in two unrelated patients using array CGH. Am J Med Genet A. 2008, 146A: 2431-2434. 10.1002/ajmg.a.32458.
Kitsiou-Tzeli S, Sismani C, Koumbaris G, Ioannides M, Kanavakis E, Kolialexi A, Mavrou A, Touliatou V, Patsalis PC: Distal del(4) (q33) syndrome: detailed clinical presentation and molecular description with array-CGH. Eur J Med Genet. 2008, 51: 61-67. 10.1016/j.ejmg.2007.09.004.
Chien WH, Gau SS, Wu YY, Huang YS, Fang JS, Chen YJ, Soong WT, Chiu YN, Chen CH: Identification and molecular characterization of two novel chromosomal deletions associated with autism. Clin Genet. 2010, 78: 449-456. 10.1111/j.1399-0004.2010.01395.x.
Xu W, Ahmad A, Dagenais S, Iyer RK, Innis JW: Chromosome 4q deletion syndrome: narrowing the cardiovascular critical region to 4q32.2-q34.3. Am J Med Genet A. 2012, 158A: 635-640. 10.1002/ajmg.a.34425.
Youngs EL, Henkhaus RS, Hellings JA, Butler MG: 12-year-old boy with a 4q35.2 microdeletion and involvement of MTNR1A, FAT1, and F11 genes. Clin Dysmorphol. 2012, 21: 93-96. 10.1097/MCD.0b013e32834e9216.
Wu D, Wang M, Wang X, Yin N, Song T, Li H, Zhang F, Zhang Y, Ye Z, Yu J, Wang DM, Zhao Z: Maternal transmission effect of a PDGF-C SNP on nonsyndromic cleft lip with or without palate from a Chinese population. PLoS One. 2012, 7: e46477-10.1371/journal.pone.0046477.
Ding H, Wu X, Bostrom H, Kim I, Wong N, Tsoi B, O’Rourke M, Koh GY, Soriano P, Betsholtz C, Hart TC, Marazita ML, Field LL, Tam PP, Nagy A: A specific requirement for PDGF-C in palate formation and PDGFR-alpha signaling. Nat Genet. 2004, 36: 1111-1116. 10.1038/ng1415.
Stańczak P, Witecka J, Szydło A, Gutmajster E, Lisik M, Auguściak-Duma A, Tarnowski M, Czekaj T, Czekaj H, Sieroń AL: Mutations in mammalian tolloid-like 1 gene detected in adult patients with ASD. Eur J Hum Genet. 2009, 17: 344-351. 10.1038/ejhg.2008.175.
Clark TG, Conway SJ, Scott IC, Labosky PA, Winnier G, Bundy J, Hogan BL, Greenspan DS: The mammalian Tolloid-like 1 gene, Tll1, is necessary for normal septation and positioning of the heart. Development. 1999, 126: 2631-2642.
Sasaki MM, Nichols JT, Kimmel CB: Edn1 and hand2 interact in early regulation of pharyngeal arch outgrowth during zebrafish development. PLoS One. 2013, 8: e67522-10.1371/journal.pone.0067522.
Tamura M, Hosoya M, Fujita M, Iida T, Amano T, Maeno A, Kataoka T, Otsuka T, Tanaka S, Tomizawa S, Shiroishi T: Overdosage of Hand2 causes limb and heart defects in the human chromosomal disorder partial trisomy distal 4q. Hum Mol Genet. 2013, 22: 2471-2481. 10.1093/hmg/ddt099.
Pashmforoush M, Pomies P, Peterson KL, Kubalak S, Ross J, Hefti A, Aebi U, Beckerle MC, Chien KR: Adult mice deficient in actinin-associated LIM-domain protein reveal a developmental pathway for right ventricular cardiomyopathy. Nat Med. 2001, 7: 591-597. 10.1038/87920.
Kakimoto Y, Ito S, Abiru H, Kotani H, Ozeki M, Tamaki K, Tsuruyama T: Sorbin and SH3 domain-containing protein 2 is released from infarcted heart in the very early phase: proteomic analysis of cardiac tissues from patients. J Am Heart Assoc. 2013, 2: e000565-10.1161/JAHA.113.000565.
Vawter MP, Atz ME, Rollins BL, Cooper-Casey KM, Shao L, Byerley WF: Genome scans and gene expression microarrays converge to identify gene regulatory loci relevant in schizophrenia. Hum Genet. 2006, 119: 558-570. 10.1007/s00439-006-0172-7.
Abou Jamra R, Becker T, Georgi A, Feulner T, Schumacher J, Stromaier J, Schirmbeck F, Schulze TG, Propping P, Rietschel M, Nöthen MM, Cichon S: Genetic variation of the FAT gene at 4q35 is associated with bipolar affective disorder. Mol Psychiatry. 2008, 13: 277-284. 10.1038/sj.mp.4002111.
Roberts JL, Hovanes K, Dasouki M, Manzardo AM, Butler MG: Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders or learning disability presenting for genetic services. Gene. 2014, 535: 70-78. 10.1016/j.gene.2013.10.020.
Bruder-Nascimento T, Chinnasamy P, Riascos-Bernal DF, Cau SB, Callera GE, Touyz RM, Tostes RC, Sibinga NE: Angiotensin II induces Fat1 expression/activation and vascular smooth muscle cell migration via Nox1-dependent reactive oxygen species generation. J Mol Cell Cardiol. 2013, 66: 18-26.
Mitchell M, Dai L, Savidge G, Alhaq A: An Alu-mediated 31.5-kb deletion as the cause of factor XI deficiency in 2 unrelated patients. Blood. 2004, 104: 2394-2396. 10.1182/blood-2004-04-1318.
Duga S, Salomon O: Congenital factor XI deficiency: an update. Semin Thromb Hemost. 2013, 39: 621-631. 10.1055/s-0033-1353420.
Koumandou VL, Scorilas A: Evolution of the plasma and tissue kallikreins, and their alternative splicing isoforms. PLoS One. 2013, 8: e68074-10.1371/journal.pone.0068074.
Häfner FM, Salam AA, Linder TE, Balmer D, Baumer A, Schinzel AA, Spillmann T, Leal SM: A novel locus (DFNA24) for prelingual nonprogressive autosomal dominant nonsyndromic hearing loss maps to 4q35-qter in a large Swiss German kindred. Am J Hum Genet. 2000, 66: 1437-1442. 10.1086/302865.
Santos RL, Häfner FM, Huygen PL, Linder TE, Schinzel AA, Spillmann T, Leal SM: Phenotypic characterization of DFNA24: prelingual progressive sensorineural hearing impairment. Audiol Neurootol. 2006, 11: 269-275. 10.1159/000093525.
Vona B, Müller T, Nanda I, Neuner C, Hofrichter MAH, Schröder J, Bartsch O, Läßig A, Keilmann A, Schraven S, Kraus F, Shehata-Dieler W, Haaf T: Targeted next-generation sequencing of deafness genes in hearing-impaired individuals uncovers informative mutations. Genet Med. 2014, PMID 24875298:Epub ahead of print
Morishita H, Makishima T, Kaneko C, Lee YS, Segil N, Takahashi K, Kuraoka A, Nakagawa T, Nabekura J, Nakayama K, Nakayama KI: Deafness due to degeneration of cochlear neurons in caspase-3-deficient mice. Biochem Biophys Res Comm. 2001, 284: 142-149. 10.1006/bbrc.2001.4939.
Parker A, Hardisty-Hughes RE, Wisby L, Joyce S, Brown SD: Melody, an ENU mutation in caspase 3, alters the catalytic cysteine residue and causes sensorineural hearing loss in mice. Mamm Genome. 2010, 21: 565-576. 10.1007/s00335-010-9306-2.
Makishima T, Hochman L, Armstrong P, Rosenberger E, Ridley R, Woo M, Perachio A, Wood S: Inner ear dysfunction in caspase-3 deficient mice. BMC Neurosci. 2011, 12: 102-10.1186/1471-2202-12-102.
Ockey CH, Feldman GV, Macaulay ME, Delaney MJ: A large deletion of the long arm of chromosome no. 4 in a child with limb abnormalities. Arch Dis Child. 1967, 42: 428-434. 10.1136/adc.42.224.428.
Kauppi L, Jeffreys AJ, Keeney S: Where the crossovers are: recombination distributions in mammals. Nat Rev Genet. 2004, 5: 413-424. 10.1038/nrg1346.
Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, Jonasdottir A, Walters GB, Jonasdottir A, Gylfason A, Kristinsson KT, Gudjonsson SA, Frigge ML, Helgason A, Thorsteinsdottir U, Stefansson K: Fine-scale recombination rate differences between sexes, populations and individuals. Nature. 2010, 467: 1099-1103. 10.1038/nature09525.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/15/72/prepub
Acknowledgements
The authors are grateful for the participation of the family for their willingness to engage in this study. We also thank Prof. Dr. Holger Thiele from the Cologne Center for Genomics for constructive dialogue about HaploPainter, Dr. Christopher Riley at Phoenix Children’s Hospital for clinical assessment, and Andrea Hörning for assistance with karyotype analysis. This work was supported by the German Research Foundation (grant no. HA 1374/7-2) and the University of Würzburg in the funding programme Open Access Publishing.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
No potential competing interest as well as commercial interests were disclosed.
Authors’ contributions
BV, IN and CN carried out the molecular cytogenetic and data analyses. JS and WSD performed a clinical analysis of the patient. VK provided materials. BV and TH researched the literature and wrote the manuscript. All authors have critically reviewed and approved the manuscript.
Electronic supplementary material
12881_2014_1216_MOESM1_ESM.doc
Additional file 1: Table S1: Summary of disease-relevant genes in the deletion region with functions, phenotypes and cases with agreeable phenotypes. (DOC 75 KB)
12881_2014_1216_MOESM2_ESM.doc
Additional file 2: Table S2: Summary of our proband and cases from DECIPHER and the literature with deletions exclusively residing in the 4q31.1qter region. (DOC 125 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Vona, B., Nanda, I., Neuner, C. et al. Terminal chromosome 4q deletion syndrome in an infant with hearing impairment and moderate syndromic features: review of literature. BMC Med Genet 15, 72 (2014). https://doi.org/10.1186/1471-2350-15-72
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-15-72