
Abstract
Background
Spinal muscular atrophy (SMA type I, II and III) is an autosomal recessive neuromuscular disorder caused by mutations in the survival motor neuron gene (SMN1). SMN2 is a centromeric copy gene that has been characterized as a major modifier of SMA severity. SMA type I patients have one or two SMN2 copies while most SMA type II patients carry three SMN2 copies and SMA III patients have three or four SMN2 copies. The SMN1 gene produces a full-length transcript (FL-SMN) while SMN2 is only able to produce a small portion of the FL-SMN because of a splice mutation which results in the production of abnormal SMNΔ7 mRNA.
Methods
In this study we performed quantification of the SMN2 gene copy number in Russian patients affected by SMA type II and III (42 and 19 patients, respectively) by means of real-time PCR. Moreover, we present two families consisting of asymptomatic carriers of a homozygous absence of the SMN1 gene. We also developed a novel RT-qPCR-based assay to determine the FL-SMN/SMNΔ7 mRNA ratio as SMA biomarker.
Results
Comparison of the SMN2 copy number and clinical features revealed a significant correlation between mild clinical phenotype (SMA type III) and presence of four copies of the SMN2 gene. In both asymptomatic cases we found an increased number of SMN2 copies in the healthy carriers and a biallelic SMN1 absence. Furthermore, the novel assay revealed a difference between SMA patients and healthy controls.
Conclusions
We suggest that the SMN2 gene copy quantification in SMA patients could be used as a prognostic tool for discrimination between the SMA type II and SMA type III diagnoses, whereas the FL-SMN/SMNΔ7 mRNA ratio could be a useful biomarker for detecting changes during SMA pharmacotherapy.
Similar content being viewed by others

Background
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder characterized by degeneration of alpha motor neurons in the anterior horns of the spinal cord. SMA is one of the leading genetic causes of infant mortality, with an incidence of about 1 in 8000 live births and with a carrier frequency of about 1 in 40 [1]. SMA subdivides into types I, II, and III based on age of onset and clinical severity [2], and are all caused by mutations within the survival motor neuron gene (SMN1) located on chromosome 5q13. Approximately 95 percent of these patients have a homozygous deletion of exon 7 in the SMN1 gene. Small intragenic mutations have been found in the remaining affected individuals who do not lack both copies of SMN1 [3]. SMN2 is an almost identical centromeric SMN1 copy, with 8 nucleotide substitutions: five in the introns and three in exons 6, 7 and 8 [4]. Single-nucleotide transition of C to T at position 6 of SMN2 exon 7 does not change an amino acid, but causes disruption of a splicing enchancer site, which results in mRNA lacking exon 7 (SMNΔ7) [5]. Most of the SMN1 transcripts are full-length whereas the majority of the SMN2 transcripts lack exon 7. Consequently, SMN2 is not able to compensate for the loss of exon 7 in SMN1 of the SMA patients. Nevertheless, the SMN2 copy number is considered to be a modifying factor for the clinical severity of SMA. SMA type I patients have one or two SMN2 copies while most SMA type II patients carry three SMN2 copies and SMA III patients have three or four SMN2 copies [6–8].
The SMN1 gene normally produces full-length SMN mRNA, whereas conversely approximately only 20 percent of the full-length SMN mRNA is produced from the SMN2 gene [9]. This observation suggests a potential significant distinction in the FL-SMN transcript levels among healthy individuals, carriers and different types of SMA patients. However, there are conflicting results concerning the differences between the levels of FL-SMN and Δ7SMN mRNA in SMA patients, carriers and healthy people [10–12]. Decreased FL-SMN expression has been shown only for type I SMA patients. Fluctuations in transcript levels throughout drug treatment were also observed in recent clinical trials [13]. However, it remains unclear whether the levels of FL-SMN and Δ7SMN mRNA are associated with the type of disease or the copy number of SMN2.
In this study, we performed SMN2 gene copy number determination for patients affected by SMA type II and III (42 and 19 patients, respectively). We also present two case-reports: one SMA type III patient and his asymptomatic sister, and one SMA type II patient and his asymptomatic mother being homozygous for the absence of the SMN1 gene. To evaluate the importance of the FL-SMN/SMNΔ7 mRNA ratio as a SMA biomarker, we carried out RT-qPCR based quantification of FL-SMN and SMNΔ7 mRNA levels in a small group of type II and III SMA patients.
Methods
Patients
Genetic analysis was approved by the ethics committee at Ott's Institute of Obstetrics and Gynecology RAMS. The adult patients and parents of all children gave written informed consent to the diagnostic procedures. Molecular testing for deletion in the SMN1 gene was performed in 190 Russian families from the North-Western region of Russia with SMA including 172 affected individuals, 243 parents, 14 siblings and 44 other relatives (totally 301 relatives were tested). SMN2 gene copy number determination was performed for 42 type II SMA-patients and 19 type III SMA-patients. SMN2 gene copy number was also determined in several clinically healthy family members including four parents, two siblings and one grandmother. Blood samples for the determination of SMN gene expression were obtained from five SMA patients, one SMA carrier and two healthy individuals (Table 1).
DNA isolation
Genomic DNA was extracted from peripheral blood leukocytes of SMA patients, their relatives and healthy individuals using the phenol-chloroform method [14].
Polymerase chain reaction - restriction fragment length polymorphism (PCR-RFLP) and single strand conformation analysis (SSCA)
All SMA patients and their relatives were tested for SMN1 exon 7 and 8 deletions by means of SSCA (for exon 7) and PCR-RFLP analysis (for exon 8). PCR-RFLP and SSCA methods were performed as described previously [15].
Real-time PCR for SMN2gene copy number determination
We applied the TaqMan real-time PCR method as previously described [16]. The DNA sample of the SMA patient with four SMN2 gene copies was used as standard in all amplification reactions. The presence of four copies was established earlier using reference DNA samples kindly provided by prof. B. Wirth (Institute of Human Genetics, University of Cologne). In each run we also used DNA samples from SMA patients with two and three copies as additional standards. The β-globine (HBB) gene was used as a reference locus in line with previously published primers and probe [17].
PCR was performed using the thermocycler Rotor-Gene 3000 (Corbett Life Science, Australia). PCR was carried out in a total volume of 20 μl and contained 10-160 ng of genomic DNA, 1 μl of each primers (1 A260 o.u.) (Syntol, Russia), 2 μl of 2 μM SMN2 MGB (Minor Groove Binder) probe (Applied Biosystems, USA), 2 μl of 2 μM of HBB probe (Syntol, Russia), 2.5 μl of 2 mM dNTP mix (Fermentas, Lithuania), 2 μl of 25 mM MgCl2, 2 μl of 10× polymerase buffer and 0.3 μl of Taq DNA polymerase (Sileks, Russia).
We amplified 160 ng, 80 ng, 40 ng, 10 ng of standard DNA and 50 ng of each tested DNA in one run. PCR conditions were as follows: 94°C for 10 minutes, 40 cycles consisting of 95°C for 20 seconds, 60°C for 1 minute. Data was analyzed by Rotor-Gene version 6.1.71 software. All samples were measured three times and a final result was inferred by averaging data. The value ranges for two, three and four SMN2 gene copy number were estimated. Confidence of differences between the values range was confirmed using unpaired Student's t-test (p < 0.01), using GraphPad Prism v5.03 (GraphPad, USA).
RNA isolation and cDNA synthesis
Peripheral blood was collected into Vacuette tubes (Greiner Bio One, USA). 2 ml of blood was lysed with hemolysis buffer (8.4 g/l NH4Cl) for 30 minutes. The leukocyte phase was obtained by centrifuging the blood sample at 1500 rpm for 20 minutes. Collected cells were stored in RNAlater solution (Quiagen, Germany) for up to twelve months at -80°C. RNA isolation was performed simultaneously from all samples using RiboPure - Blood Kit (Applied Biosystems). For cDNA synthesis 12 μl of each RNA sample was incubated with 0.5 μl of 25 mM dNTP mix (Fermentas) and 0.5 μl of hexaprimers (50 A260 o.e.) (Roche) at 65°C for 5 minutes. Samples were kept on ice for 1 minute. 4 μl of 5× FS buffer, 2 μl 0, 1 M DTT and 1 μl of M-MLV Reverse transcriptase (Invitrogen) were added to samples. Reactions were incubated at 25°C for 10 minutes, then at 37°C for 1 hour, then at 95°C for 15 minutes.
Quantitative reverse transcriptase real-time PCR
The assay was carried out on a MyiQ thermal cycler (Bio-Rad Laboratories, Sweden), using 96-well plates. The primers were designed using Beacon Primer Design 4.0 software (Premier Biosoft, USA). Primers for full-length SMN transcripts amplification were forward 5'-CTGATGCTTTGGGAAGTAT-3', reverse 5'-GCCAGCATTTCTCCTTAA-3', for Δ7SMN transcripts amplification were forward 5'-GTCCAGATTCTCTTGATGAT-3', reverse 5'-GCCAGCATTTCCATATAATAG-3'. We also amplified two reference genes - glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and histone H3b. Primers sequences were following: for GAPDH locus forward 5'-CGCCAGCCGAGCCACATC-3', reverse 5'-CGCCCAATACGACCAAATCCG-3'; for H3b locus forward 5'-ATCCGCCGCTACCAAAAG-3', reverse 5'-CGAAGATCGGTCTTGAAGTC-3'. Reactions were performed in the final volume of 20 μl and contained 5 μl of cDNA (5 ng/μl), 0.05 μl of each primer (100 pmol/μl), 1 μl DMSO, 0.5 μl of SYBR GREEN I (1:50000; Invitrogen, Sweden) in TE buffer (pH 7.8), 0.2 μl of 25 mM dNTP mix (Fermentas), 2 μl 10× buffer, 1.6 μl of 50 mM MgCl2, 0.08 μl of Taq DNA polymerase (Biotools, Spain). Cycling conditions were as follows: 3-minutes initial denaturation step, followed by 40 cycles of 95°C for 20 seconds, 20 seconds at 55-60.5 (optimal annealing temperature of primers), 30 seconds at 72°C. Fluorescence was measured after the elongation phase. 81 cycles of 10 sec at 55°C with increasing increments of 0.5°C per cycle was performed for melting curve analysis. A negative control for each primer's pair was included on each plate. All samples were run in duplicate. MyiQ software v 1.04 (Bio-Rad Laboratories, Sweden) was used to process real-time PCR data and determine threshold cycle (Ct) values. Melting curve analysis was performed to confirm that only one product was amplified and there were not any products in negative controls. LinRegPCR was used to calculate PCR efficiencies for each sample. Outliers were excluded and the average PCR efficiency for each primers pair was calculated using Grubbs' test for outliers (GraphPad, USA). Relative quantities with standard deviations were calculated using the delta Ct method. Normalization was performed via the GeNorm method using expression levels of GAPDH and H3b.
Results
Quantification of SMN2gene copy number in patients with spinal muscular atrophy
In this study we performed SMN2 dosage analysis in 42 type II-, and 19 type III SMA patients. The SMN2 gene copy number was determined using a real-time PCR quantification method with MGB-TaqMan probes [16]. DNA samples from the SMA patients carrying between one and four SMN2 copies were used as controls for the analysis.
We found that the sample-to-sample variation after relative quantification analysis for the two SMN2 gene copies per genome was in the range of 0.73-1.32; for three copies per genome it was in range of 1.27-1.72; and for four copies per genome it was in range of 1.71-2.2. These values were multiplied by a factor of two to obtain the gene copy number. These results are in agreement with previously published data obtained by real-time PCR approaches using TaqMan or LightCycler techniques [8, 16, 18].
The results of the SMN2 gene copy number quantification in the SMA patients are presented in Figure 1. It can be seen that most of the SMA type II patients have three copies of SMN2 gene per genome (76.2%), whereas two and four copies were found in equal numbers of the patients (11.9%). For the SMA type III group, we found three copies of SMN2 gene for lesser number of patients (57.9%) while four copies were found in 7 of 19 patients (36.8%). It should be noted that we found one patient affected by SMA type III bearing two copies of the SMN2 gene (5.3%). The average SMN2 copy number in type II and type III patients was found to be similar (3.0 ± 0.49 vs 3.32 ± 0.58; p > 0.01). We were not able to reveal significant differences between the SMA type II and III patients bearing two copies of the SMN2 gene (5 of 42 type II patients and one of 19 type III patients) (p > 0.05). This was likely due to the low number of individuals in each group. The difference in frequency of three copies of SMN2 gene between type II and III patients was also not found to be significant (32 of 42 type II patients and 11 of 19 type III patients) (p > 0.05). However, we did find a significant difference in the frequency of having four SMN2 gene copies between type II and type III patients (p = 0.0233, χ2 = 5.148).

Frequencies (%) of the SMN2 gene copy numbers in patients with SMA type II and III. * - χ2-test was used for the comparison of SMN2 gene copy number between type II and type III patients (p = 0.0233, χ2 = 5.148).
Asymptomatic family members lacking both copies of the SMN1gene
We carried out routine molecular genetic diagnoses in 301 clinically healthy members of Russian SMA families, including obligatory deletion carriers such as parents and siblings who have a chance of being heterozygous carriers, and other relatives. In each family, deletions of exons 7 and 8 of SMN1 and SMN2 were determined by means of SSCP analysis and RFLP analysis [15]. As a result of the diagnosis, we revealed two families of asymptomatic individuals who were homozygous for the absence of exons 7 and 8 of SMN1. Therefore, the prevalence of asymptomatic cases in Russian families may be estimated at approximately 0.7% of first-degree relatives of SMA patients.
Family 1 (no. S265)
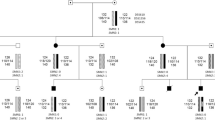
There were two half-siblings in family 1 (Figure 2a). The younger brother was affected with SMA type II. The diagnosis was confirmed by molecular testing of deletions of the SMN1 exon 7 and 8. Molecular diagnostics showed a heterozygous deletion carrier state in a healthy stepbrother (III:1). The father (II:2) of the affected boy (III:2) was found to be heterozygous for the deletion, whereas the 45 year old mother (II:1) carried a biallelic deletion of exon 7 and 8 of SMN1. Quantification of SMN2 gene copy number in the members of family 1 allowed us to explain the absence of SMA symptoms in the mother. It was shown that the father and the SMA type II affected son carried two copies of SMN2. Three copies of SMN2 were found in the healthy stepbrother. Furthermore, five copies of the SMN2 gene were found in the asymptomatic mother.

Pedigrees of spinal muscular atrophy (SMA) families with biallelic absence of the SMN1 gene in unaffected subjects. (a) family 1, (b) family 2; circles represent females, squares represent males, black symbols represent biallelic SMN1 absence in affected individuals, dot symbols represent heterozygous SMA carriers, vertical line symbols represent biallelic SMN1 absence in unaffected individuals.
Family 2 (no. S280)
There were two siblings in family 2 (Figure 2b). The younger brother was affected with SMA type IIIb. The actual SMA symptoms debuted at age 21. Molecular diagnostics confirmed the original diagnosis and revealed a homozygous deletion of exon 7 and 8 of SMN1. The same analysis showed biallelic deletion in a healthy 25 years old sister (II:1). Both parents were found to be deletion carriers. The number of SMN2 gene copies was determined in the members of family 2. We found two copies of SMN2 in the father and four copies in the mother. The analysis revealed four copies of SMN2 gene in the proband and his asymptomatic sister.
Measurement of SMN baseline expression levels
In the present study we have developed a novel assay based on real-time PCR, which allows the quantification of FL-SMN and SMNΔ7 transcripts. The assay was evaluated by characterization of the SMN baseline expression level in blood samples taken from eight individuals - two patients affected by SMA type II, two patients affected by SMA type III, one asymptomatic individual, one SMA carrier and two healthy controls (Table 1). The assay was used to investigate whether FL-SMN mRNA levels are reduced in the patients compared with healthy individuals.
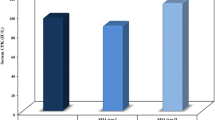
Baseline level of the FL-SMN mRNA expression varied greatly in the SMA patients in range 0.85 - 3.23 (Figure 3a). Mean ± SEM was found to be 1.97 ± 0.43. In addition, sustained variability was observed in the baseline level of SMNΔ7 mRNA expression in range 0.83 - 2.78. Mean ± SEM was found to be 1.89 ± 0.37 (Figure 3b). Thus the mRNA levels of FL-SMN and SMNΔ7 in the control subjects overlapped with the values for the SMA patients, which represented similar, slightly lower (for FL-SMN) or slightly higher (for SMNΔ7) values. It is suggested that the similarity in expression levels between control individuals and the patients may be due to increased SMN2 gene copy number. Three SMA patients from the studied cohort were found to have three SMN2 gene copies whereas the other two had four SMN2 gene copies (Table 1).

SMN gene baseline expression variability in SMA patients and healthy individuals. (a) the variability of baseline full-length SMN transcript level; (b) the variability of baseline SMNΔ7 transcript level; (c) the variability of baseline FL-SMN/SMNΔ7 mRNA ratio.
Previously it has been shown that FL-SMN/SMNΔ7 mRNA ratio can be used in the SMN expression analysis in order to avoid confounding effects of FL-SMN and SMNΔ7 mRNA level fluctuations [13]. Taking this approach we analysed FL-SMN/SMNΔ7 mRNA ratio in the patients and control individuals. It can be seen in Figure 3c that the quantification of FL-SMN/SMNΔ7 mRNA ratio revealed a difference between SMA patients and healthy controls. This parameter varied in the range of 0.56 - 2.10. The mean ± SEM was found to be 1.15 ± 0.27.
Discussion
By comparing the SMN2 gene copy number and clinical features of patients with SMA, we have revealed, for the first time in a Russian population, a significant correlation between mild clinical phenotype (SMA type III) and a presence of four copies of SMN2 gene. The SMN2 gene is considered to be a major modifier of SMA. Despite the presence of the splice mutation in exon 7, the SMN2 gene is able to produce up to 10% of FL-SMN mRNA. Multiplication of SMN2 gene copies in the genome of SMA patients can therefore lead to variability in the SMA phenotype [8, 19, 20]. Quantification of the SMN2 gene copy number has been suggested to be an important prognostic criterion for SMA diagnosis [8]. In our study, we suggest that the SMA patients bearing four copies of the SMN2 gene can be considered as type III patients rather than the II type. This is because, according to the data obtained (Figure 1) 12 percent of SMA type II patients were found to have four copies. Thus, the specificity of the SMN2 gene copy number as a prognostic tool can be estimated as an 88 percent risk for a child with four SMN2 gene copies to have SMA type III. For example in a previous study, the risk of developing SMA type III in a child with four SMN2 copies was estimated to be 83.6 percent [8]. This finding is in concordance with previously published data concerning genotype-phenotype correlation in SMA patients [8, 19]. However, it should be noted that this genotype-phenotype correlation is not absolute and can be influenced by modifying genes, point mutations or conversions of SMN2 [8]. Our results suggest that the SMN2 gene copy measurements in SMA patients can be used as a prognostic tool for discrimination between the SMA type II and SMA type III diagnosis.
The importance of the SMN2 gene copy number determination can be further illustrated by our findings in families that have unaffected relatives with a homozygous loss of the SMN1 gene. Being an exceptionally rare event, homozygous deletion of SMN1 exon 7 and 8 in unaffected relatives of SMA patients has been reported several times in American, German and Polish SMA families [21–24]. In this study we were able, for the first time, to report two cases of asymptomatic individuals from Russian SMA families. In both of the cases of asymptomatic loss of the SMN1 gene the influence of SMN2 copy number on the SMA severity was demonstrated. In family 1, we see that the higher SMN2 copy number completely abolishes the SMA phenotype in a mother of a child severely affected by SMA type II. The second case demonstrates that four copies of SMN2 results in a very mild SMA phenotype in a man homozygous for absence of SMN1. Conversely, no symptoms were observed in the sister who had the same genotype. This can only be explained by influence on SMA phenotype by genetic factors other than the SMN2 gene copy number. Family 2 represents a rare case of siblings with identical SMN1 mutations, identical SMN2 copy number, but discrepancy in phenotype. This finding suggests that there are additional factors, such as mutations, genetic and epigenetic modifiers to influence the severity of SMA. For instance, higher expression of plastin 3 has been observed in asymptomatic SMN1-deleted females, which has been shown to rescue axon growth defects in animal models for SMN1 deficiency [25]. The c.859G > C substitution in SMN2 gene has also been suggested as a positive modifier of SMA phenotype [26]. This substitution forms a new exonic splicing enhancer that increases the amount of full-length SMN2 mRNA. The c.859G > C variant was detected in SMA patients discordant for the disease severity and the SMN2 gene copy number [27]. Variations in the disease progression between SMA patients with identical number of SMN2 gene can also be explained by differences in DNA methylation. Methylation of a small part of CpG islands in the region of upstream and downstream of the translational SMN2 start site has been identified to correlate with the disease severity [28]. Gender could be suggested as SMA severity modifiyng factor; to date most of asymptomatic persons with homozygous loss of the SMN1 gene are females [29].
The number of SMN2 gene copies in SMA patients has been studied intensively in several previous studies including different populations. However, to date no previous publications concerning Russian populations have been analysed. Here we present SMN2 dosage analysis in 61 SMA patients who originated from North-Western Russia. Individuals affected by SMA type II and III were subjected to the analysis because it has been suggested that their milder phenotype is a result of an increase of the SMN2 gene copy number. The distribution of SMN2 gene copy number observed in the SMA patients from the northwestern region of Russia was compared with the distributions reported for the SMA patients from some other populations. We did not observe any difference in SMN2 gene copy number distribution between Russian and German, Spanish and Vietnamese SMA patients [8, 18, 19].
Recent clinical trials on SMA treatment by histone deacetylation inhibitors such as valproic acid and phenylbutyrate have revealed some promising results. These trials focused on children with SMA types II and III [13, 30–32]. Our clinical experience, from a study of how valproic acid can influence the symptoms of SMA, highlights the necessity to elaborate with reliable and efficient biomarkers on the progress of the SMA treatment [33].
In the present study we developed a novel RT-qPCR-based assay to determine baseline level of FL-SMN and SMNΔ7 mRNA expression in SMA patients and normal individuals. The assay was evaluated on a small group of type II and III SMA patients. A sustained variability in the relative amount both FL-SMN and SMNΔ7 transcripts among these patients was found, which prevents the use of these parameters as biomarkers of SMA treatment. The mRNA levels of FL-SMN and SMNΔ7 in the control subjects overlapped with the values for the SMA patients, which represented similar or slightly lower (for FL-SMN) or slightly higher (for SMNΔ7) values. Our data on the SMN baseline expression levels are similar to the results published by others. For example, in intial studies of SMN2 gene expression in SMA patients an overlap between SMN/GAPDH ratios in patients and controls was found [34]. In the study of Simard and co-authors, FL-SMN mRNA and SMNΔ7 mRNA significantly varied in the range of 0.30 - 1.22 and 0.61 - 2.97 [11]. Absolute quantification of FL-SMN mRNA in SMA patients revealed a broad range of variability - 33.75 - 123.00 [12]. The relatively high level of SMNΔ7 mRNA found in our patients compared to controls is an interesting feature that has also been described previously [10, 35]. The role of SMNΔ7 mRNA in the development of SMA is unclear. Higher SMNΔ7 mRNA level has been shown to negatively influence the SMA development via neurotoxicity mediated by a pro-apoptotic action of the truncated SMN protein [36]. However, an increased expression of SMNΔ7 mRNA has been suggested to lead to a higher life expectancy in an SMA mouse model [37].
In accordance with the literature, FL-SMN/SMNΔ7 mRNA ratio was used in our analysis in order to avoid confounding effects of FL-SMN and SMNΔ7 mRNA level fluctuations [13]. It can be seen in Figure 3c that the quantification of FL-SMN/SMNΔ7 mRNA ratio revealed a difference between SMA patients and healthy controls. This parameter varied in the range of 0.56 - 2.10 and does not overlap with control values. Thus, we suggest that the FL-SMN/SMNΔ7 mRNA ratio could be used as a diagnostic biomarker for discrimination between SMA patients and normal individuals. It could also be used to determine the effect of the SMA treatment by drugs which are able to correct exon 7 splicing, e.g. histone deacetylation inhibitors. It should be noted that the mechanism of action of some histone deacetylation inhibitors is complex, for example it seems that valproic acid affects both SMN gene expression and splicing [38]. However, the clear difference in FL-SMN/SMNΔ7 mRNA ratio between SMA patients and normal individuals, as found in this study and also shown by others, suggests the usefulness of this biomarker for detecting changes during SMA pharmacotherapy.
Conclusions
The SMN2 gene copy quantification in SMA patients can be used as a prognostic tool for discrimination between the SMA type II and SMA type III diagnosis. A novel RT-qPCR-based assay can be used to determine the FL-SMN/SMNΔ7 mRNA ratio. This ratio could be a diagnostic biomarker for monitoring changes during SMA pharmacotherapy.
References
Biros I, Forrest S: Spinal muscular atrophy: untangling the knot?. Med Genet. 1999, 36: 1-8.
Munsat TL, Davies KE: International SMA consortium meeting. Neuromuscul Disord. 1992, 2: 423-428. 10.1016/S0960-8966(06)80015-5.
Parsons DW, McAndrew PE, Monani UR, Mendell JR, Burghes AH, Prior TW: An base pair duplication in exon 6 of the SMN produces a type I spinal muscular atrophy (SMA) phenotype: further evidence for SMN as the primary SMA-determining gene. Hum Mol Genet. 1996, 5: 1727-1732. 10.1093/hmg/5.11.1727.
Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al: Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995, 80: 155-165. 10.1016/0092-8674(95)90460-3.
Cartegni L, Krainer AR: Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002, 30: 377-384. 10.1038/ng854.
McAndrew PE, Parsons DW, Simard LR, Rochette C, Ray PN, Mendell JR, Prior TW, Burghes AH: Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet. 1997, 60: 1411-1422. 10.1086/515465.
Mailman MD, Heinz JW, Papp AC, Snyder PJ, Sedra MS, Wirth B, Burghes AH, Prior TW: Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med. 2002, 4: 20-26. 10.1097/00125817-200201000-00004.
Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B: Quantitative analysis of SMN1 and SMN2 based on real-time lightcycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002, 70: 358-368. 10.1086/338627.
Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J: Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997, 16: 265-269. 10.1038/ng0797-265.
Sumner CJ, Huynh TN, Markowitz JA, Perhac JS, Hill B, Coovert DD, Schussler K, Chen X, Jarecki J, Burghes AH, Taylor JP, Fischbeck KH: Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann Neurol. 2003, 54: 647-654. 10.1002/ana.10743.
Simard LR, Bélanger MC, Morissette S, Wride M, Prior TW, Swoboda KJ: Preclinical validation of a multiplex real-time assay to quantify SMN mRNA in patients with SMA. Neurology. 2007, 68: 451-456. 10.1212/01.wnl.0000252934.70676.ab.
Tiziano FD, Pinto AM, Fiori S, Lomastro R, Messina S, Bruno C, Pini A, Pane M, D'Amico A, Ghezzo A, Bertini E, Mercuri E, Neri G, Brahe C: SMN transcript levels in leukocytes of SMA patients determined by absolute real-time PCR. Eur J Hum Genet. 2010, 18: 52-58. 10.1038/ejhg.2009.116.
Swoboda KJ, Scott CB, Reyna SP, Prior TW, LaSalle B, Sorenson SL, Wood J, Acsadi G, Crawford TO, Kissel JT, Krosschell KJ, D'Anjou G, Bromberg MB, Schroth MK, Chan GM, Elsheikh B, Simard LR: Phase II Open Label Study of Valproic Acid in Spinal Muscular Atrophy. PloS One. 2009, 4: e5268-10.1371/journal.pone.0005268.
Sambrook J, Fitsch EF, Maniatis T: Molecular Cloning: A Laboratory Manual. 1989, Cold Spring Harbor, Cold Spring Harbor Press
Glotov AS, Kiselev AV, Ivashchenko TE, Baranov VS: Analysis of deletional damage in SMN1, SMN2, and NAIP genes in patients with spinal muscular atrophy in the northwestern region of Russia. Genetika. 2001, 37: 1156-1159.
Anhuf D, Eggermann T, Rudnik-Schöneborn S, Zerres K: Determination of SMN1 and SMN2 Copy Number using Taqman Technology. Hum Mutat. 2003, 22: 74-78. 10.1002/humu.10221.
Lo YM, Tein MS, Lau TK, Haines CJ, Leung TN, Poon PM, Wainscoat JS, Johnson PJ, Chang AM, Hjelm NM: Quantitative Analysis of Fetal DNA in Maternal Plasma and Serum: Implications for Noninvasive Prenatal Diagnosis. Am J Hum Genet. 1998, 62: 768-775. 10.1086/301800.
Tran VK, Sasongko TH, Hong DD, Hoan NT, Dung VC, Lee MJ, Takeshima Y, Matsuo M, Nishio H: SMN2 and NAIP gene dosages in Vietnamese patients with spinal muscular atrophy. Pediat Int. 2008, 50: 346-351. 10.1111/j.1442-200X.2008.02590.x.
Cuscó I, Barceló MJ, Rojas-García R, Illa I, Gámez J, Cervera C, Pou A, Izquierdo G, Baiget M, Tizzano EF: SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J Neurol. 2006, 253: 21-25. 10.1007/s00415-005-0912-y.
Rudnik-Schöneborn S, Berg C, Zerres K, Betzler C, Grimm T, Eggermann T, Eggermann K, Wirth R, Wirth B, Heller R: Genotype-phenotype studies in infantile spinal muscular atrophy (SMA) type I in Germany: implications for clinical trials and genetic counseling. Clin Genet. 2009, 76: 168-178. 10.1111/j.1399-0004.2009.01200.x.
Wang CH, Xu J, Carter TA, Ross BM, Dominski MK, Bellcross CA, Penchaszadeh GK, Munsat TL, Gilliam TC: Characterization of survival motor neuron (SMNT) gene deletions in asymptomatic carriers of spinal muscular atrophy. Hum Mol Genet. 1996, 5: 359-365. 10.1093/hmg/5.3.359.
Prior T, Swoboda K, Scott H, Hejmanowski A: Homozygous SMN1 deletion in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet. 2004, 130A: 307-310. 10.1002/ajmg.a.30251.
Wirth B, Brichta L, Schrank B, Lochmüller H, Blick S, Baasner A, Heller R: Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet. 2006, 119: 422-428. 10.1007/s00439-006-0156-7.
Jedrzejowska M, Borkowska J, Zimowski J, Kostera-Pruszczyk A, Milewski M, Jurek M, Sielska D, Kostyk E, Nyka W, Zaremba J, Hausmanowa-Petrusewicz I: Unaffected patients with a homozygous absence of the SMN1 gene. Eur J Hum Genet. 2008, 16: 930-934. 10.1038/ejhg.2008.41.
Oprea GE, Kröber S, McWhorter ML, Rossoll W, Müller S, Krawczak M, Bassell GJ, Beattie CE, Wirth B: Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008, 320: 524-527. 10.1126/science.1155085.
Prior TW, Krainer AR, Hua Y, Swoboda KJ, Snyder PC, Bridgeman SJ, Burghes AH, Kissel JT: A Positive Modifier of Spinal Muscular Atrophy in the SMN2 Gene. Am J Hum Genet. 2009, 85: 408-413. 10.1016/j.ajhg.2009.08.002.
Bernal S, Alías L, Barceló MJ, Also-Rallo E, Martínez-Hernández R, Gámez J, Guillén-Navarro E, Rosell J, Hernando I, Rodríguez-Alvarez FJ, Borrego S, Millán JM, Hernández-Chico C, Baiget M, Fuentes-Prior P, Tizzano EF: The c.859G > C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J Med Genet. 2010, 47: 640-642. 10.1136/jmg.2010.079004.
Hauke J, Riessland M, Lunke S, Eyüpoglu IY, Blümcke I, El-Osta A, Wirth B, Hahnen E: Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum Mol Genet. 2009, 18: 304-317.
Wirth B, Brichta L, Hahnen E: Spinal Muscular Atrophy: From Gene to Therapy. Semin Pediatr Neurol. 2006, 13: 121-131. 10.1016/j.spen.2006.06.008.
Weihl CC, Connolly AM, Pestronk A: Valproate may improve strength and function in patients with type III/IV spinal muscle atrophy. Neurology. 2006, 67: 500-501. 10.1212/01.wnl.0000231139.26253.d0.
Mercuri E, Bertini E, Messina S, Solari A, D'Amico A, Angelozzi C, Battini R, Berardinelli A, Boffi P, Bruno C, Cini C, Colitto F, Kinali M, Minetti C, Mongini T, Morandi L, Neri G, Orcesi S, Pane M, Pelliccioni M, Pini A, Tiziano FD, Villanova M, Vita G, Brahe C: Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology. 2007, 68: 51-55. 10.1212/01.wnl.0000249142.82285.d6.
Liang WC, Yuo CY, Chang JG, Chen YC, Chang YF, Wang HY, Ju YH, Chiou SS, Jong YJ: The effect of hydroxyurea in spinal muscular atrophy cells and patients. J Neurol Sci. 2008, 268: 87-94. 10.1016/j.jns.2007.11.012.
Baranov VS, Kiselev AV, Vakharlovskiĭ VG, Zhelezniakova GIu, Komantsev VN, Malysheva OV, Glotov AS, Ivashchenko TE, Baranov AN: Molecular genetic basis of proximal spinal muscular atrophy and experience in its pharmaceutical treatment. Genetika. 2008, 44: 1325-1337.
Nishio H, Ishikawa Y, Lee MJ, Fujii M, Kanda F, Jinnai K, Takahashi K, Takeshima Y, Wada H, Takada S, Nakamura H, Matsuo M, Sumino K: Decreased expression of full-length mRNA for cBCD541 does not correlate with spinal muscular atrophy phenotype severity. Neurology. 1997, 48: 1266-1270.
Brichta L, Holker I, Haug K, Klockgether T, Wirth B: In vivo activation of SMN in spinal muscular atrophy carriers and patients treated with valproate. Ann Neurol. 2006, 59: 970-975. 10.1002/ana.20836.
Kerr DA, Nery JP, Traystman RJ, Chau BN, Hardwick JM: Survival motor neuron protein modulates neuron-specific apoptosis. Proc Natl Acad Sci USA. 2000, 97: 13312-13317. 10.1073/pnas.230364197.
Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH: SMN Delta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005, 14: 845-857. 10.1093/hmg/ddi078.
Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, Eyupoglu IY, Wirth B: Valproic acid increases the SMN2 protein level: a well-know drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003, 12: 2481-2489. 10.1093/hmg/ddg256.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/12/96/prepub
Acknowledgements
This work was supported by grant of Saint-Petersburg City Administration. Partial support by RFBR grant 10-04-01236-a is also acknowledged. HBS was supported by the Swedish Research Council. The authors thank Prof. Tatiana Ivaschenko and Dr. Olga Malysheva for helpful advice. Also, we are gratefrul to Dr. Samantha Brooks for English correction of the article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors read and approved the final manuscript. The contributors are listed in the parentheses: study concept and design (AVK and VGV); acquisition of data (GYZ, AVK, RC and AAE); analysis and interpretation of data (GYZ, AVK and MRA); drafting of the manuscript (GYZ and AVK); critical revision of the manuscript (MRA, HBS and VSB); obtaining of funding (AVK and HBS); clinical support (VGV); study supervision (AVK and VSB).
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zheleznyakova, G.Y., Kiselev, A.V., Vakharlovsky, V.G. et al. Genetic and expression studies of SMN2 gene in Russian patients with spinal muscular atrophy type II and III. BMC Med Genet 12, 96 (2011). https://doi.org/10.1186/1471-2350-12-96
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-12-96