
Abstract
Background
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disease representing the most prevalent monogenic cause of infant mortality. It results from the loss of SMN1 gene, but retention of its paralog SMN2 whose copy number can modulate the disease severity and guide the therapeutic regimen.
Methods
For SMA molecular analysis, 236 unrelated Egyptian patients were enrolled at our institution. The Multiplex ligation-dependent probe amplification analysis (MLPA) was applied to investigate the main genetic defect in the enrolled patients (SMN1 loss) and to determine a possible genotype–phenotype correlation between the copy number of other genes in the SMN locus (5q13.2) and disease severity in Egyptian patients with SMA. A small cohort of healthy subjects (n = 57) was also included to investigate the possible differences in the distributions of SMN2 and NAIP genes between patients and healthy individuals.
Results
Disease diagnosis was confirmed in only 148 patients (62.7%) highlighting the clinical overlapping of the disease and emphasizing the importance of molecular diagnosis. In patients with homozygous SMN1 loss, the disease was mediated by gene deletion and conversion in 135 (91.2%) and 13 (8.8%) patients, respectively. In the study cohort, SMN2 and NAIP copy numbers were inversely correlated with disease severity. However, no significant association was detected between GTF2H2A and SERF1B copy numbers and patient phenotype. Significant differences were demonstrated in the copy numbers of SMN2 and NAIP between SMA patients and healthy subjects.
Conclusion
Molecular analysis of SMA is essential for disease diagnosis. Consistent with previous studies on other populations, there is a close relationship between SMN2 and NAIP copy numbers and clinical phenotype. Additionally, potential differences in these two genes distributions are existing between patients and healthy subjects. National program for carrier screening should be established as a preventive disease strategy. On the other hand, neonatal testing would provide accurate estimation for disease incidence.
Similar content being viewed by others


Introduction
Spinal muscular atrophy (SMA) is a major genetic cause of infant mortality worldwide [1]. It is characterized by degeneration of α-motor neurons in the anterior horn of the spinal cord, i.e., lower motor neurons, leading to progressive muscle weakness and atrophy [2]. The incidence of SMA is 1 in 6000–10,000 live births. In the Middle East, SMA incidence is up to 40-fold higher than the Western world due to the increased rate of consanguineous marriages [3]. In the Egyptian population, consanguinity was reported in about 50% of SMA patients [4]. On the other hand, SMA has a carrier frequency of 1in 25–50 in most populations, with lower rate in some ethnicities. For example, the frequency of SMA carriers in the Chinese population was estimated as low as about 3% only [5]. However, the carrier rate in the Middle East seems to be relatively higher (1 in 20) [3].
Disease symptoms vary greatly among different patients. In general, the disease is classified into four types according to the age of onset and the ultimately gained motor function. Type 1 (infantile SMA/Werdnig-Hoffmann disease, OMIM #253300): Symptoms begin within the first six months of life. Affected infants are unable to sit without support, and have trouble breathing, feeding, and swallowing. They often pass away by the age of 2 years. Type 2 (intermediate SMA/Dubowitz disease, OMIM #253500): Manifestations start between 6 and 18 months of age. Children with this type cannot walk independently. They might survive into adulthood by virtue of improved standards of healthcare. Type 3 (juvenile SMA/Kugelberg-Welander disease, OMIM #253400): Symptoms usually appear around 18 months of age or in early childhood. These patients have walking difficulties and might eventually require the use of wheelchairs. They generally have an almost normal life span. Type 4 (late SMA, OMIM #271150): It is a very rare type that usually starts in young adulthood resulting in mild motor impairment [6]. Some classifications tend to categorize patients with significant muscle weakness and respiratory distress at birth into a distinct type designated as SMA type 0/prenatal SMA. Indeed, such infants presented with reduced fetal movements in utero and they rapidly progress to respiratory failure often by the first month of life [7].
SMA is caused by homozygous mutations in the SMN1 gene (survival motor neuron 1, OMIM #600354), at the 5q13.2 locus, where SMN protein produced at low insufficient levels. In more than 95% of cases, the disease results from the loss of SMN1 gene. However, intragenic gene mutations, including missense, nonsense, frameshift and splice-site variations, account for the remaining 5% of cases [6, 8]. The SMN1 is located within a genomic segment of inverted duplication. This segment is unique to human lineages and contains 4 main genes: SMN1 (survival motor neuron 1), NAIP (neuronal apoptosis inhibitor protein, OMIM#606831), GTF2H2A (general transcription factor IIH, p44, OMIM#601748) and SERF1A (small EDRK-rich factor 1A, H4F5A, OMIM# 603011). The duplicated genes are either identical to their partner gene (SERF1B), differ by a small number of nucleotides but still produce some functional protein molecules (SMN2, OMIM#601627) or are pseudogenes (ΨGTF2H2B and ΨNAIP∆5) [9]. Importantly, numerous studies have elaborated that SMN2, NAIP, GTF2H2A and SERF1A can affect SMA severity by certain degrees, where SMN2 is the primary disease-modifying gene [10].
The major clinically relevant difference between SMN1 and SMN2 is the C‐to‐T transition (c.850C > T) in exon 7. This position is located in the middle of the exonic splicing enhancer (ESE) sequence affecting the inclusion of exon 7 in SMN transcripts and leads to exon exclusion (SMNΔ7) from the majority (~ 90%) of SMN2-derived mRNAs. This negatively affects protein stability and self-oligomerization [11]. Noteworthy, when exon 8 of SMN1 is retained in SMA patients, SMN2 exon 7 recombines with SMN1 exon 8, forming a hybrid SMN gene. This phenomenon is known as gene conversion representing one of the mechanisms that accounts for increased SMN2 copy number and associated decreased disease severity [12].
Three gene-targeting SMN replacement therapies; nusinersen (Spinraza; Biogen), onasemnogene abeparvovec (Zolgensma; Novartis), and risdiplam (Evrysdi; Roche) have been currently approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Onasemnogene abeparvovec replaces the full-length SMN mRNA and protein, while nusinersin and risdiplam act by increasing exon 7 inclusion in SMN2 transcripts emphasizing the need for SMN2 copy number investigation to instruct therapeutic plans [13].
Multiplex ligation–dependent probe amplification (MLPA) analysis greatly improves SMA diagnostics. Single reaction can simply and rapidly detect the main genetic defect of SMA, evaluate the SMN2 copy number and determine the extent of deletion in the 5q13.2 region [14]. In the current study, MLPA was applied for molecular analysis of Egyptian patients with provisional diagnosis of SMA, and to investigate a possible genotype–phenotype correlation between the copy numbers of other 5q13.2 genes and disease severity in patients with confirmed diagnosis. A small cohort of healthy subjects was also analyzed to assess the possible differences in SMN2 and NAIP distributions between subjects with and without SMA.
Subjects and methods
Subjects
From January 2017 to December 2020, 236 unrelated Egyptian patients were recruited from Clinical Genetics Department at National Research Centre (NCR) and Neuromuscular Unit at Faculty of Medicine—Ain Shams University for molecular diagnosis of SMA. The study also included 57 healthy individuals. For patient selection and clinical classification, the International Spinal Muscular Atrophy Consortium criteria were applied. These criteria include hypotonia, which is typically symmetrical, more proximal than distal, and with preserved sensation, as well as EMG, which indicates a motor neuron disease [15].
Three milliliters of peripheral blood were withdrawn from all studied subjects on 0.5 M EDTA tubes.
Molecular analysis
DNA was extracted from peripheral blood leukocytes using PAXgene DNA blood extraction kit (Qiagen, Germany); according to the manufacturer’s protocol. The MLPA assay was performed using one of two SALSA MLPA Probemixes; P021-A2 or P021-B1 (MRC-Holland, Amsterdam, Netherlands) as instructed by the manufacturer. Both probemixes include four probes specific for sequences in exon 7 or 8 of either SMN1 or SMN2 genes. In addition, the 2 probemixes contain some probes detecting sequences that are present in both SMN1 and SMN2. The probemix P021-A2 contains 4 general probes for both SMN genes; one for exons 1, 4, 6 & 8. However, the probemix P021-B1 holds 17 general probes; at least one for exons 1, 2a, 2b, 3, 4, 5, 6 & 8 and introns 6 & 7 with one additional probe for exons 1, 2b, 3 & 8 and three additional ones for intron 7. On the other hand, they contain one probe specific for exon 5 of NAIP which is absent in NAIPΨ. Furthermore, P021-A2 probemix also includes one probe for exon 13 of both NAIP and NAIPΨ genes, one probe for exon1 of SERF1B gene and three probes for exons 4, 7 and 10 of GTF2H2 genes. The MLPA products were analyzed using ABI 3500 genetic analyzer (Applied Biosystems), with GeneScan™ 500 LIZ™ Dye Size Standard (ThermoFisher Scientific, UK). Data analysis and interpretation were done by Coffyalyser.Net software (www.mlpa.com).
Statistical analysis
Statistical analyses were performed using the statistical package for the social sciences (SPSS Statistics for Windows, Version 25.0; IBM Corp., Armonk, NY, USA). Qualitative variables were reported as the number of cases (percentage) and compared using the Pearson’s chi-square (χ2) test. Quantitative variables were expressed as mean ± standard deviation (SD) and compared using one-way analysis of variance (ANOVA). The correlation between different types of SMA and copy numbers of various modifier genes was assessed using Spearman’s rank correlation coefficient. A two-sided probability (P) value was used for all statistical analyses and a P value of < 0.05 was considered statistically significant.
Results
The study included 236 patients with provisional diagnosis of SMA (132 males and 104 females) whose age ranged from 2 months to 55 years. According to their age of onset, they were classified as 32 (13.6%) type 1, 78 (33.1%) type 2, 114 (48.3%) type 3 and 12 (5.1%) type 4 (Table 1). MLPA analysis revealed homozygous deletions of SMN1 in 148 patients (62.7%). Among them, 13/148 patients (8.8%) demonstrated the absence of exon 7 only, while exon 8 is retained indicating the occurrence of gene conversion in those patients. While, normal SMN1 copy numbers, ranging from 2 to 4 copies, were indicated in 74 patients (31.6%). The remaining 9 patients (3.8%) had heterozygous deletion (1 copy) of SMN1. Among these patients, one patient showed heterozygous deletion of exons 1 to 6 of either SMN1 or SMN2 (Fig. 2A). Thus, there would be 4 expected genotypes for this patient, where only one would confirm SMA diagnosis (Fig. 2B).
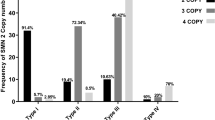
Patients with homozygous gene deletions (n = 148) were classified into 25 (16.9%) type 1, 52 (35.1%) type 2, 69 (46.6%) type 3 and 2 (1.4%) type 4. They had SMN2 copies ranging from 2 to 6, with no heterozygous or homozygous SMN2 deletions (Table 2 & Fig. 1). Using Spearman correlation test, an inverse significant correlation was observed between phenotype severity and SMN2 copy number in terms of both exons 7 & 8 (Additional file 1: Table 2). On the other hand, the distribution of NAIP copy number in patients with homozygous SMN1 deletions were as follows: 49 showed homozygous deletion (33.1%), 51 revealed heterozygous deletion (34.5%), and 47 had 2 copies (31.7%). Only one type III patient (0.7%) had 3 copies (Table 2 & Fig. 1). A significant inverse correlation was also demonstrated between NAIP copy number and the disease severity (r = 0.377, p = < 0.001) (Additional file 1: Table 2). Furthermore, the copy numbers of GTF2H2 and SERF1B were estimated in 63 patients with homozygously deleted SMN1 identified using SALSA MLPA Probemix P021-A2. However, no significant association was detected between both genes copy numbers and the clinical severity (Additional file 1: Table 1). Simultaneous deletion of all 5q13.2 main genes was not detected in any patient of the studied cohort.

Bar charts showing Copy number variations of SMN2 and NAIP genes in SMA patients (n = 148) with homozygous deletion of exon 7 of SMN1 gene
Most of the healthy subjects (n = 30) typically had 2 copies of SMN1 exon 7 (52.6%). Only one copy was detected in 18 subjects (31.6%). Other subjects (n = 9) hold 3 or 4 copies (15.8%). The distribution of SMN2 exon 7 copy numbers among healthy subjects was as follows: 0 copies in 6 individuals (10.5%), 1 copy in 11 (19.4%), 2 copies in 24 (42.1%), 3 copies in 10 (17.5%) and 4 copies in 6 (10.5%). On the other hand, the majority of healthy subjects (52; 92.9%) possessed 2 or 3 copies of NAIP, where only 4 subjects had homozygous (n = 1) or heterozygous deletion (n = 4) of NAIP (7%). Interestingly, statistically significant difference was revealed in the copy numbers of SMN2 and NAIP between patients and healthy subjects (P < 0.001) (Table 2).
Discussion
Spinal muscular atrophy (SMA) is a common autosomal recessive neuromuscular disorder. Survival Motor Neuron 1 gene (SMN1) located on 5q13 (SMA locus) is the main disease associated gene. The majority of SMA cases (94%) results from complete or partial deletion of SMN1 gene. The most common partial deletion encompasses exons 7 and 8 (SMN1∆78). However, other partial deletions have been identified, including exon 7 only (SMN1∆7), exons 5 and 6 (SMN1∆56), exons 2a through 5 (SMN1∆2a5) and exons 1 through 6 (SMN∆16) and exon 8 (SMN1∆8). Even though exon 8 is downstream of the protein-encoding region, it may affect mRNA stability as well as post-transcriptional gene regulation [9]. Moreover, the launching of SMA genetic therapy targeting SMN2 gene increases the demand on molecular testing of both genes (SMN1 & SMN2). Variable methods have been used as PCR-RFLP, quantitative PCR (qPCR), multiple ligation probe amplification (MLPA) and multiplex droplet digital PCR (ddPCR). PCR-RFLP technique has limited sensitivity and reproducibility. qPCR requires normalization with standard curves. Whereas, MLPA is a comprehensive single reaction test for detecting CNVs in several SMA-related genes fragments. It can consistently distinguish between CNVs of SMN1 and SMN2 accurately. However, MLPA is a multi-step relatively time-consuming technique. Recently, ddPCR has overcome the pitfalls of MLPA, but it is a high-cost method that requires a dedicated instrument platform [16].
Herein, 236 patients were studied, the most prevalent SMA type was type III, followed by type II, then type I, where type IV was much lower. However, SMA type I is the most common type, these patients have high mortality rate. In other words, although they have the highest incidence rate; they have the lowest prevalence frequency [17]. Its relatively low predominance among the study cohort would reflect the high mortality rate of these patients, and emphasizing the significance of neonatal screening of SMA, to conquer the missed diagnosis of SMA at that early age. In this study, MLPA assay confirmed SMA diagnosis in 148 patients (62.7%). In two previous studies on Egyptian patients with provisional SMA diagnosis, homozygous absence of exon 7 of SMN1 gene was found in 54.5% and 80% of patients, respectively [18, 19].
About 5% of SMA patients harbor compound heterozygous mutations of SMN1 gene [20, 21]. In our cohort, 3.8% (9/236) of patients had heterozygous deletions. Importantly, one of these patients showed heterozygous deletion of exons 1 to 6. Where the MLPA probemix analyzes the copy numbers of exons 1–6 of both SMN1 and SMN2 genes simultaneously, such result could not assure SMA diagnosis in this patient (Fig. 2b). In most cases, compound heterozygosity results from the deletion of one SMN1 allele and an intragenic mutation within the other allele. More than 100 subtle pathogenic variations have been identified in the SMN1 gene [9]. However, due to the great homology between SMN1 and SMN2, combination of long-range PCR (LR-PCR) and nested PCR should be considered in the analysis of SMN1 intragenic mutations [22].

Analysis of SMA patient with type III showing heterozygous deletion of exons 7 and 8 of SMN1 gene, and expected heterozygous deletion of exons 1–6 of SMN1 gene. A MLPA analysis showing median value of 0.755 for the 10 probes detecting exon 1–6 of both SMN1 and SMN2 genes (3 copies) and median values of exons 7 and 8 of SMN1 gene and SMN2 gene between 0.52–0.63 (1 copy of each). B Expected distributions of SMN1 & SMN2 genes exons showing (a) heterozygous deletion of the whole SMN1 gene from one allele, which doesn’t explain the phenotype, (b) heterozygous deletion of exons 7 & 8 of SMN1 gene from one allele and heterozygous deletion of exons 1–6 from the other allele, which could be pathogenic for SMA., (c &d) heterozygous deletion of exons 1–6 of SMN2 gene
Patients with two or more copies of exon7 of the SMN1 gene may have causative pathogenic mutations in non-5q genes. This group accounts for about 6% of all SMA patients where, at least 16 different genes have been found to be associated with these non-5q forms with considerable phenotypic variability and diverse inheritance patterns [23]. Recently, SMN1 deletions or mutations in suspected SMA patients are detected in about 50% of them only. This could be due to clinical overlapping with other neuromuscular disorders, e.g., hereditary motor neuropathies (HMN), myasthenic syndromes [24]. Whole exome sequencing (WES) for this group of patients would be the best choice for detecting the causative gene.
Homozygous deletion of SMN1 exon 7 with the retaining of SMN1 exon 8 is known as hybrid SMN gene or gene conversion [12, 25]. It was reported that the frequencies of hybrid SMN gene in SMA patients vary from 5 to 30% among different ethnic groups [12]. Combining data from the current study and other previous research [19], it has been determined that the frequency of the hybrid SMN gene in Egyptian patients seems to be between low (< 10%) and medium (10–20%) frequencies, with a total of 9.8% (16/164) and only one patient of Type I SMA. Reduced phenotype in case of gene conversion might be justified by the rise of the CNVs of SMN2 which is considered the most reliable modifier gene. It is a highly homologous paralog of SMN1 gene acting as a hypomorphic allele [26]. SMN2 produces non-functioning truncated SMN protein, as exon 7 is mainly missed from its transcript (SMNΔ7). However, approximately 10% of SMN2 transcripts encodes functioning full-length SMN protein due to alternative splicing. Thus, the higher the number of copies of SMN2, the larger the amount of the full length SMN protein produced, and thus the less SMA phenotype severity [27]. Therefore, the majority of gene conversion is associated with less severe phenotypes, as observed in Egyptian SMA patients who were mostly Type II or III.
The majority of SMA patients have 2 copies or more of SMN2 [28] and numerous studies have demonstrated a strong inverse relationship between SMN2 copy number and clinical severity [13]. In the current study, SMN2 copy number ranged from 2 to 6 copies in SMA patients and significant inverse correlation in terms of both exons 7 and 8 was estimated between phenotype and CNVs. Interestingly, increased CNVs of SMN2 have been identified in a number of asymptomatic subjects who had homozygous deletion of SMN1 [29,30,31]. It is noteworthy that the copy number variant (CNV) is not the only modifying factor of SMN2. Certain structural intronic variants in SMN2, such as c.835-44G˃A and c.888 + 100A˃G can affect exon 7 inclusion in SMN transcripts producing greater amounts of SMN protein and resulting in milder phenotype [32]. Furthermore, SMN2 is a primary target for the development of therapeutics for SMA. Thereby determination of SMN2 copy number becomes a crucial criterion for therapeutic application in SMA patients. Hence, analysis of SMN2 copy number is used not only as a prognostic tool, but also to guide gene therapy based therapeutic strategies. Importantly, the Egyptian National Drug Authority has registered Risdiplam in June 2021 as the first SMA therapy. It has been approved to treat SMA patients starting from age of 2 months, with a once daily dose determined by patient’s age and bodyweight [33].
Other genes in the SMN gene locus might also affect clinical phenotype. In the current study, simultaneous deletions of SMN1 and NAIP were detected in 49 patients (33.1%). In patients with type I SMA, all patients had either homozygous (n = 17) or heterozygous (n = 8) NAIP deletion. Several studies have also demonstrated that lower copy numbers of NAIP were associated with more severe phenotype [10, 34, 35]. Consistently, we revealed an inversely significant correlation between NAIP copy number and disease severity (p = < 0.001). On the other hand, large-scale deletions extending to SERF1 and GTF2H2 genes are also observed in patients with type I SMA [36]. In the current study, none of SMN1 deleted patients showed simultaneous deletion of SERF1 and GTF2H2. In the context of these 2 distributions, Amara et al. [37] reported significant association between SMA disease severity and SERF1 but not GTF2H2 in Tunisian patients. SERF1 is more closely related to SMN1 on the genomic organization, enhancing its possible frequency for deletion with SMN1 gene in SMA patients. Conversely, no relationship was observed between the 2 genes and SMA clinical severity in other study cohorts [38, 39]. Here, we also failed to estimate any significant association between GTF2H2 or SERF1 copy numbers and SMA phenotype. Generally, weak correlation seems to be existing between these two genes and disease phenotype, so that they have been currently excluded from the prognostic molecular testing.
Among healthy subjects, we identified 18 subjects with a single SMN1 copy, with a carrier frequency of 31.6%. The great majority of SMA carriers can be identified by the presence of a single copy of SMN1 exon 7. However, about 5% of SMA carriers have two SMN1 copies on one chromosome and zero copies on the other [40]. In other words, these so-called silent carriers have a cis allelic distribution (2 + 0), rather than a trans allelic distribution (1 + 1). Standard assays including MLPA cannot distinguish '1 + 1' from '2 + 0' (silent carriers). On the other hand, differences in the copy numbers of exons 7 and 8 in both SMN1 (n = 23) and SMN2 (n = 29) have been detected in significant proportion of healthy subjects. This would be mainly attributed to the conversions between the 2 genes. Importantly, six possible hybrid SMN genes were determined [41]. Although, homozygous and heterozygous deletions of exons 7 and 8 of SMN genes were up to 20% in Caucasian populations [42]. Combined CNVs of SMN1 and SMN2 genes were consistent within other populations ranging from 3.6 copies in Asians to 3.8 copies in Africans. The median CNVs of SMN2 in European and Asian populations were two copies, with ratios of 49.2% and 55.3%, respectively. Whereas, in Africans the median CNVs was 1 copy of SMN2 (50.3%), while only 24.9% had 2 copies. The median CNVs of SMN2 in this study was 2 copies of 42.1% [43].
In the current study, the distributions of SMN2 and NAIP were significantly different between SMA patients and healthy individuals (P < 0.001) with increased copy numbers in patients. Prior studies among various ethnicities consistently revealed that CNVs of the SMN2 gene were significantly lower in control subjects. [44, 45]. Additionally, both SMN2 and NAIP genes copy numbers were significantly different (P < 0.001) between Chinese SMA patients and healthy subjects [28]. The proportion of SMA patients (33.1%) lacking NAIP gene was dramatically higher than that of healthy individuals (1.8%). In fact, NAIP gene is the gene that precedes SMN1, so that co-deletion may occur identifying a potential cause of direct correlation between CNVs of SMN1 and NAIP genes.
Conclusions
Molecular analysis of SMA is essential to ensure diagnosis and guide therapeutic plans. MLPA is the gold standard of SMA diagnosis. It can detect SMN1 copy number, investigate the mechanism responsible for SMN1 loss (i.e., deletion or conversion to SMN2), evaluate the SMN2 copy number and determine the extent of deletion in the 5q13.2 region. Improvements in molecular diagnostic tools are associated with enhanced detection rate and accurate estimation for disease incidence. However, other disease causative variations rather than the most common SMN1 deletion should be studied through either SMN1 gene sequencing or whole exome sequencing. Neonatal screening would overcome the missed diagnosis of SMA, particularly for type I patients with short life expectancies. Carrier screening should be implemented as a national program in Egypt as a preventive disease strategy.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Tisdale S, Pellizzoni L (2015) Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J Neurosci 35(23):8691–8700
Verhaart IEC, Robertson A, Leary R, McMacken G, Konig K, Kirschner J et al (2017) A multi-source approach to determine SMA incidence and research ready population. J Neurol 264(7):1465–1473
Ali HG, Ibrahim K, Elsaid MF, Mohamed RB, Abeidah MIA, Al Rawwas AO et al (2021) Gene therapy for spinal muscular atrophy: the Qatari experience. Gene Ther 28(10):676–680
Shawky R, Elsayed N (2011) Clinico-epidemiologic characteristics of spinal muscular atrophy among Egyptians. World Pumps 12:25–30
Chen TH, Tzeng CC, Wang CC, Wu SM, Chang JG, Yang SN et al (2011) Identification of bidirectional gene conversion between SMN1 and SMN2 by simultaneous analysis of SMN dosage and hybrid genes in a Chinese population. J Neurol Sci 308(1–2):83–87
Farooq F, Holcik M, Mackenzie A (2013) Spinal muscular atrophy: classification, diagnosis, background, molecular mechanism and development of therapeutics. In: Kishore U, editor. Neurodegenerative Diseases [Internet]. London: IntechOpen; 2013.
MacLeod MJ, Taylor JE, Lunt PW, Mathew CG, Robb SA (1999) Prenatal onset spinal muscular atrophy. Eur J Paediatr Neurol 3(2):65–72
Ross LF, Kwon JM (2019) Spinal muscular atrophy: past, present, and future. NeoReviews 20(8):e437–e451
Butchbach MER (2021) Genomic variability in the survival motor neuron genes (SMN1 and SMN2): implications for spinal muscular atrophy phenotype and therapeutics development. Int J Mol Sci 22(15):7896
He J, Zhang Q-J, Lin Q-F, Chen Y-F, Lin X-Z, Lin M-T et al (2013) Molecular analysis of SMN1, SMN2, NAIP, GTF2H2, and H4F5 genes in 157 Chinese patients with spinal muscular atrophy. Gene 518(2):325–329
Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH et al (1999) A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 8(7):1177–1183
Niba ETE, Nishio H, Wijaya YOS, Lai PS, Tozawa T, Chiyonobu T et al (2021) Clinical phenotypes of spinal muscular atrophy patients with hybrid SMN gene. Brain Dev 43(2):294–302
Cusco I, Bernal S, Blasco-Perez L, Calucho M, Alias L, Fuentes-Prior P et al (2020) Practical guidelines to manage discordant situations of SMN2 copy number in patients with spinal muscular atrophy. Neurol Genet 6(6):e530
Arkblad E, Darin N, Berg K, Kimber E, Brandberg G, Lindberg C et al (2007) Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul Disord NMD 16:830–838
Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B et al (2007) Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol 22(8):1027–1049
Wang KC, Fang CY, Chang CC, Chiang CK, Chen YW (2021) A rapid molecular diagnostic method for spinal muscular atrophy. J Neurogenet 35(1):29–32
Okamoto K, Fukuda M, Saito I, Urate R, Maniwa S, Usui D et al (2019) Incidence of infantile spinal muscular atrophy on Shikoku Island of Japan. Brain Dev 41(1):36–42
Shawky RM, Abd el-Aleem K, Rifaat MM, Moustafa A (2001) Molecular diagnosis of spinal muscular atrophy in Egyptians. East Mediterr Health J 7(1–2):229–237
Essawi ML, Effat LK, Shanab GM, Al-Ettribi GM, El-Haronui AA, Karim AM (2007) Molecular analysis of SMN1 and NAIP genes in Egyptian patients with spinal muscular atrophy. Bratisl Lek Listy 108(3):133–137
Alias L, Bernal S, Fuentes-Prior P, Barcelo MJ, Also E, Martinez-Hernandez R et al (2009) Mutation update of spinal muscular atrophy in Spain: molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Hum Genet 125(1):29–39
Markowitz JA, Singh P, Darras BT (2012) Spinal muscular atrophy: a clinical and research update. Pediatr Neurol 46(1):1–12
Bai JL, Qu YJ, Cao YY, Li EZ, Wang LW, Li Y et al (2014) Subtle mutation detection of SMN1 gene in Chinese spinal muscular atrophy patients: implication of molecular diagnostic procedure for SMN1 gene mutations. Genet Test Mol Biomark 18(8):546–551
Peeters K, Chamova T, Jordanova A (2014) Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain 137(Pt 11):2879–2896
Karakaya M, Storbeck M, Strathmann EA, Delle Vedove A, Holker I, Altmueller J et al (2018) Targeted sequencing with expanded gene profile enables high diagnostic yield in non-5q-spinal muscular atrophies. Hum Mutat 39(9):1284–1298
Hahnen E, Schonling J, Rudnik-Schoneborn S, Zerres K, Wirth B (1996) Hybrid survival motor neuron genes in patients with autosomal recessive spinal muscular atrophy: new insights into molecular mechanisms responsible for the disease. Am J Hum Genet 59(5):1057–1065
Wirth B, Garbes L, Riessland M (2013) How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr Opin Genet Dev 23(3):330–338
Cho S, Dreyfuss G (2010) A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev 24(5):438–442
Fang P, Li L, Zeng J, Zhou W, Wu W-Q, Zhong Z-Y et al (2015) Molecular characterization and copy number of SMN1, SMN2 and NAIP in Chinese patients with spinal muscular atrophy and unrelated healthy controls. BMC Musculoskelet Disord. https://doi.org/10.1186/s12891-015-0457-x
Eissa NR, Hassan HA, Senousy SM, Soliman HN, Essawi ML (2022) SMA carrier testing using Real-time PCR as a potential preconception screening tool. Egypt J Med Hum Genet 23(1):24
Jędrzejowska M, Borkowska J, Zimowski J, Kostera-Pruszczyk A, Milewski M, Jurek M et al (2008) Unaffected patients with a homozygous absence of the SMN1 gene. Eur J Hum Genet 16(8):930–934
Prior TW, Swoboda KJ, Scott HD, Hejmanowski AQ (2004) Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet A 130A(3):307–310
Wu X, Wang SH, Sun J, Krainer AR, Hua Y, Prior TW (2017) A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Hum Mol Genet 26(14):2768–2780
Ratni H, Scalco RS, Stephan AH (2021) Risdiplam, the first approved small molecule splicing modifier drug as a blueprint for future transformative medicines. ACS Med Chem Lett 12(6):874–877
Hassan HA, Zaki MS, Issa MY, El-Bagoury NM, Essawi ML (2020) Genetic pattern of SMN1, SMN2, and NAIP genes in prognosis of SMA patients. Egypt J Med Hum Genet 21(1):1–7
Omrani O, Bonyadi M, Barzgar M (2009) Molecular analysis of the SMN and NAIP genes in Iranian spinal muscular atrophy patients. Pediatr Int 51(2):193–196
Butchbach ME (2016) Copy number variations in the survival motor neuron genes: implications for spinal muscular atrophy and other neurodegenerative diseases. Front Mol Biosci 3:7
Amara A, Adala L, Ben Charfeddine I, Mamaï O, Mili A, Lazreg TB et al (2012) Correlation of SMN2, NAIP, p44, H4F5 and Occludin genes copy number with spinal muscular atrophy phenotype in Tunisian patients. Eur J Paediatr Neurol 16(2):167–174
Arkblad E, Tulinius M, Kroksmark AK, Henricsson M, Darin N (2009) A population-based study of genotypic and phenotypic variability in children with spinal muscular atrophy. Acta Paediatr 98(5):865–872
Karasu N, Acer H, Akalin H, Demir M, Sahin IO, Gokce N et al. (2022) Molecular analysis of SMN2, NAIP and GTF2H2 gene deletions and relation with clinical subtypes of spinal muscular atrophy
Alías L, Barceló MJ, Bernal S, Martínez-Hernández R, Also-Rallo E, Vázquez C et al (2014) Improving detection and genetic counseling in carriers of spinal muscular atrophy with two copies of the SMN1 gene. Clin Genet 85(5):470–475
Wang CC, Jong YJ, Chang JG, Chen YL, Wu SM (2010) Universal fluorescent multiplex PCR and capillary electrophoresis for evaluation of gene conversion between SMN1 and SMN2 in spinal muscular atrophy. Anal Bioanal Chem 397(6):2375–2383
Sugarman EA, Nagan N, Zhu H, Akmaev VR, Zhou Z, Rohlfs EM et al (2012) Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet 20(1):27–32
Vijzelaar R, Snetselaar R, Clausen M, Mason AG, Rinsma M, Zegers M et al (2019) The frequency of SMN gene variants lacking exon 7 and 8 is highly population dependent. PLoS ONE 14(7):e0220211
Crawford TO, Paushkin SV, Kobayashi DT, Forrest SJ, Joyce CL, Finkel RS et al (2012) Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS ONE 7(4):e33572
Hendrickson BC, Donohoe C, Akmaev VR, Sugarman EA, Labrousse P, Boguslavskiy L et al (2009) Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Genet 46(9):641–644
Acknowledgments
The authors would like to thank all of the patients and their families who provided samples for this study and agreed to have their data shared.
Funding
The research was funded by NRC (National Research Centre in Egypt) through the project entitled “Multiplex Ligation-dependent Probe Amplification (MLPA) approach as a rapid and sensitive molecular tool for diagnosis and prognosis of Spinal Muscular Atrophy (SMA)” Grant number: 11010168.
Author information
Authors and Affiliations
Contributions
All authors have read and approved the manuscript. HH contributed in molecular studies and writing and revising the article. WS contributed in molecular studies, statistical analysis and writing the article. NRE contributed in molecular studies and writing the article. MZ contributed in clinical diagnosis and assessment of patients. NF contributed in clinical diagnosis and assessment of patients. NME contributed in molecular studies and writing the article. MI contributed in clinical diagnosis and assessment of patients. ME contributed in the study design, molecular studies and revising the article. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study protocol was reviewed and approved by Medical Research Ethics Committee (MREC) of the NRC [registration number: 16-100].
Consent to participate
According to Medical Research Ethics Committee (MREC) of the NRC, written informed consents were taken from all subjects or from their guardians approving to take part of this study and to publish their data in scientific journal.
Competing interests
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supplementary tables 1 and 2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hassan, H.A., Fahmy, N.A., El-Bagoury, N.M. et al. MLPA analysis for molecular diagnosis of spinal muscular atrophy and correlation of 5q13.2 genes with disease phenotype in Egyptian patients. Egypt J Med Hum Genet 23, 156 (2022). https://doi.org/10.1186/s43042-022-00373-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00373-y