
Abstract
Background
Auxin/Indole-3-Acetic Acid (Aux/IAA) and Auxin Response Factor (ARF) transcription factors are key regulators of auxin responses in plants. We identified the suites of genes in the two gene families in Populus and performed comparative genomic analysis with Arabidopsis and rice.
Results
A total of 35 Aux/IAA and 39 ARF genes were identified in the Populus genome. Comparative phylogenetic analysis revealed that several Aux/IAA and ARF subgroups have differentially expanded or contracted between the two dicotyledonous plants. Activator ARF genes were found to be two fold-overrepresented in the Populus genome. PoptrIAA and PoptrARF gene families appear to have expanded due to high segmental and low tandem duplication events. Furthermore, expression studies showed that genes in the expanded PoptrIAA3 subgroup display differential expression.
Conclusion
The present study examines the extent of conservation and divergence in the structure and evolution of Populus Aux/IAA and ARF gene families with respect to Arabidopsis and rice. The gene-family analysis reported here will be useful in conducting future functional genomics studies to understand how the molecular roles of these large gene families translate into a diversity of biologically meaningful auxin effects.
Similar content being viewed by others


Background
Aux/IAAs are auxin response genes that code for nuclear localized proteins [1]. Aux/IAA proteins generally have four characteristic domains; an N-terminal repression domain, an adjacent domain involved in protein stability, and two C-terminal domains (CTD), III and IV, through which Aux/IAA proteins form homo- and heterodimers with Aux/IAA or ARF proteins [2]. Most ARF proteins contain an N-terminal B3-like DNA binding domain that includes an ARF family-specific domain, a variable middle region that confers activator or repressor activity, and domains III and IV that are also found in Aux/IAA [3]. ARF proteins are capable, irrespective of auxin status, of binding to auxin responsive cis-elements (AuxRE; TGTCTC) present upstream to the coding sequence of auxin responsive genes [4, 5]. Aux/IAA proteins bind to the DNA-bound ARF partner proteins via domains III and IV and repress ARF activity. In the auxin activated status, Aux/IAA proteins are ubiquitinated via interactions with the auxin-modified SCFTIR1complex and subsequently degraded by 26S proteasome action [6–8]. While some ARFs possess a characteristic glutamine (Q)-rich middle region which confers an activator activity, ARFs with a proline, serine and threonine-rich middle region are found to be associated with repressor activity [3, 9]. The complexity of auxin regulatory activity is due in part to the large sizes of the ARF and Aux/IAA gene families, as well as variations in activation or repression activity among ARFs, heterodimerization affinities, expression patterns, and auxin-mediated transcriptional and posttranscriptional regulation.
Despite the knowledge that Aux/IAAs and ARFs influence apical dominance, vascular development, tropic movements, root growth, tissue and organ patterning, and flower and fruit development [10], many questions still linger. For example, these gene families remain largely uncharacterized in forest tree species and the degree of conservation of gene families between annual and perennial woody plants is unknown. Furthermore, the mechanisms of Aux/IAA and ARF interaction and regulation are not completely understood and much remains to be learned about their roles in the contexts of a cell and the whole organism. Identification of Aux/IAA and ARF gene families from distinct model plants is a necessary step in formulating better hypotheses related to physiological and developmental processes. The recent sequencing of the Populus genome has provided an additional reference genome for testing inferences on auxin signal transduction events obtained previously through functional genomic studies of Arabidopsis.
The present paper summarizes findings from bioinformatics-based comparative genomic studies to identify the total number of Aux/IAA and ARF genes in Populus, to predict the protein domain architectures, and to assess the extent of conservation and divergence between Populus, Arabidopsis and rice gene families. Targeted RT-PCR and whole genome microarray analyses, and EST database surveys were also undertaken to explore differential expression of closely grouping co-orthologs. Furthermore, we have reflected on the possible implications of differential patterns of retention, loss, and expansion of duplicated homeologous genes.
Results and discussion
Identification and sequence analysis of Populus Aux/IAAgenes
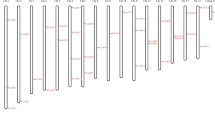
The Populus genome has 35 predicted Aux/IAA genes (henceforth referred to as PoptrIAA) compared to 29 genes in the Arabidopsis genome (henceforth referred to as AtIAA) [11] and to 31 genes in the rice genome (henceforth referred to as OsIAA) [12]. In silico mapping of the gene loci showed that PoptrIAA genes are present on 10 of the 19 chromosomes (Figure 1; See Additional file 1).

Chromosomal positions of PoptrIAA and PoptrARF genes. Scale represents a 5 Mb chromosomal distance. Colors indicate the chimeric nature of most linkage groups. Common colors refer to homeologous genome blocks, presumed to have arisen from the salicoid genome duplication 65 Mya and shared by two chromosomes [13]. Chromosome numbers (linkage group number I-XIX) and sizes (Mb) are indicated at the bottom end of each chromosome. PoptrIAA and PoptrARF genes are represented in blue and black font colors, respectively. 4 PoptrIAA and 11 PoptrARF genes reside on unassembled scaffolds (See Additional file 1).
Any analysis of Populus gene family evolution must take into account the most significant event in the recent evolution of the genus; a genome-wide duplication event that occurred approximately 65 Mya and which is still detectable over approximately 92% of the genome [13]. Based on the age estimates of duplicate genes and homology-microsynteny analysis (See Additional file 2), 14 PoptrIAA genes pairs (80% of total genes) are represented within segmental duplication regions associated with the recent salicoid duplication event. It is intriguing that 6 pairs of tandem duplicates are represented as 'ancient tandem genes' within more recent whole genome or segmental duplicate clusters. These 15 PoptrIAA genes (~43% of total) are represented in 7 distinct tandem duplicate gene clusters, with 6 clusters containing two tandem genes and 1 cluster containing 3 tandem genes (PoptrIAA15, PoptrIAA19.1 and PoptrIAA19.2).
It is possible that some of the apparently closely-related genes are in fact alleles from unassembled haplotypes, which are potential artifacts from shotgun assembly of this highly heterozygous genome. For example, PoptrIAA3.1 and PoptrIAA3.2 group closely with their tandem duplicates, PoptrIAA16.1 and PoptrIAA16.2, and PoptrIAA3.1 and PoptrIAA16.1 genes are located on a presently unassembled scaffold (scaffold_70). However, the apparent co-orthologs are divergent at the amino acid level as well as in the flanking gene order and identity in the syntenic blocks, which argue against the classification of the scaffold as a haplotype.
Further analyses of conserved domains and multiple sequence alignments of predicted proteins showed that some Populus genes contain modifications in the conserved domain architecture (See Additional files 3, 4 and 5). Based on MEME-MAST protein motif analysis, domain I was found to be missing in PoptrIAA29.1, PoptrIAA29.2 and PoptrIAA29.3 and also in the Arabidopsis ortholog, AtIAA29. Similarly, domains I and II appear to be missing in PoptrIAA33.1, PoptrIAA33.2 and PoptrIAA34 as well as AtIAA33 and AtIAA34. Multiple sequence alignment shows that the "idLgLsLrt" sequence reported as domain I motif in AtIAA34 (LxLxL) [14] is conserved with AtIAA14, AtIAA15, AtIAA16 and AtIAA17 (See Additional file 3) in which the consensus sequence of the reported domain I is "TELxLxLPG" [14].
Domain II enables the SCFTIR1-dependent proteasome-mediated degradation of Aux/IAA [15]. Rapid basal degradation rate and auxin responsiveness of Aux/IAA proteins are found to be associated with alterations in the highly conserved domain II motif, 'VGWPPI/V' [15] and the 'KR' motif between domains I and II [16]. Studies in Arabidopsis show that mutations in the VGWPPV motif render the repressor protein stable [17–23]. Multiple sequence alignments of full-length amino acid sequences revealed deviations in both the 'VGWPPI/V' and 'KR' motif sequences in PoptrIAA7.1, PoptrIAA20.1, PoptrIAA 20.2, PoptrIAA 33.1, PoptrIAA 33.2, PoptrIAA 34 and AtIAA20, AtIAA30, AtIAA31, AtIAA32, AtIAA33 and AtIAA34. Deviations in the conserved 'KR' motif alone were observed in PoptrIAA27.1, PoptrIAA26.2 and AtIAA28 sequences. However, PoptrIAA7.1 was found to be unique in that it contains a tandem duplication of the domain II region (See Additional file 5). This unique feature has not been detected in Aux/IAA genes reported from any other genera including Arabidopsis, Oryza, Vitis, Nicotiana and Coffea, but was found in GenBank ESTs derived from P. trichocarpa, P. deltoides, P. tremuloides, P. tremula × tremuloides [24], P. alba × tremula, P. trichocarpa × deltoides and P. fremontii × angustifolia, suggesting that this is a Populus-specific acquisition.
Comparative analysis of Populus and Arabidopsis Aux/IAAgene families
Phylogenetic reconstruction at the molecular level using all available and predicted Populus, Arabidopsis and rice Aux/IAA amino acid sequences shows that four groups of PoptrIAAs (PoptrIAA3, 16, 27 and 29) have expanded to contain three or more members each (Figure 2). Three AtIAAs did not have a representative sequence ortholog in Populus, indicating acquisition or perpetuation of distinct Aux/IAAs unique to Arabidopsis and its relatively recent ancestors.

Phylogenetic analysis of predicted full-length Aux/IAA protein sequences using Neighbor-Joining method. Amino acid sequences of full-length predicted proteins were aligned using MUSCLE program. Tree was produced as described in methods. Bootstrap support is indicated at each node. Green boxes represent sustained expansion of subgroups in Populus.
There are four homologs of AtIAA16, PoptrIAA16.1–16.4, predicted in the Populus genome. Phylogenetic groupings suggest absence of AtIAA16 orthologs from rice. PoptrIAA16.3 and PoptrIAA16.4 are likely co-orthologs of an ancestral gene lost in Arabidopsis. These co-orthologs have intact conserved domains and EST evidence supporting their functionality (See Additional files 1 and 5). Based on EST data, PoptrIAA16.1 appears to express during wood formation (in cambial zone and tension wood), whereas PoptrIAA16.3 and 16.4 appear to express in young leaves (See Additional file 6). A PoptrIAA16.1-like EST was previously reported from hybrid aspen to be expressed in the context of wood formation and a PoptrIAA16.3-like EST was shown to be expressed in dividing and expanding cells [24]. These findings are partially supported by our whole genome microarray data, which showed that PoptrIAA16.1 was expressed in xylem, phloem, cortex, root and seed (See Additional files 7, 8 and 9).
However, there was no array evidence to support expression of PoptrIAA16.3 and PoptrIAA16.4 in young leaf tissues, though PoptrIAA16.3 was expressed at the stem apex, in apical vegetative buds, newly set reproductive buds of both genders, and in older leaves.
Two Aux/IAA proteins, IAA26/PAP1 (Phytochrome associated protein1) and IAA27 are known to interact with phytochrome A (PHYA) [25, 26] and TMV replicase [1, 27, 28]. Populus has three IAA27-like Aux/IAA genes. EST and microarray data suggest expression in shoot meristem, floral buds and dormant and active cambia (See Additional files 6, 7, 8 and 9). Possible affinity for interaction with PHYA protein suggests a role for PoptrIAA27 in external-stimuli-dependent cambium activation or growth status. PoptrIAA26.1 and PoptrIAA26.2 genes cluster closely with AtIAA18 and AtIAA26 and are expressed in shoot meristem, young and senescing leaves, male catkins, and floral buds. RT-PCR survey of PoptrIAA26.1 and PoptrIAA26.2 genes shows highest expression in young leaves. Since AtIAA26 and AtIAA27 proteins display binding affinity towards PHYA, it is likely that these Populus sequence orthologs are also involved in mediating the photoregulation of various tree developmental processes.
PoptrIAA3.1–3.6 represent a six-member PoptrIAA3 subgroup that groups closely with AtIAA1–4. This subgroup provides striking evidence for functional divergence following selective retention of duplicated genes. While the Arabidopsis shy2 mutant, carrying a gain-of-function mutation in AtIAA3, has upcurled leaves, slower gravitropic response, shorter hypocotyls and fewer lateral roots [22], a functional role for AtIAA4 is yet to be assigned. Gene-specific real-time RT-PCR showed that genes in the PoptrIAA3 subgroup display differential expression between leaf, stem and root tissues (Figure 3). PoptrIAA3.2 was found to have a higher expression level by several fold in stem than in roots. In an earlier study of aspen Aux/IAA genes, the PoptrIAA3.2-like gene had highest expression in developing xylem [24], which is also supported by our microarray results (Figure 4). EST data suggest that PoptrIAA3.1 and 3.2 appear to be preferentially expressed in the cambial zone and during wood formation (See Additional file 6), and the microarray data suggest that these genes are also strongly expressed in newly germinated seedlings. PoptrIAA3.6-like ESTs are found in libraries of dormant buds and senescing leaves. PoptrIAA3.4 has a distinct expression pattern compared to other PoptrIAA3 genes, with detectable ESTs in male and female catkins, and highest expression in floral buds as determined by the microarray (Figure 4).

Expression analysis of PoptrIAA3 subgroup genes using real-time RT- PCR. Fold Change (*) is represented relative to lowest value observed for the gene. Lowest value was determined by comparison of relative threshold cycle values for a specific gene across leaf, stem and root samples. Fold change was calculated by the formula 2-ΔΔCt, where ΔΔCt is the difference between ΔCt of a gene is a given tissue and the lowest value ΔCt observed for that gene in any of the three tissue types. ΔCt was estimated by the formula; (Ct of gene of interest) – (geometric mean of ΔCt of 18S RNA gene, control gene). Note that the relative fold change in expression of PoptrIAA3.2 gene in stem with respect to root (tissue with lowest PoptrIAA3.2 expression level) is 128, which has been represented on a discontinuous y-axis to capture the other lower fold change values observed.

Microarray expression support for PoptrIAA3 subgroup. Data are expressed as fold change from negative controls, which consisted of the 95th percentile signal from presumably unexpressed transposable element target sequences. Error bars represent standard errors from two biological replicates. G43h, germinant 43 hours after imbibition; ApB, apical bud; AxB, axillary bud; YFB, young female bud; YMB, young male bud; F, female catkin, post-fertilization; LPI1, leaf plastochron index 1; LPI2, leaf plastochron index 2; LPI5, leaf plastochron index 5; PC, phloem plus cortex. Samples are further defined in Additional file 17.
Identification and sequence analysis of Populus ARFgenes
A total of 39 predicted ARF genes (henceforth referred to as PoptrARF) were found in the Populus genome, compared to 23 genes reported from the Arabidopsis genome (henceforth referred to as AtARF) [2] and to 25 genes reported from the rice genome (henceforth referred to as OsARF) [29]. In silico mapping of gene loci showed PoptrARF genes are present on all chromosomes except VII, XIII, XVII and XIX (Figure 1). Eleven out of 39 genes lie within unassembled scaffolds. Conserved domain evaluations showed that four gene models (PoptrARF3.3, 6.3, 16.5 and 16.6) appear to lack one or more domains that are otherwise conserved in their closest sequence ortholog (See Additional files 10, 11 and 12).
Sixteen PoptrARF gene pairs (82% of total) are estimated to be represented in chromosomal segmental duplications arising out of the salicoid whole genome duplication event. Two genes, PoptrARF16.4 and PoptrARF16.5, (5% of total) are represented as one tandem duplication pair. The Arabidopsis genome contains a group of seven tandemly duplicated ARF genes that thus far have not been observed in other plant species including Populus and rice.
Comparative analysis of Populus and Arabidopsis ARFgene families
Phylogenetic analysis using known and predicted Populus, Arabidopsis and rice ARF protein sequences shows distinct gene family histories even between the two dicots (Figure 5). The ratio of activator ARFs (defined by the Q-rich middle region) in Arabidopsis and Populus is 1:2.6 whereas the ratio of repressor and other ARFs is 1:1.4, indicating a two-fold enrichment of activator ARFs during Populus evolution.

Phylogenetic analysis of predicted full-length ARF protein sequences using Neighbor-Joining method. Amino acids sequences of full-length predicted proteins were aligned using MUSCLE program. Tree was produced as described in methods. Bootstrap support is indicated at each node. Green and orange boxes represent sustained expansion of subgroups in Populus and Arabidopsis, respectively. Grey box represents the Q-rich activator ARF subgroup.
A pair of Populus genes, PoptrARF7.3 and PoptrARF 7.4, was found to group closely with three OsARF genes but had no obvious Arabidopsis orthologs. The loss-of-function ARF7 mutant displays altered leaf expansion, lateral root formation [30] and hypocotyl phototropism [31] in Arabidopsis. PoptrARF7.3 has EST support from a tension wood library (See Additional file 6), and microarray data indicated that PoptrARF7.3 has higher expression in xylem tissue (See Additional files 7, 8 and 9). It is possible that analogous to AtARF7's role in auxin dependent differential growth in aerial plant form [31], PoptrARF7.3 may be involved in differential growth in woody stems in response to tension stress.
At least three different singleton ArabidopsisARF genes (AtARF2, 6 and 16) were found to cluster with four or more Populus genes each. PoptrARF6.4 and PoptrARF6.5 are likely co-orthologs of an ancestral gene lost in Arabidopsis. AtARF 6 and its Populus co-orthologs have Q-rich middle regions. Potato ARF6, which is similar to AtARF6, is reported to be involved in meristem activation [32]. Sprouting buds, apical meristem and leaf tips were reported to have the highest ARF6 transcript levels. While PoptrARF6.1 has EST support from cambial zone and tension wood tissue libraries, PoptrARF6.4, has expression support from a dormant bud library and PoptrARF6.2 has EST support from floral bud libraries. Furthermore, our microarray experiments indicated that the five members of the PoptrARF6 subgroup showed differential patterns of expression. PoptrARF6.1 and 6.4 were expressed at low levels, with peaks in mature leaves and phloem-cortex samples. In contrast, PoptrARF6.2 and 6.3 were strongly expressed across most tissue types, with particularly strong expression in xylem, phloem, and vegetative and reproductive meristems. Finally, PoptrARF6.5 was not significantly expressed in any of the tissues tested (See Additional files 7 and 8). Interestingly, AtARF6 and 8 (double mutant) are associated with floral development in Arabidopsis [33]. They also display reduced stature, possibly due to reduced apical meristem activity. Though further experimental validation is required, preliminary information suggests that this subgroup has functional roles in controlling meristematic activity in distinct tissues and developmental stages.
AtARF2 negatively regulates differential growth in Arabidopsis hypocotyls [34]. AtARF2 mutants are reported to have defective floral structures [35]. T-DNA insertion mutants of AtARF2 have larger rosette leaves and reduced number and size of other aerial organs including inflorescences [33]. AtARF2 T-DNA insertion mutants also exhibit extra cell division and expansion in seeds and other vegetative and floral organs[35]. Populus has four putative AtARF2 orthologs (Figure 5). PoptrARF2.5 and PoptrARF2.6 are likely co-orthologs of an ancestral gene lost in Arabidopsis. Based on microarray and EST support, PoptrARF2.1 and 2.2 appear to express almost ubiquitously, with particularly strong expression in xylem and phloem. PoptrARF2.3 and PoptrARF2.4 were expressed in vegetative and floral buds, as well as in the cambial (cell division) zone. Tissue distribution of Populus ESTs and array results suggest that these sequence orthologs may possess similar functional contexts in Populus and Arabidopsis.
Characterization of the ettin (ett) mutant revealed that AtARF3 is involved in floral meristem, gynoecium, stamen and perianth patterning [36]. Populus has three ARF3-like genes out of which, PoptrARF3.1 and 3.2 had some EST and microarray expression support from vegetative and reproductive buds, but surprisingly these genes were most strongly expressed in xylem and phloem. PoptrARF3.3 was not included on the microarray because the gene model was artificially truncated in the initial annotation.
Monopteros (mp), a loss-of-function AtARF5 mutant, has a severely malformed embryonic axis and vascular system, and defective inflorescences where lateral flowers are completely lacking or reduced in number [37]. Populus has two putative orthologs of AtARF5, PoptrARF5.1 and 5.2, and RT-PCR and microarray results indicate slightly higher expression in roots compared to stem and leaves (See Additional files 7, 8 and 13). Populus EST and microarray data show that PoptrARF5.2 is highly expressed in floral buds. It remains to be determined if these co-orthologs could be considered as sub-functionalized with respect to AtARF5 (or common ancestor) or if they play additional roles in tree development.
Evolution, divergence and regulation of Aux/IAA and ARFgene families
Genome-wide duplications followed by a series of reciprocal tandem terminal fusions have resulted in a dramatic reorganization of the duplicated genome segments in Populus. Ensuing gene loss and expansion events have contributed toward divergence in gene family structures between the dicots, Arabidopsis and Populus [13, 38]. Moreover, the modes of expansion, either through tandem or segmental duplication, differ between members of each gene family.
Phylogenetic analysis revealed a few subgroups such as IAA16 and ARF4 that contained sequence representatives in Populus and Arabidopsis but not in rice indicating that these subgroups were acquired or differentially retained in dicots post-divergence from monocots.
Contrary to the ARF gene family, Aux/IAA gene family was observed to have expanded largely due to segmental duplications in Arabidopsis [38]. Segmental duplications have also contributed to expansion of both these gene families in rice [12, 29]. Our study indicates that PoptrIAA as well as PoptrARF genes have been largely retained at a higher than average rate following the salicoid genome-wide duplication and rearrangement events. On a genome-wide scale, approximately 14,000 of the 45,000 (~32%) predicted genes are retained in duplicated pairs resulting from the salicoid duplication event [13]. The retention rates for PoptrIAA and PoptrARF families are 80% and 82% respectively. This is in line with the expectation that genes involved in transcription regulation and signal transduction are preferentially retained following duplication [39–41]. The low proportion of retained tandem duplicates (5%) in the PoptrARF gene family compared to the PoptrIAA family is potentially due to constraints associated with dimer stoichiometry maintenance for ARF transcription factor activity as tandem clusters do exist for other Populus gene families [13]. Even though 43% of PoptrIAA genes are represented in tandem clusters of two to three genes each, nearly 86% of these tandem clusters have likely expanded and been retained following chromosome-level segmental duplication or whole-genome duplication events. These observations suggest that PoptrIAA and PoptrARF transcription factor families consist of genes originating primarily from high segmental (large-scale) and secondarily from low tandem (small-scale) duplication events. Sub-functionalization, neo-functionalization and non-functionalization events associated with duplicate transcription factor genes carry a greater weight for functional ramifications such as the ability to differentially regulate auxin signal transduction pathways. It could be speculated, based primarily on our understanding of the functional role of the nearest Arabidopsis ortholog [33–35, 42], that the presence of multiple ARF2 co-orthologs in the Populus genome could reflect the greater need for temporal control on cell division and expansion in the context of flowering, senescence and abscission in Populus, a perennial deciduous tree that exhibits seasonal dynamics and transitioning between juvenile to mature to reproductive stages across a time-span of years as opposed to days as in the annual Arabidopsis. Considering the complexity of ARF- and Aux/IAA- mediated regulation, the reasons for and implications of diversification will require further understanding using evolutionary systems biology studies [43].
Closely-related ARFs that represent pairs of sister loci have been found to have strong double-mutant phenotypes and overlapping expression domains [33]. Furthermore, interaction affinities were found to be stronger among intra-group (activator or repressor ARF groups) members when compared to inter-group members [44]. Moreover, single and double mutants resulting from AtIAA12 gain-of-function and ARF5 loss-of-function mutants have similar mutant phenotypes. It is proposed that ARF5 may act in a positive and IAA12 in negative regulatory way to control embryogenesis and root meristem development in an auxin-dependent manner [21]. RT-PCR results show that PoptrARF5 and PoptrIAA12 genes display contrasting expression patterns in roots (See Additional file 13). The high expression of PoptrARF5 in roots and low expression of PoptrIAA12 suggests that they may co-regulate root development in Populus in an auxin dependent manner. The potential number of heterotypic interactions between Populus ARFs and Aux/IAAs are likely several times greater than the number of members in these two large gene families. This may contribute to higher-order auxin signal and response mechanisms needed by perennial plants to achieve greater developmental plasticity.
The auxin response mechanism has recently been shown to be also regulated through small noncoding RNA species; microRNA (miRNA) and trans-acting short-interfering RNA (ta-siRNA). AtARF 10, 16 and 17 are known to be regulated by miR160, a miRNA group that is highly conserved across the plant kingdom [45, 46] and AtARF6 and AtARF8 have shown to be regulated by miR167 [47]. Both miR160 and miR167 families are predicted to be two-fold overrepresented in the Populus genome when compared to Arabidopsis [13]. Target sequences for miR160 have been detected in PoptrARF10.1-10.2, PoptrARF16.1–16.5 and PoptrARF17.1–17.2 and miR167 targets have been found in PoptrARF6.1–6.3 and PoptrARF8.1–8.2.
TAS3 ta-siRNA has been shown to mediate post-transcriptional regulation of ARF3 and ARF4 gene expression [48, 49] and to play a key role in juvenile to adult phase transition in Arabidopsis [50, 51]. The ta-siRNA target site sequences were reported to be conserved among AtARF2, AtARF3 and AtARF4 and related rice and wheat sequences [49]. Homologous conserved sites were found to occur once in PoptrARF2.2 and PoptrARF2.6 and twice in PoptrARF3.1, PoptrARF3.2 and PoptrARF4 genes. GenBank database searches showed that grape, tobacco, medicago and tomato ARF3-like sequences also carry two conserved ta-siRNA target sites. The individual PoptrARF co-orthologs as well as the different genes represented in the miR and tasiR groups provide myriad opportunities for complex regulatory interactions of auxin-related transcription in Populus.
Conclusion
This study identified the suites of Aux/IAA and ARF genes in the Populus genome and revealed their gene family structures. Comparative genomics with Arabidopsis and rice suggested that the two gene families have a common origin, including conservation of activator groups in ARF gene families of all three genomes, but have experienced separate evolutionary histories, diverging in important ways, including the lack of large clusters of tandemly duplicated AtARF genes from Populus and rice, lack of PoptrARF7.3 and PoptrARF7.4 orthologs in Arabidopsis and substantial expansion of certain PoptrIAA and PoptrARF subgroups through large-scale segmental duplications. Overall, the gene family structures of Populus and Arabidopsis displayed a greater degree of conservation with each other than in comparison with the monocot plant, rice.
The Populus genome sequence and the findings reported here provide new opportunities to facilitate postulation and exploration of hypotheses linking auxin response regulatory genes to conserved core plant processes, as well as perennial plant features such as wood development, long-distance nutrient and water movement, seasonal dynamics, and disease resistance. Targeted reverse genetics studies, high-resolution spatiotemporal expression surveys as well as investigations of in vivo homo- and hetero-dimerization affinities of the various Aux/IAA and ARF gene family members among taxa will need to be carried out to understand how the molecular functions of these genes translate into a diverse suite of auxin-mediated effects at the whole-plant level. Further studies on basic aspects of the functional contexts of Aux/IAA and ARF proteins will open opportunities for applications in agricultural, forestry, environmental and energy sectors. One such application includes ongoing functional genomics studies to investigate the roles of PoptrIAA and PoptrARF genes in carbon allocation and carbon sequestration.
Methods
Identification of Aux/IAA and ARF gene families in Populus
Genes were initially identified using Pfam domain IDs assigned to predicted Populus gene models in the JGI (DOE Joint Genome Institute, CA) annotation pipeline [13], and by using Arabidopsis ARF and Aux/IAA proteins [2, 11] as queries in BLASTP searches of predicted Populus proteins (Populus Genome v 1.1, January 2007). Populus proteins identified in this initial search were used as query sequences in additional BLASTP searches of the predicted Populus protein set for exhaustive identification of divergent Populus gene family members. Redundant and invalid gene models were verified based on gene structure, intactness of conserved motifs, EST support and synteny analysis. We have also included in our study an incomplete gene model (fgenesh4_pg.C_scaffold_1006000001) representative of PoptrARF6.3 because this gene model is flanked by a sequence gap followed by the conserved ARF amino terminus domains, which could potentially be corrected into a complete gene model in the upcoming version of the genome. Furthermore, this gene showed very strong evidence of expression based on microarray analyses. Sequence conservation and microsynteny analysis of Populus gene models with Populus homeologous (duplicated) genomic regions and the Arabidopsis genome was conducted using the Vista Browser tool with default curve calculation parameters; nucleotide sequence 'conservation identity' of 70% and 'minimum conservation width' of 100 bp [52]. One gene model per locus, which included some JGI annotated models representing the promoted or reference set, were used in this study. It should also be noted that in the current version of the genome (v 1.1), some of the scaffolds could potentially represent haplotypes and not unique unassembled genomic regions. Gene nomenclature is based on the consensus standard established by the International Populus Genome Consortium (IPGC) to distinguish Populus trichocarpa from other Populus species.
Chromosomal mapping of PoptrIAA and PoptrARFgenes and estimation of duplicate genes
Information on chromosomal location was gathered from the Populus genome browser [53]. Chromosomal locations were determined by integrating the genome assembly with a microsatellite-based genetic map for P. trichocarpa × P. deltoides [13, 54]. The physical chromosomal location was represented in a graphical output by scaling the 19 chromosomes (depicting regions of genome-wide duplications as in Tuskan et al., 2006) followed by scale-guided positioning or mapping of the loci. Identification of homeologous chromosome segments resulting from whole-genome duplication events was accomplished as described in Tuskan et al., 2006. Briefly, global alignments were performed using the double-affine Smith-Waterman algorithm. Genetic distances between pairs were calculated as the proportion of four-fold degenerate nucleotide sites that underwent transversions (4DTV distance). 4DTV stands for four-fold synonymous (degenerative) third-codon transversion. It represents a transversion in the third nucleotide position within four codons that does not result in a change in corresponding amino acid identity within the protein it codes for. Such an estimate of synonymous mutation rate within a transcribed region of a gene but not in region that experiences selection is a conserved means of estimating divergence within the more recent evolutionary past. Distances corresponding to the 'salicoid' whole-genome duplication events were delineated based on discrete peaks in 4DTV distributions. Duplicated segments were defined as regions on different linkage groups or scaffolds containing six or more homeologous pairs with similar 4DTV values, with fewer than 25 nonhomeologous genes intervening. Gene pairs resulting from the 'salicoid' duplication (apparently common to the Salicaceae) were defined by 4DTV values between 0.04 and 0.17. Microsynteny in flanking regions of segmental duplicates was verified using the Genome Browser. Tandemly duplicated genes that matched the same homeolog were only counted once for this analysis.
Sequence and phylogenetic analysis
Conserved protein motifs were determined from CD-searches [55] and using MEME-MAST programs [56, 57]. Sequence identity between two genes was determined using the bl2seq tool [58]. Multiple sequence alignments were performed using MUSCLE sequence alignment program [59]. Phylogenetic trees were constructed in two ways using amino acid sequence alignments of conserved regions or full-length sequences of all predicted proteins in Aux/IAA and ARF gene families of Populus, Arabidopsis [2, 9, 11] and rice [12, 29]. Arabidopsis and rice sequences were obtained from TAIR and TIGR databases. Unrooted PHYLIP trees with 1000 bootstraps were generated by Neighbor-Joining method using ClustalX 1.83 program. Phylograms were visualized in TreeView v1.6.6. Bayesian phylogenetic analysis of conserved, collated and aligned Aux/IAA (See Additional file 14) and ARF amino acid sequences (See Additional file 15) was performed using the MRBAYES (version 3.1.2) package [60, 61]. We used the WAG substitution frequency matrix [62] with among-sites rate variation modeled by means of a discrete γ distribution with four equally probable categories. Two independent runs of 1–2 million Monte Carlo Markov Chain generations with four chains each were run. Trees were sampled every 100 generations and stationary phase and burnin value was determined by plotting the likelihood scores against number of generations. Posterior probabilities calculated from consensus are shown on branches.
RNA isolation and real-time PCR analysis
Leaf, stem and root tissue material were collected from greenhouse-grown P. trichocarpa (Nisqually-1)plants and immediately frozen in liquid nitrogen. Total RNA was extracted from plant samples using plant RNeasy kit (Qiagen, CA) followed with DNase I treatment. The quality and quantity of RNA was evaluated using a Nanodrop spectrophotometer and gel electrophoresis. cDNA was synthesized from two micrograms of RNA using SuperScriptIII (Invitrogen, CA) according to manufacturers' instructions. Real-time PCR was carried out using cDNA, primer pairs (See Additional file 16) and iQ SYBR mix (BioRad, CA). Samples were run in triplicate in an iCycler real-time PCR machine (BioRad, CA) and normalized with respect to18S threshold cycle values. Fold change was calculated by the formula 2-ΔΔCt, where ΔΔCt is the difference between ΔCt of a gene in a given tissue and the lowest value ΔCt for that gene in any of the three tissue types. ΔCt was estimated by the formula; Ct of gene of interest – geometric mean of ΔCt of 18S RNA gene (control gene). EST information was obtained from GenBank and the Populus EST database, PopulusDB [63].
Microarray analysis
We used a whole genome microarray that we designed together with NimbleGen Systems, Inc. The array targets 55,794 predicted transcripts from the poplar genome sequencing project. There are three different isothermal 60 mer probes per target, designed for maximum specificity. All probes are replicated once on the array for a total of over 385,000 probes synthesized in situ on glass slides [64].
Tissues for microarray hybridizations were obtained from field and greenhouse-grown trees, and tissue collection methods varied depending on the tissue type. All tissue was obtained from Populus trichocarpa clone Nisqually-1, except for floral tissue and seeds, which were collected from trees growing in the wild near Corvallis, Oregon. Tissues and abbreviations are listed in Additional file 17. Two biological replicates were used for each of 12 tissue types, and three biological replicates were used for xylem and phloem samples. RNA was extracted as described for RT-PCR analyses. Labeling, hybridization, and scanning were carried out by NimbleGen using their standard expression array protocols.
Array data (See Additional file 18) were normalized using the NimbleGen microarray data processing pipeline (NMPP) [65]. We used a two step normalization procedure starting with an initial quantile normalization among replicates within each tissue, and then a global normalization to adjust all tissues to a similar baseline. Gene-level analyses were performed using the mean normalized fluorescence values for all probes and replicates. For negative controls we used probes targeting 3,149 transposable elements that were contaminants in the initial release of Populus gene models. This approach is warranted because the vast majority of these elements will be quiescent at any particular time, an assumption that is supported by the significantly lower hybridization signals from these probes compared to all other groups of targets on the array (data not shown). For each experiment we defined the expression threshold as the 95th percentile of the fluorescence values for all negative control targets. Relative expression of individual genes was assessed as fold-change from this negative control threshold value. A two-fold change was considered to be evidence of significant expression.
References
Abel S, Theologis A: A polymorphic bipartite motif signals nuclear targeting of early auxin-inducible proteins related to PS-IAA4 from pea (Pisum sativum). Plant J. 1995, 8 (1): 87-96. 10.1046/j.1365-313X.1995.08010087.x.
Hagen G, Guilfoyle T: Auxin-responsive gene expression: genes, promoters and regulatory factors. Plant Mol Biol. 2002, 49 (3–4): 373-385. 10.1023/A:1015207114117.
Ulmasov T, Hagen G, Guilfoyle TJ: Dimerization and DNA binding of auxin response factors. Plant J. 1999, 19 (3): 309-319. 10.1046/j.1365-313X.1999.00538.x.
Ulmasov T, Hagen G, Guilfoyle TJ: ARF1, a transcription factor that binds to auxin response elements. Science. 1997, 276 (5320): 1865-1868. 10.1126/science.276.5320.1865.
Ulmasov T, Hagen G, Guilfoyle TJ: Activation and repression of transcription by auxin-response factors. Proc Natl Acad Sci USA. 1999, 96 (10): 5844-5849. 10.1073/pnas.96.10.5844.
Dharmasiri N, Dharmasiri S, Estelle M: The F-box protein TIR1 is an auxin receptor. Nature. 2005, 435 (7041): 441-445. 10.1038/nature03543.
Kepinski S, Leyser O: Auxin-induced SCFTIR1-Aux/IAA interaction involves stable modification of the SCFTIR1 complex. Proc Natl Acad Sci USA. 2004, 101 (33): 12381-12386. 10.1073/pnas.0402868101.
Kepinski S, Leyser O: The Arabidopsis F-box protein TIR1 is an auxin receptor. Nature. 2005, 435 (7041): 446-451. 10.1038/nature03542.
Tiwari SB, Hagen G, Guilfoyle T: The roles of auxin response factor domains in auxin-responsive transcription. Plant Cell. 2003, 15 (2): 533-543. 10.1105/tpc.008417.
Woodward AW, Bartel B: Auxin: regulation, action, and interaction. Ann Bot (Lond). 2005, 95 (5): 707-735. 10.1093/aob/mci083.
Liscum E, Reed JW: Genetics of Aux/IAA and ARF action in plant growth and development. Plant Mol Biol. 2002, 49 (3–4): 387-400. 10.1023/A:1015255030047.
Jain M, Kaur N, Garg R, Thakur JK, Tyagi AK, Khurana JP: Structure and expression analysis of early auxin-responsive Aux/IAA gene family in rice (Oryza sativa). Functional & integrative genomics. 2006, 6 (1): 47-59. 10.1007/s10142-005-0005-0.
Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J, Sterck L, Aerts A, Bhalerao RR, Bhalerao RP, Blaudez D, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M, Chapman J, Chen GL, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q, Cunningham R, Davis J, Degroeve S, Dejardin A, Depamphilis C, Detter J, Dirks B, Dubchak I, Duplessis S, Ehlting J, Ellis B, Gendler K, Goodstein D, Gribskov M, Grimwood J, Groover A, Gunter L, Hamberger B, Heinze B, Helariutta Y, Henrissat B, Holligan D, Holt R, Huang W, Islam-Faridi N, Jones S, Jones-Rhoades M, Jorgensen R, Joshi C, Kangasjarvi J, Karlsson J, Kelleher C, Kirkpatrick R, Kirst M, Kohler A, Kalluri U, Larimer F, Leebens-Mack J, Leple JC, Locascio P, Lou Y, Lucas S, Martin F, Montanini B, Napoli C, Nelson DR, Nelson C, Nieminen K, Nilsson O, Pereda V, Peter G, Philippe R, Pilate G, Poliakov A, Razumovskaya J, Richardson P, Rinaldi C, Ritland K, Rouze P, Ryaboy D, Schmutz J, Schrader J, Segerman B, Shin H, Siddiqui A, Sterky F, Terry A, Tsai CJ, Uberbacher E, Unneberg P, Vahala J, Wall K, Wessler S, Yang G, Yin T, Douglas C, Marra M, Sandberg G, Van de Peer Y, Rokhsar D: The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science. 2006, 313 (5793): 1596-1604. 10.1126/science.1128691.
Tiwari SB, Hagen G, Guilfoyle TJ: Aux/IAA proteins contain a potent transcriptional repression domain. Plant Cell. 2004, 16 (2): 533-543. 10.1105/tpc.017384.
Ramos JA, Zenser N, Leyser O, Callis J: Rapid degradation of auxin/indoleacetic acid proteins requires conserved amino acids of domain II and is proteasome dependent. Plant Cell. 2001, 13 (10): 2349-2360. 10.1105/tpc.13.10.2349.
Dreher KA, Brown J, Saw RE, Callis J: The Arabidopsis Aux/IAA protein family has diversified in degradation and auxin responsiveness. Plant Cell. 2006, 18 (3): 699-714. 10.1105/tpc.105.039172.
Nagpal P, Walker LM, Young JC, Sonawala A, Timpte C, Estelle M, Reed JW: AXR2 encodes a member of the Aux/IAA protein family. Plant Physiol. 2000, 123 (2): 563-574. 10.1104/pp.123.2.563.
Reed JW: Roles and activities of Aux/IAA proteins in Arabidopsis. Trends Plant Sci. 2001, 6 (9): 420-425. 10.1016/S1360-1385(01)02042-8.
Rogg LE, Lasswell J, Bartel B: A gain-of-function mutation in IAA28 suppresses lateral root development. Plant Cell. 2001, 13 (3): 465-480. 10.1105/tpc.13.3.465.
Fukaki H, Tameda S, Masuda H, Tasaka M: Lateral root formation is blocked by a gain-of-function mutation in the SOLITARY-ROOT/IAA14 gene of Arabidopsis. Plant J. 2002, 29 (2): 153-168. 10.1046/j.0960-7412.2001.01201.x.
Hamann T, Benkova E, Baurle I, Kientz M, Jurgens G: The Arabidopsis BODENLOS gene encodes an auxin response protein inhibiting MONOPTEROS-mediated embryo patterning. Genes Dev. 2002, 16 (13): 1610-1615. 10.1101/gad.229402.
Tian Q, Uhlir NJ, Reed JW: Arabidopsis SHY2/IAA3 inhibits auxin-regulated gene expression. Plant Cell. 2002, 14 (2): 301-319. 10.1105/tpc.010283.
Tatematsu K, Kumagai S, Muto H, Sato A, Watahiki MK, Harper RM, Liscum E, Yamamoto KT: MASSUGU2 encodes Aux/IAA19, an auxin-regulated protein that functions together with the transcriptional activator NPH4/ARF7 to regulate differential growth responses of hypocotyl and formation of lateral roots in Arabidopsis thaliana. Plant Cell. 2004, 16 (2): 379-393. 10.1105/tpc.018630.
Moyle R, Schrader J, Stenberg A, Olsson O, Saxena S, Sandberg G, Bhalerao RP: Environmental and auxin regulation of wood formation involves members of the Aux/IAA gene family in hybrid aspen. Plant J. 2002, 31 (6): 675-685. 10.1046/j.1365-313X.2002.01386.x.
Choi G, Yi H, Lee J, Kwon YK, Soh MS, Shin B, Luka Z, Hahn TR, Song PS: Phytochrome signalling is mediated through nucleoside diphosphate kinase 2. Nature. 1999, 401 (6753): 610-613. 10.1038/44176.
Soh MS, Hong SH, Kim BC, Vizir I, Park DH, Choi G, Hong MY, Chung YY, Furuya M, Nam HG: Regulation of both light- and auxin-mediated development by the Arabidopsis IAA3/SHY2 gene. Journal of Plant Biology. 1999, 42: 239-246.
Padmanabhan MS, Goregaoker SP, Golem S, Shiferaw H, Culver JN: Interaction of the tobacco mosaic virus replicase protein with the Aux/IAA protein PAP1/IAA26 is associated with disease development. Journal of virology. 2005, 79 (4): 2549-2558. 10.1128/JVI.79.4.2549-2558.2005.
Padmanabhan MS, Shiferaw H, Culver JN: The Tobacco mosaic virus replicase protein disrupts the localization and function of interacting Aux/IAA proteins. Mol Plant Microbe Interact. 2006, 19 (8): 864-873. 10.1094/MPMI-19-0864.
Wang D, Pei K, Fu Y, Sun Z, Li S, Liu H, Tang K, Han B, Tao Y: Genome-wide analysis of the auxin response factors (ARF) gene family in rice (Oryza sativa). Gene. 2007, 394 (1–2): 13-24. 10.1016/j.gene.2007.01.006.
Wilmoth JC, Wang S, Tiwari SB, Joshi AD, Hagen G, Guilfoyle TJ, Alonso JM, Ecker JR, Reed JW: NPH4/ARF7 and ARF19 promote leaf expansion and auxin-induced lateral root formation. Plant J. 2005, 43 (1): 118-130. 10.1111/j.1365-313X.2005.02432.x.
Harper RM, Stowe-Evans EL, Luesse DR, Muto H, Tatematsu K, Watahiki MK, Yamamoto K, Liscum E: The NPH4 locus encodes the auxin response factor ARF7, a conditional regulator of differential growth in aerial Arabidopsis tissue. Plant Cell. 2000, 12 (5): 757-770. 10.1105/tpc.12.5.757.
Faivre-Rampant O, Cardle L, Marshall D, Viola R, Taylor MA: Changes in gene expression during meristem activation processes in Solanum tuberosum with a focus on the regulation of an auxin response factor gene. J Exp Bot. 2004, 55 (397): 613-622. 10.1093/jxb/erh075.
Okushima Y, Overvoorde PJ, Arima K, Alonso JM, Chan A, Chang C, Ecker JR, Hughes B, Lui A, Nguyen D, Onodera C, Quach H, Smith A, Yu G, Theologis A: Functional genomic analysis of the AUXIN RESPONSE FACTOR gene family members in Arabidopsis thaliana: unique and overlapping functions of ARF7 and ARF19. Plant Cell. 2005, 17 (2): 444-463. 10.1105/tpc.104.028316.
Li H, Johnson P, Stepanova A, Alonso JM, Ecker JR: Convergence of signaling pathways in the control of differential cell growth in Arabidopsis. Dev Cell. 2004, 7 (2): 193-204. 10.1016/j.devcel.2004.07.002.
Schruff MC, Spielman M, Tiwari S, Adams S, Fenby N, Scott RJ: The AUXIN RESPONSE FACTOR 2 gene of Arabidopsis links auxin signalling, cell division, and the size of seeds and other organs. Development. 2006, 133 (2): 251-261. 10.1242/dev.02194.
Sessions A, Nemhauser JL, McColl A, Roe JL, Feldmann KA, Zambryski PC: ETTIN patterns the Arabidopsis floral meristem and reproductive organs. Development. 1997, 124 (22): 4481-4491.
Hardtke CS, Berleth T: The Arabidopsis gene MONOPTEROS encodes a transcription factor mediating embryo axis formation and vascular development. Embo J. 1998, 17 (5): 1405-1411. 10.1093/emboj/17.5.1405.
Remington DL, Vision TJ, Guilfoyle TJ, Reed JW: Contrasting modes of diversification in the Aux/IAA and ARF gene families. Plant Physiol. 2004, 135 (3): 1738-1752. 10.1104/pp.104.039669.
Blanc G, Wolfe KH: Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell. 2004, 16 (7): 1679-1691. 10.1105/tpc.021410.
Maere S, De Bodt S, Raes J, Casneuf T, Van Montagu M, Kuiper M, Van de Peer Y: Modeling gene and genome duplications in eukaryotes. Proc Natl Acad Sci USA. 2005, 102 (15): 5454-5459. 10.1073/pnas.0501102102.
Seoighe C, Gehring C: Genome duplication led to highly selective expansion of the Arabidopsis thaliana proteome. Trends Genet. 2004, 20 (10): 461-464. 10.1016/j.tig.2004.07.008.
Ellis CM, Nagpal P, Young JC, Hagen G, Guilfoyle TJ, Reed JW: AUXIN RESPONSE FACTOR1 and AUXIN RESPONSE FACTOR2 regulate senescence and floral organ abscission in Arabidopsis thaliana. Development. 2005, 132 (20): 4563-4574. 10.1242/dev.02012.
Hughes AL: Gene duplication and the origin of novel proteins. Proc Natl Acad Sci USA. 2005, 102 (25): 8791-8792. 10.1073/pnas.0503922102.
Hardtke CS, Ckurshumova W, Vidaurre DP, Singh SA, Stamatiou G, Tiwari SB, Hagen G, Guilfoyle TJ, Berleth T: Overlapping and non-redundant functions of the Arabidopsis auxin response factors MONOPTEROS and NONPHOTOTROPIC HYPOCOTYL 4. Development. 2004, 131 (5): 1089-1100. 10.1242/dev.00925.
Mallory AC, Bartel DP, Bartel B: MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell. 2005, 17 (5): 1360-1375. 10.1105/tpc.105.031716.
Axtell MJ, Bartel DP: Antiquity of microRNAs and their targets in land plants. Plant Cell. 2005, 17 (6): 1658-1673. 10.1105/tpc.105.032185.
Wu MF, Tian Q, Reed JW: Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development. 2006, 133 (21): 4211-4218. 10.1242/dev.02602.
Allen E, Xie Z, Gustafson AM, Carrington JC: microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005, 121 (2): 207-221. 10.1016/j.cell.2005.04.004.
Williams L, Carles CC, Osmont KS, Fletcher JC: A database analysis method identifies an endogenous trans-acting short-interfering RNA that targets the Arabidopsis ARF2, ARF3, and ARF4 genes. Proc Natl Acad Sci USA. 2005, 102 (27): 9703-9708. 10.1073/pnas.0504029102.
Hunter C, Willmann MR, Wu G, Yoshikawa M, de la Luz Gutierrez-Nava M, Poethig SR: Trans-acting siRNA-mediated repression of ETTIN and ARF4 regulates heteroblasty in Arabidopsis. Development. 2006, 133 (15): 2973-2981. 10.1242/dev.02491.
Fahlgren N, Montgomery TA, Howell MD, Allen E, Dvorak SK, Alexander AL, Carrington JC: Regulation of AUXIN RESPONSE FACTOR3 by TAS3 ta-siRNA affects developmental timing and patterning in Arabidopsis. Curr Biol. 2006, 16 (9): 939-944. 10.1016/j.cub.2006.03.065.
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I: VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, W273-279. 10.1093/nar/gkh458. 32 Web Server
Yin TM, DiFazio SP, Gunter LE, Riemenschneider D, Tuskan GA: Large-scale heterospecific segregation distortion in Populus revealed by a dense genetic map. Theor Appl Genet. 2004, 109 (3): 451-463. 10.1007/s00122-004-1653-5.
Marchler-Bauer A, Anderson JB, Derbyshire MK, DeWeese-Scott C, Gonzales NR, Gwadz M, Hao L, He S, Hurwitz DI, Jackson JD, Ke Z, Krylov D, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Thanki N, Yamashita RA, Yin JJ, Zhang D, Bryant SH: CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007, D237-240. 10.1093/nar/gkl951. 35 Database
Bailey TL, Elkan C: Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994, 2: 28-36.
Bailey TL, Gribskov M: Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998, Oxford, England, 14 (1): 48-54. 10.1093/bioinformatics/14.1.48.
Tatusova TA, Madden TL: BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 1999, 174 (2): 247-250. 10.1111/j.1574-6968.1999.tb13575.x.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32 (5): 1792-1797. 10.1093/nar/gkh340.
Huelsenbeck JP, Ronquist F: MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001, Oxford, England, 17 (8): 754-755. 10.1093/bioinformatics/17.8.754.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, Oxford, England, 19 (12): 1572-1574. 10.1093/bioinformatics/btg180.
Whelan S, Goldman N: A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol. 2001, 18 (5): 691-699.
Sterky F, Bhalerao RR, Unneberg P, Segerman B, Nilsson P, Brunner AM, Charbonnel-Campaa L, Lindvall JJ, Tandre K, Strauss SH, Sundberg B, Gustafsson P, Uhlen M, Bhalerao RP, Nilsson O, Sandberg G, Karlsson J, Lundeberg J, Jansson S: A Populus EST resource for plant functional genomics. Proc Natl Acad Sci USA. 2004, 101 (38): 13951-13956. 10.1073/pnas.0401641101.
Groover AT, Mansfield SD, DiFazio SP, Dupper G, Fontana JR, Millar R, Wang Y: The Populus homeobox gene ARBORKNOX1 reveals overlapping mechanisms regulating the shoot apical meristem and the vascular cambium. Plant Mol Biol. 2006, 61 (6): 917-932. 10.1007/s11103-006-0059-y.
Wang X, He H, Li L, Chen R, Deng XW, Li S: NMPP: a user-customized NimbleGen microarray data processing pipeline. Bioinformatics. 2006, Oxford, England, 22 (23): 2955-2957. 10.1093/bioinformatics/btl525.
Acknowledgements
The authors would like to thank the International Populus Genome Consortium for sequencing, assembling and annotating the Populus genome and for sharing the Populus EST database and the Vista browser tool. We also thank S. Mane, P. Dharmawardhana and O. Crasta for help with microarray data analysis and the three anonymous reviewers for their helpful comments. This research was supported by the U.S Department of Energy, Office of Science, Biological and Environmental Research (project title; "Genome-Enabled Discovery of Carbon Sequestration Genes in Populus") to GAT and DOE/BER grant no. DE-FG02-06ER64185 to AMB. Oak Ridge National Laboratory is managed by UT-Battelle, LLC, for the U.S. Department of Energy under contract DE-AC05-00OR22725.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
UCK carried out the sequence, phylogenetic and expression analyses and drafted the manuscript. SPD contributed to sequence analysis and together with AMB generated the microarray data and carried out data analysis. GAT contributed to the conception of the study. All authors read and approved the final manuscript.
Electronic supplementary material
12870_2007_200_MOESM2_ESM.xls
Additional File 2: Summary of Vista conservation- microsynteny analysis. "yes" and "no" refer to existence of at least 70% identity within a 100 bp sliding window between Poplar v.1.0, Poplar duplicates and Arabidopsis thaliana sequences. (XLS 18 KB)
12870_2007_200_MOESM3_ESM.pdf
Additional File 3: Multiple sequence alignment of full-length amino acid sequences of predicted Populus, Arabidopsis and rice Aux/IAA proteins. Sequences were aligned using MUSCLE program. Consensus sequence is indicated at the bottom of the alignment. (PDF 60 KB)
12870_2007_200_MOESM4_ESM.pdf
Additional File 4: Multiple sequence alignment of conserved regions of predicted Populus, Arabidopsis and rice Aux/IAA protein sequences. Sequences were aligned using MUSCLE program. Consensus sequence is indicated at the bottom of the alignment. (PDF 22 KB)
12870_2007_200_MOESM5_ESM.pdf
Additional File 5: Prediction of conserved domains in full-length amino acid sequences of predicted Populus, Arabidopsis and rice Aux/IAA proteins. Conserved domains were predicted using the MEME and MAST programs. Motif number 6 represents the conserved domain I, Motif number 3 represents the conserved domain II, Motif number 2 represents the conserved domain III and Motif numbers 1, 4 and 5 represent the conserved domain IV. (PDF 51 KB)
12870_2007_200_MOESM6_ESM.xls
Additional File 6: EST support for Populus Aux/IAA and ARF genes. Numbers indicate the number of ESTs represented in each tissue library. Source: the Populus EST database, PopulusDB. (XLS 28 KB)
12870_2007_200_MOESM7_ESM.xls
Additional File 7: Fold change data from microarray expression analyses for PoptrIAA and PoptrARF genes. Data are presented as normalized expression values of target genes relative to the 95th percentile of expression values for transposable element targets (presumably unexpressed). Genes with a greater than 2-fold difference from negative controls are highlighted in bold. Sample abbreviations are defined in Additional file 17. (XLS 34 KB)
12870_2007_200_MOESM8_ESM.xls
Additional File 8: Normalized raw data from microarray expression analyses for PoptrIAA and PoptrARF genes. Data were normalized using quantile normalization within replicates and then across tissue types. Data are presented as relative expression values. Also included are 95th percentile of normalized expression for negative control probes. Sample abbreviations are defined in Additional file 17. (XLS 56 KB)
12870_2007_200_MOESM9_ESM.pdf
Additional File 9: Temperature diagram representation of microarray results. Figures are temperature diagrams with intensity of signal calculated in columns, which are derived from means for all data for a particular tissue type. The bottom line in each figure represents the negative control. Red means higher expression, dark blue means lower expression. Sample abbreviations are defined in Additional file 17. (PDF 28 KB)
12870_2007_200_MOESM10_ESM.pdf
Additional File 10: Multiple sequence alignment of full-length amino acid sequences of predicted Populus, Arabidopsis and rice ARF proteins. Sequences were aligned using MUSCLE program. Consensus sequence is indicated at the bottom of the alignment. (PDF 185 KB)
12870_2007_200_MOESM11_ESM.pdf
Additional File 11: Multiple sequence alignment of conserved regions of predicted Populus, Arabidopsis and rice ARF protein sequences. Sequences were aligned using MUSCLE program. Consensus sequence is indicated at the bottom of the alignment. (PDF 45 KB)
12870_2007_200_MOESM12_ESM.pdf
Additional File 12: Prediction of conserved domains in full-length amino acid sequences of predicted Populus, Arabidopsis and rice ARF proteins. Conserved domains were predicted using the MEME and MAST programs. Motif numbers 1, 2, 3, 4, 7 and 11 represent the conserved B3 domain. Motif numbers 6, 8 and 12 represent the conserved auxin response domain and Motif numbers 5 and 10 represent the conserved C-terminal Aux/IAA domain (III and IV). (PDF 126 KB)
12870_2007_200_MOESM13_ESM.pdf
Additional File 13: Expression analysis of PoptrARF5 and PoptrIAA12 genes using real-time RT-PCR. * Fold Change is represented relative to lowest value observed for the gene. Lowest value was determined by comparison of relative threshold cycle values for a specific gene across leaf, stem and root samples. Fold change was calculated by the formula 2-ΔΔCt, where ΔΔCt is the difference between ΔCt of a gene is a given tissue and the lowest value ΔCt for that gene in any of the three tissue types. ΔCt was estimated by the formula; (Ct of gene of interest) – (geometric mean of ΔCt of 18S RNA gene, control gene). (PDF 12 KB)
12870_2007_200_MOESM14_ESM.pdf
Additional File 14: Bayesian phylogenetic analysis of conserved regions of predicted Aux/IAA protein sequences using MRBAYES. Amino acid sequences of full-length predicted proteins were aligned using MUSCLE program. Tree was produced using conserved collated regions (See Additional file 4) as described in methods. Posterior probabilities calculated from consensus are shown on branches. Green boxes represent sustained expansion of subgroups in Populus. (PDF 21 KB)
12870_2007_200_MOESM15_ESM.pdf
Additional File 15: Bayesian phylogenetic analysis of conserved regions of predicted ARF protein sequences using MRBAYES. Amino acid sequences of full-length predicted proteins were aligned using MUSCLE program. Tree was produced using conserved collated regions (See Additional file 11) as described in methods. Posterior probabilities calculated from consensus are shown on branches. Green and orange boxes represent sustained expansion of subgroups in Populus and Arabidopsis, respectively. Grey box represents the Q-rich activator ARF subgroup. (PDF 39 KB)
12870_2007_200_MOESM17_ESM.xls
Additional File 17: Tissues used for microarray experiments. A description of sources of RNA and collection methods for array experiments. (XLS 18 KB)
12870_2007_200_MOESM18_ESM.xls
Additional File 18: Microarray data for PoptrIAA and PoptrARF gene families from the NimbleGen Populus whole genome microarray version 1.0. Data are means among all probes for the specified target, normalized as described in the text. Different columns for the same tissue type are biological replicates. Negative controls are 95th percentile of expression levels for 3155 Transposable Elements from the Populus genome. (XLS 30 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kalluri, U.C., DiFazio, S.P., Brunner, A.M. et al. Genome-wide analysis of Aux/IAA and ARF gene families in Populus trichocarpa. BMC Plant Biol 7, 59 (2007). https://doi.org/10.1186/1471-2229-7-59
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-7-59