
Abstract
Background
Identifying the target genes of transcription factors is important for unraveling regulatory networks in all types of organisms. Our interest was precisely to uncover the spectrum of loci regulated by a widespread plant transcription factor involved in physiological adaptation to drought, a type of stress that plants have encountered since the colonization of land habitats 400 MYA. The regulator under study, named ASR1, is exclusive to the plant kingdom (albeit absent in Arabidopsis) and known to alleviate the stress caused by restricted water availability. As its target genes are still unknown despite the original cloning of Asr1 cDNA 20 years ago, we examined the tomato genome for specific loci interacting in vivo with this conspicuous protein.
Results
We performed ChIP followed by high throughput DNA sequencing (ChIP-seq) on leaves from stressed tomato plants, using a high-quality anti-ASR1 antibody. In this way, we unraveled a novel repertoire of target genes, some of which are clearly involved in the response to drought stress. Many of the ASR1-enriched genomic loci we found encode enzymes involved in cell wall synthesis and remodeling as well as channels implicated in water and solute flux, such as aquaporins. In addition, we were able to determine a robust consensus ASR1-binding DNA motif.
Conclusions
The finding of cell wall synthesis and aquaporin genes as targets of ASR1 is consistent with their suggested role in the physiological adaptation of plants to water loss. The results gain insight into the environmental stress-sensing pathways leading to plant tolerance of drought.
Similar content being viewed by others


Background
Plant species in arid zones are constantly exposed to drought stress [1]. Tolerance to such water deficits most likely occurred in organisms like bryophyte mosses and was evolutionarily important during the conquest of land by plants [2, 3]. Proteins of the LEA superfamily are part of the molecular response to this stressful environment [4] and are classified into groups based on amino acid sequence motifs [5]. Despite the various roles suggested for LEA proteins, their precise functions have not been fully revealed.
The widespread (albeit absent in Arabidopsis) ASR proteins (Abscisic, Stress, Ripening) are considered to be a subgroup of the LEA superfamily [6, 7]. From an evolutionary standpoint, we previously reported the locus Asr2 to have been a target of positive selection in dry habitats, at least in species of the Solanum genus [8, 9]. Regarding biochemical function, the paralogous Asr1, cloned as long as 20 years ago (GenBank accession number L08255) [10], encodes a 14-kDa polypeptide (ASR1) proposed to act as both a chaperone [11] and a transcription factor (TF) [12]. However, no target genes from tomato have been reported for ASR1 albeit at the beginning and even at completion of this work, two target genes from other species had been identified: i) a sugar transport gene in Vitis vinifera (grape) [13] and ii) ABI4 in transgenic Arabidopsis thaliana[14]. Given the complexity of the drought stress response in general [15, 16], we have long suspected that a great deal of targets exist in the large tomato genome, which contains as many as 34,771 protein-coding genes [17].
Therefore, we were convinced that it was worthwhile pursuing the challenge to identify the direct target genes of ASR1, our regulator of interest, which is thought to control the downstream network necessary for cellular adjustment to water loss. We believed that knowledge on this particular “targetome” would generate valuable mechanistic insights into the genetic program leading to such a physiological adaptation. To achieve this goal, we carried out ChIP-sequencing (ChIP-seq), a strategy that combines chromatin immunoprecipitation (ChIP) with massively parallel (throughput) DNA sequencing to identify the in vivo binding sites of DNA-associated proteins, including TFs. As it proved to be useful to map global binding sites precisely for any nuclear protein of interest believed to associate with chromatin, ChIP-seq has emerged as a powerful tool in eukaryotes, particularly in mammals, including humans [18, 19], and plants [18–20]. In this way and using a high-quality anti-ASR1 antibody and advanced bioinformatics tools, we generated ChIP-seq data that allowed us to assemble a genome-wide high-resolution DNA-binding map of ASR1, highlighting plant genes that appear to be logically associated with the drought stress response, namely those encoding aquaporins and those associated with the cell wall.
Results
The size of the immunoprecipitated fragments (input for ChIP) and quality assessment of the affinity-purified anti-ASR1 antibody
After shearing DNA through sonication of lysed nuclei, we determined the average size of the resulting DNA fragments by means of gel electrophoresis. They were approximately 400 bp (Figure 1A), a suitable size for input DNA for subsequent ChIP and library construction.

Library construction and quality assessment of the antibody. A) Determination of the DNA fragment sizes in input chromatin samples by agarose gel electrophoresis. The gel was stained with ethidium bromide and the band intensity was quantified with ImageJ software. M indicates molecular weight markers (100-bp ladder). B) Western blot of the ASR1-immunoprecipitated DNA samples subjected to the ChIP protocol (without crosslinking) using the pure specific anti-ASR1 antibody (ASR1) or an irrelevant, non-specific antibody (NS) and a secondary antibody that recognizes only native immunoglobulins to prevent the undesired detection of immunoglobulins previously used for immunoprecipitation (which denatured on the gel during electrophoresis). INP indicates the input chromatin sample.
After the ASR1 protein was successful purified (Additional file 1: Figure S1), an anti-ASR1 antibody was raised in rabbits, affinity-purified and checked via a dot blot (Additional file 1: Figure S2). The immunoprecipitation (IP) ability of this polyclonal anti-ASR1 antibody was tested by performing a preliminary IP assay followed by SDS-PAGE and a Western blot. As expected, we were able to detect a clear single band corresponding to ASR1 (14 kDa) both in samples precipitated with the specific antibody alone as well as in whole chromatin (Figure 1B).
Once the quality of the antibody and the size of the sheared DNA fragments were assessed, we performed the ChIP protocol (see Methods section for details).
Anti-ASR1 ChIP followed by deep sequencing
We performed ChIP, using stressed tomato leaves as the starting tissue and purified anti-ASR1 antibody for the IP assay. The recovered DNA was subjected to high throughput sequencing on Illumina Hiseq 2000 equipment. To identify immuno-enriched regions, we made use of the Macs software program [21] (settings described in Additional file 1: List 1). Macs generated a list of 225 regions enriched in the immunoprecipitated sample; the most statistically relevant are shown in Table 1 (a complete list is given in Additional file 2: Data set 1). To corroborate the informatics analysis, the peaks were manually visualized using the Integrative Genomics Viewer (IGV) genome browser [22] (Figure 2A). Analysis was also performed with the software program Cisgenome [23] and the statistical package CSAR [24], but these programs yielded false-positive peaks (present in both precipitated and INPUT samples) and were thus not used for further analysis. The software program readingExtension was used to extend the reads of the SAM file from approximately 51 to 400 nt (Additional file 3: TomatoProgramCode zip file).

Visual verification of the peaks and individual validation. A) A histogram indicating the number of reads of 150-bp window DNA sequences along five selected genomic regions (3–830, 9–950, 10–800, 10–810 and 10–820) where statistically significant peaks were observed. The corresponding gene organization in exons (boxes) and introns is also shown. The +1 arrow indicates the sense of transcription. ST-3 yielded no peaks. B) qPCR for the same five selected sequences present in peaks from ChIP-seq. For each locus, the % INPUT is shown. The ST-3 amplicon was used as a negative control. Amplicon names are abbreviations for gene codes (see Table 2, which also shows gene function). Primers are listed in Additional file 1: Table S1. *p < 0.05, **p < 0.01, ***p < 0.005 (a one-tailed Student’s t-student compared against ST-3).
Validation of the immuno-enriched sequences revealed by high throughput sequencing
To validate the accepted peaks resulting from our genome-wide approach, we performed qPCR designed to individually amplify several previously ChIP-enriched regions, chosen due to either the known function of the genes present in the peaks or a high statistical value (Figure 2B, Table 2, Additional file 1: Table S1). An additional peak-free region was used as a negative control. We used a duplicate of the sample subjected to deep sequencing as a DNA template for PCR amplification, in addition to two other independent immunoprecipitates obtained under the same conditions. In all three samples, the five selected amplicons were found to be significantly enriched (using a one-tailed T-student test) when compared to the negative control ST-3 (Figure 2B).
Overall bioinformatics analysis
Once we confirmed that our ChIP data were reproducible and confident, we carried out a deeper inspection of the data. We observed that 70% of the 225 regions studied localize to gene-containing regions (the complete list is in Additional file 4: Data set 2). We defined these “genic regions” as spanning from 3 kb upstream of the transcription initiation site to 1 kb downstream of the end of transcription. By further analyzing the peaks found within genes, we observed that 42% of them localized to the upstream regulatory region of the corresponding gene (up to 3 kb upstream of the transcription start point) and 45% of them localized to coding regions plus introns. The small remaining fraction (13%) of peaks fell within 1 kb of the downstream sequences (Figure 3A).

Analysis of ASR1-bound genomic regions. A) Distribution of ASR1-binding regions along the genome. The percentage of peaks located 5’ (up to 3 kb upstream of the start point of transcription), within the gene bodies and 3’ (down to 1 kb downstream of the end of transcription). B) Cellular functions of the enriched genes present in peaks. Classification was based on reported gene functions. Small categories comprising less than 2% of the genes were grouped and referred to as “other categories” instead of “miscellaneous”. C-D-E) Average distribution of leaf ASR1 protein upon stress along distinct tomato gene regions. For each region displayed in A, we considered only those genes that gave reads mainly in each visualized category (66 genes for “promoter” (C), 72 genes for “exons” (D) and 21 genes for “downstream regions” (E)). Each gene region was subdivided into 100 consecutive segments. The X-axis represents the segment number. The number of reads in each segment (for each gene) was relativized to the number of reads of the gene that showed the highest peak. Normalized values from all the genes for each segment were then averaged (Y-axis) to display any bias in the location of ASR1 binding within each region.
Average distribution maps of leaf ASR1 along the tomato genome upon stress
To get an overall picture of the distribution of ASR1 binding along different gene zones, we constructed average distribution maps for target genes by using the software program “averageDistribution” (Additional file 3: TomatoProgramCode zip file). While the reads were evenly distributed amongst the promoter regions, the peaks were centered when present in exons or downstream regions (Figure 3C).
Sorting out ASR1 target genes according to function
Genes associated with ASR1 were classified using the software program Mapman [25]. The sorting was based on possible gene functions, mainly according to sequence homology and the presence of domains of known functions in other organisms. We observed a great diversity of gene groups involved with most of the cell functions (Figure 3B, the complete list is in Additional file 5: Data set 3). We also used Mapman software to define which gene groups were significantly over-represented compared with their relative amount in the whole genome. Over-represented categories included genes encoding cell wall-related proteins and major intrinsic proteins (MIPs), as well as miscellaneous functions (Table 3, Additional file 5: Data set 3).
Identification of the consensus ASR1-binding motif
The consensus ASR1-binding motif was determined using the software program Gimmemotif [26] that identifies similar redundant motifs using a “weighted information content” based on similarity scores and clustering using an iterative procedure. Using this computational tool, we found a robust consensus DNA motif for ASR1 (Figure 4A). Gimmemotif yielded several consensus sequences, and we chose the one with the best performance.

Consensus ASR1-binding DNA motif. The consensus sequence was determined by Gimmemotif [26]. A) The best consensus sequence motif obtained. The size of each letter is proportional to the frequency of each nucleotide in that position within the consensus motif. B) Specificity and sensitivity of consensus sequences tested with a ROC curve using both genomic and random background types. Values of the area under the ROC curve (AUC) near 1 indicate better consensus sequences while values near 0.5 indicate a lack of consensus. C) Frequency distribution of hits for the consensus motif relative to the center of the input peaks. The graph indicates that the consensus motif is well centered.
To assess performance, Gimmemotif provides a ROC curve [26] that plots the number of true positives (sensitivity) as a function of false positives (specificity) (Figure 4B). The performance is measured as the area below the ROC curve (ABC-ROC), which ranges from 0.5 for the lowest performance to 1 for perfect performance. The ROC curve is obtained by using either a “genomic” background with the same dinucleotide frequency as the input values or a “random” matched background composed of genomic sequences randomly taken from a similar distance to the transcription start site as the input peaks [26]. Finally, the software plotted the location of the obtained consensus motif relative to the center of each peak and showed that the obtained motif is predominantly located in the middle of the reads (Figure 4C).
Additionally, we used the software program "consensusCounter" (Additional file 3: TomatoProgramCode zip file) to determine that the frequency of the consensus motif in all peaks was 1.45 × 10-2 (Additional file 6: Data set 4) compared with the 2.64 × 10-4 frequency (50-fold difference) expected by chance using the 33.2% GC content in the tomato genome [27].
The impact of ASR1 binding on target gene expression
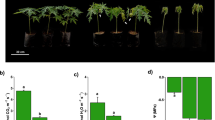
To determine whether the regulation of the obtained target genes is ASR1-dependent, we compared the expression of two representative target genes in the leaves of Asr1-silenced transgenic plants (two different lines) after a 6-hr water stress treatment (as a 3-hr period was not sufficient to yield significant changes in expression, data not shown). Silenced plants showed a marked decrease (p < 0.0001) in Asr1 mRNA in comparison with WT plants (Figure 5A). When normalizing against the house-keeping gene Ubi3, the mRNA levels of gene 10–820 (an aquaporin gene, complete name: Solyc10g054820.1) from both transgenic lines were significantly lower (p < 0.01) in comparison with WT plants (Figure 5B). On the other hand, the expression levels of gene 3–200 (a glucan endo-1 3-beta-glucosidase gene, complete name: Solyc03g115200.2) were decreased only in one Asr1-silenced line (Figure 5B). Similar results were obtained when mRNA levels were normalized to EF-1, a house-keeping gene (Additional file 1: Figure S3 A and B).

Stress-driven changes in the expression of identified target genes are ASR1-dependent. A) Expression levels of Asr1 in WT and two different transgenic ASR1-silenced lines (L-5 and L-17). B) Expression levels of 3–200 (Solyc03g115200.2, Glucan endo-1,3-beta-glucosidase-1) and 10–820 (Solyc10g054820.1, Aquaporin) in WT and ASR1-silenced lines. Transcript levels were normalized to the house-keeping gene Ubi3. *p < 0.05, **p < 0.01, ***p < 0.0001 (ANOVA and Bonferroni post-test compared against WT). Two technical replicates and five biological replicates were done.
These results, obtained with representatives of cell wall- and aquaporin-associated genes, indicate that the binding of ASR1 to its target genes is truly productive and results in gene regulation upon the induction of water stress, confirming previous hints of ASR1 being a direct transcriptional activator [12, 13, 28].
The identified consensus motif sequence (Figure 4A) was consistently found three times at the single exon of gene 10–820 and 7 times at the promoter of gene 3–200.
Discussion and conclusions
For our genome-wide analysis, we carefully followed the current guidelines for ChIP-seq [29] and assessed the quality of the antibody and the robustness of our bioinformatics tools to interpret our high throughput DNA sequencing results. Our ChIP-seq data showed 225 peaks with different values of statistical significance. The results of ChIP-seq were also analyzed with Cisgenome [30] and CSAR [24], but these results gave false peaks and were thus discarded. Artifacts can arise because the annotated sequence of the tomato genome [17] is not error-free, particularly regarding the copy number of repetitive elements and ribosomal genes, which may have been underestimated.
ASR1 showed a binding preference (70%) for gene regions, which is expected for a TF. Considering that tomato genes (including their introns) represent only approximately 13% of the whole genome (even considering non-protein-coding RNA genes) [31], this proportion is even more striking. When we performed a more in-depth analysis of the location of the immuno-enriched sequences that fell into the “gene region” category in the genome, we observed that the majority of them were either upstream of the genes or in the body of the genes, rather than 3’downstream of them. This was not surprising as it is widely accepted that regulatory regions can be found not only at 5’ upstream regions and sizeable distances from the coding region, but also within protein-coding exons as is the case of enhancers of zebra fish developmental genes [32].
A comprehensive analysis of the target genes was made difficult by the diversity of functional categories (at the biochemical or cellular level) encountered. For this reason, we explored over-represented genes whose molecular or cellular functions seemed to be more influenced by ASR1 a priori, for example, those encoding cell wall proteins and aquaporins (AQP), which are related to the physiological response to limited water accessibility [33].
Aquaporin genes belong to the Major Intrinsic Protein (MIP) gene family. In plants, the number of AQP genes present in a single species is rather high, with more than 30 AQP genes frequently found in each genome [34]. Plant AQPs are classified into seven different subfamilies according to sequence similarity [35–37]. Studies of AQP expression under drought stress have focused mainly on the PIP (Plasma Membrane Intrinsic Proteins) subfamily and have yielded opposite or conservative results, depending on the isoform analyzed [38–40]. Some reports show that TIPs (tonoplast intrinsic proteins) and XIPs (X intrinsic proteins) subfamilies also modify their expression under abiotic stress [41–43]. The expression of TIPs has been observed in stomatal guard cells and is in agreement with their known role in drought stress [44]. One of the AQP genes we found as target of ASR1 is an ortholog of AtPIP1;4 (NP_567178.1), frequently expressed in leaves and flowers. Consistent with our results, this Arabidopsis gene has been reported to be up-regulated during drought stress [39]. Moreover, transcript levels of the tomato aquaporin gene Solyc10g054820.1 were found lower in ASR1-silenced plants compared to WT ones, thus validating the results of ASR1 binding at a functional level. In agreement with an expected delay between TF binding and effective transcription due to chromatin remodeling [45], such differences in expression were not detected at the 3 hours of stress chosen for the ChIP experiments but after 6 hours instead.
The plant cell wall provides structural support during development and represents the first line of defense against biotic and abiotic stressors, including drought. In recent years, evidence has accumulated for a dedicated maintenance mechanism for plant cell integrity under diverse biotic and abiotic stress; however, the underlying mechanism remains to be elucidated. Thus, the discovery of cell wall-related genes as targets of ASR1 makes sense in light of the complex network of polymers essential for maintaining turgor pressure. Water loss in this matrix results in a severe disruption of cell wall integrity, which can be irreversible [33]. In addition, because turgor pressure is fundamental to plant cell growth [46], it is conceivable that tissues in active growth (i.e., apical parts) make their walls more extensible, while other tissues (i.e., cell walls) harden, allowing for continuous growth under low water potentials [47]. It is interesting that the “cell wall” gene group we identified includes genes involved in cellulose synthesis, cell wall breakdown and remodeling.
In particular, we found that ASR1 binds directly to several cell wall-related genes. One such gene is Solyc08g082650.2.1, which is annotated as a cellulose synthase-like (CSL) protein. The three closest genes related to Solyc08g082650.2.1 in Arabidopsis thaliana are the paralogous cellulose synthase-like G (CSLG) proteins 2 (AtCSLG2, At4g24000) and 3 (AtCSLG3, At4g23990), which contain a relatively high degree of identity (48-50%) in the region between aa 26–205 (p range = 5 - 6e-43). By searching the GENEVESTIGATOR platform (http://www.genevestigator.com), we found that AtCSLG2/G3 appear to be active during leaf senescence and that osmotic or water stress stimulates their expression up to 50-fold.
Another cell wall-associated gene that emerged as a direct target of ASR1 is Solyc03g115200.2.1, which is annotated as Glucan endo-1,3-β-glucosidase 1. The closest ortholog in Arabidopsis thaliana is the plasmodesmata callose-binding protein 3 (AtPDCB3) gene, with a high degree of identity (52%; p = 1.0e-43). AtPDCB3 exhibits specific callose-binding activity in vitro and localizes to plasmodesmata [48]. Deposition of callose (a β-1,3-glucan) at plasmodesmata is known to be stimulated by physical and physiological stresses [49, 50]. Interestingly, the GENEVESTIGATOR database reveals that the expression of AtPDCB3 is highest in seeds during desiccation and stratification, reinforcing the concept of a conserved response of this kind of proteins to restricted water availability in both Arabidopsis and tomato.
Another work seeking targets of tomato ASR1 was able to identify ABI4, a gene involved in seed germination [14]. However, this study suffers from the fact that it has been conducted in transgenic Arabidopsis, with a smaller genome, thus missing many possible target genes.
Our results also revealed a consensus in vivo ASR-binding DNA motif with little variation: (A/T)(A/G)GCCCA, almost identical to the one very recently found for ASR5 in rice subjected to Aluminium stress [51] and to the one described for the TF AtTCP20 [52] (Table 4), whose targets are cytochrome genes in Arabidopsis[53]. It is thus tempting to speculate that AtTCP20 may function as the counterpart to ASR1 in Arabidopsis, which would be consistent with the known connection between adaptation to abiotic stress and oxidative respiration through mitochondrial electron transport in plants [54]. Interestingly, we also identified four target paralogous genes of ASR1 encoding Cytochrome P450. A related Cytochrome P450 functioning as an ABA-8’-hydroxylase is known to inactivate ABA [55], a paradigmatic plant hormone involved in the response to drought stress. Because ABA induces ASR1 expression and the mentioned enzyme catalyzes the first step in the oxidative degradation of ABA, ASR1 might regulate ABA endogenous levels in a feedback fashion under water stress situations.
In addition, the motif we found shows partial coincidence with the one obtained by SELEX [57]. It is noteworthy that while ChIP captures sequences in vivo, SELEX is carried out in vitro with ADN devoid of histones, an artificial condition. On the other hand, our ChIP data shows no enrichment of promoters/enhancers of genes orthologous to grape VvHT1, a sugar transporter gene reported to be a target of ASR [58]. This result is consistent with the absence of the consensus motif in VvHT1 and its presence—repeatedly up to eight times—in genomic regions highly enriched by ChIP. Here, it is important to note that [13] used a heterologous system (yeast) to test a plant protein-DNA interaction and that neither the grape ASR protein nor the VvHT1 gene are identical to their counterparts in tomato.
At this point, it is worth mentioning the scope of ChIP in general. This procedure, due to its crosslinking step, is also able to detect proteins indirectly binding to DNA, for example by forming a DNA-interacting complex. Nevertheless, even in this second scenario, the information gained by ChIP is useful. The way to discriminate between direct and indirect binding is to perform in vitro assays with the purified TF, in this case ASR1, at the expense of losing the in vivo context (native chromatin), which has already been performed [14, 57].
Conclusions
In summary, we have uncovered a novel repertoire of target genes of the TF ASR1, some of which are clearly involved in the response and physiological adaptation of plants to water stress. These findings will hopefully enable us to gain additional insight into both the early environmental stress-sensing molecular events triggered by ABA and the late physiological adjustments that finally confer tolerance to drought.
Methods
WT plants
Commercial tomato (Solanum lycopersicum) seeds were germinated on blotting paper for 7 days and then transferred to pots containing soil mix in a growth chamber under a photoperiod of 16 hr light/8 hr darkness at 26°C. Plants were used 4 weeks later.
Stress conditions
Soil was carefully removed from the roots with the aid of a small amount of water to minimize damage. Plants were then stressed by being placed onto blotting paper under an incandescent lamp for 3 hr. Some plants were re-watered to confirm reversibility of stress and healthy recovery. Young leaves were then cut and immediately frozen in liquid nitrogen for subsequent purification of nuclei as the starting material for ChIP (see below). For expression analysis, the procedure was the same except that we applied a 6-hr water stress treatment, as a 3-hr period was insufficient to yield significant changes in expression.
Asr1-silenced transgenic plants
Tomato (Solanum lycopersicum) L. cv Moneymaker seeds were obtained from Meyer Beck (Berlin), and the plants were handled as previously described [59]. The 348-bp coding region of the tomato Asr1 gene (GenBank U86130.1) was cloned in antisense orientation into the multiple cloning site of the pBINAR vector [60] between the Cauliflower mosaic virus 35S promoter and the octopine synthase terminator. The construct was delivered by Agrobacterium tumefaciens into tomato cotyledons. Emerging shoots were excised and selected on Murashige and Skoog media containing kanamycin (100 mg/l). When the plants developed roots, they were transferred to soil in the greenhouse for subsequent selection. The initial screening for the 39 lines was based on a diminished expression displayed in Northern blots. For the expression assays, we used five WT tomato plants and 10 Asr1-silenced plants (five from line 5 and five from line 17).
ChIP-sequencing (ChIP-seq)
This strategy combines chromatin immunoprecipitation (ChIP) with high throughput DNA sequencing to identify the binding sites of nuclear proteins connected non-covalently to DNA. A key success factor is the generation of a high-quality antibody against the purified protein of interest. In this work, ChIP-seq was chosen to identify the targets of ASR1 starting out with the purification of ASR1, against which antibodies were produced.
Expression of recombinant ASR1 in Escherichia coliBL21
Expression of recombinant ASR1 was achieved using a plasmid (PRSET B vector, Invitrogen) that contains the tomato Asr1 cDNA driven by the prokaryote promoter sequence recognized by the RNA polymerase of bacteriophage T7 (construct T7-ASR1). The recombinant plasmid was introduced into the Escherichia coli BL21 strain, which has a lac-driven T7 RNA polymerase gene. For testing induction, low-scale cultures were started and Asr1 was indirectly induced by IPTG at different times. As a negative control, a culture of plasmid-free E. coli BL21 was used. Crude protein extracts were run in 15% PAGE, transferred to a nitrocellulose membrane and stained with Coomassie Blue.
Purification of ASR1 by affinity chromatography
Cultures induced by IPTG for 2 hr were lysed through sonication, and recombinant ASR1 protein was purified with a pre-packed Ni2+column (HisTrap Kit, Pharmacia Biotech) using its natural histidine-rich tract as a tag. The ASR1 protein appeared to be pure, with no detectable contaminant proteins, after elution through a non-linear continuous gradient of imidazol (Additional file 1: Figure S1).
Antibody raised against pure ASR1
Purified ASR1 was concentrated through 10-kDa cutoff spin columns (Vivaspin, GE Healthcare) and inoculated into rabbits. Bleedings were performed for titrations of antisera.
Purification of total immunoglobulins from rabbit antisera
Total immunoglobulins from antisera were precipitated with ammonium sulfate, dialyzed and purified with a “Hi-trap protein G” column (Pharmacia Biotech). Their presence was monitored through absorbance at 280 nm. Immunoglobulin-rich fractions were collected and concentrated by Vivaspin columns (GE Healthcare).
Purification and checking of the anti-ASR1 antibody
The purified antibodies were diluted in PBS to conduct an affinity chromatography protocol using ASR1 covalently bound to a cyanogen-activated Sepharose solid matrix (C9210 beads, Sigma). Dot blots and subsequent Ponceau Red staining were performed to visualize the eluted fractions that contained the protein material (Additional file 1: Figure 2)—the pure and specific antibody that was later tested by immunoprecipitation (see two paragraphs below).
ChIP protocol
The ChIP protocol was performed as described in [61] from nuclei of water stressed tomato leaves. For DNA fragmentation, we used a Biorruptor UCD-200 TM machine (Diagenode, Denville, NJ, USA) (30 cycles at max power, 30 sec ON/30 sec OFF).
Immunoprecipitation and Western blot
To test the quality (the specificity and precipitation capacity towards ASR1) of the antibody, we first performed a non-crosslinking ChIP procedure using fruit as the starting tissue. The protocol was stopped at the crosslinking reversal step. Precipitated proteins were eluted with 0.1 M glycine at a pH of 2.4. The sample was then transferred into a tube containing enough 1 M Tris-base to neutralize the low pH and loaded onto an SDS-PAGE gel for subsequent Western blot to detect the immunoprecipitated ASR1. Because the same primary anti-ASR1 antibody was used for both IP and Western blot, for the latter we used a secondary anti-native rabbit immunoglobulin TrueBlot antibody (Rockland, USA) to avoid the detection of immunoglobulins that had been run (denatured) on the gel and whose quality was tested in this assay.
Construction of the DNA fragment library for deep sequencing
Following post-stress leaf chromatin immunoprecipitation, the DNA was purified using “AMPXP” beads according to the manufacturer’s protocol (Ambion). The DNA was eluted in 10 μl MilliQ water followed by two repeat purifications. The DNA fragments were refilled to get blunt ends using the Klenow fragment of DNA polymerase. Fragmented ends were phosphorylated by T4 kinase and ligated to different double-stranded adaptors by concentrated DNA ligase. The adaptors had 5 nucleotides in their 3′ ends as “bar codes” for sample identification in multiplex runs. The DNA was re-purified with shorter incubation times to favor the loss of adaptor primer dimers shorter than 200 bp that were generated during ligation. PCR reactions were performed using 0.5 μl of dimer-free adaptor samples, adaptor-specific primers and high-fidelity thermostable DNA polymerase (Phusion Hot Start II, Thermo Scientific) with low cycle numbers (between 12 and 18) until a product concentration of 5 ng/μl was reached. Cycling conditions were as follows: 1 starting denaturation cycle of 30 sec at 98°C, 12–18 cycles of 10 sec at 98°C (denaturing), 30 sec at 65°C (annealing), 30 sec at 72°C (elongation) and a final elongation step for 5 min at 72°C. Amplified products with each bar code were combined and re-purified with beads and resuspended. The concentration of each sample for sequencing was 2 ng/μl.
Deep sequencing proper
DNA samples from the library were sequenced on a HiSeq Illumina® machine (Illumina, San Diego, CA, USA) at BTI, Cornell University campus, Ithaca, NY, USA).
Data access
The raw sequencing data have been uploaded at the following NCBI website, publicly available: http://www.ncbi.nlm.nih.gov/sra/?term=SRX257002.
Bioinformatics tools
The reads were processed into separate sequences according to bar codes and trimmed by using the fastx toolbox software (http://cancan.cshl.edu/labmembers/gordon/fastx_toolkit/). Sequences were aligned with those from the tomato genome (version SL2.40) available on the webpage of the Genome Project [17] (http://solgenomics.net/organism/Solanum_lycopersicum/genome) using the software program Bowtie [62]. Reads that produced two mismatches when aligned were discarded, as were reads that aligned with more than one position in the genome. The reads were sorted and indexed with the aid of SAMtools (Sequence Alignment Map format) at the Boyce Thompson Institute for Plant Research. Finally, the number of sequences that fell within each 150-bp window was counted and visualized with IGV [22]. For peak calling, data were analyzed using the Macs program [63] (parameters listed in Additional file 1: List 1), which searches for peaks (regions that yielded a high number of reads) present in the sample but not in the control (input sample). We also used the CSAR [24] and cisgenome [23] programs, which use the statistical programming language R [64]. The consensus motif was determined by the Gimmemotif program [26]. Over-represented gene categories were resolved with the Mapman program [25] (parameters listed in Additional file 1: List 1). The ASR1-binding sequence was compared to four different databases using the STAMP program [56].
The in-house software programs “averageDistribution”, “consensusCounter” and “readingExtension” were created to make our software freely available. The source code was uploaded as Additional file 3: (in the zip folder “TomatoProgramCode”). For each program, there is a folder including a readme.txt file containing a brief explanation about what it does and how to execute the program, as well as a description of the contents of each file.
We used the STAMP program [56] to search databases for additional TFs with similar DNA-binding motifs as the one recognized by ASR1.
Individual real time PCRs for the validation of deep-sequencing results
The reactions were carried out with purified immunoprecipitated DNA and Recombinant Taq DNA polymerase (Invitrogen) in a final volume of 25 μl. A DNA Engine Opticon (MJ Research Inc.) thermocycler was used with annealing temperatures set to achieve 90% amplification efficiency (between 59°C and 64°C, depending on the primers). Denaturation curves were calculated and the amplified DNA was run through an agarose gel to ensure the existence of a single product. The conditions were as follows: 1 cycle of 5 min at 94°C (initial denaturation), 35 cycles of 30 sec at 94°C (denaturation), 30 sec at 59/64°C (annealing) and 30 sec at 72°C (elongation).
Expression analysis of target genes
For RNA extraction, we used the TriReagent kit (MRC Inc.) with 300 mg of previously mortar-ground leaves as a starting material and 1.5 ml of TriReagent solution according to the manufacturer’s protocol. All RNA samples were quantified using a Nanodrop 2000 (Thermo Scientific) spectrophotometer. To eliminate contamination of the samples by residual DNA, 10 μg of each RNA sample were treated with 12.5 U DNAseI (Invitrogen). Reverse transcription was achieved using 2 μl of DNAseI-treated RNA, 50 U MMLV-RT (Promega) and oligo-dT (50 pmoles) in a 25 μl final volume for 1 hr at 42°C. To prevent RNA degradation, 10 U of RNAseOUT (Invitrogen) was added. Following reverse transcription, qPCR was carried out using 5 μl of a 1/10 dilution of the cDNA samples (obtained as described above) per PCR reaction. Reactions were performed in a DNA Engine Opticon (MJ Research Inc.) thermocycler. We used 0.625 U of Taq DNA Polymerase (Invitrogen), 3 mM magnesium chloride, 2 mM of dNTPs mixture (Fermentas) and 0.2 μM of each primer (IDT Inc.) in a final volume of 25 μl. We used Sybr Green® (Roche) as the fluorophore. Reactions were conducted under the following cycling conditions: 2 min of denaturation at 94°C, 40 cycles of 30 sec of denaturation at 94°C, 30 sec of annealing and 30 sec of elongation at 72°C. A melting curve was generated between 65 and 95°C with readings at every 0.5°C. For Asr1, Ubi3 and EF-1 quantitation we used 67°C as the annealing temperature. For the Solyc03g115200.2 gene (3–200), we used 58.6°C and for the Solyc10g054820.1 gene (10–820), we used 62.4°C. qRT-PCR was validated by a standard and melting curve. Primers are listed in Additional file 1: Table S1. For each amplicon, a standard curve was made. Quantitation of DNA in each sample was extrapolated from its respective standard curve. Values were then relativized to that of a housekeeping gene calculated in the same way [65]. Levels from Asr1, Solyc03g115200.2 (3–200) and Solyc10g054820.1 (10–820) were normalized to Ubi3 or EF-1 levels. Statistical analysis was performed with the GraphPad software program using a one-way ANOVA statistical test with a 95% confidence level.
Description of additional data files
The Data set 1 contains a table with the list of peaks found with the Macs software. Data set 2 shows a list of all the genes with peaks near them. Data set 3 shows a list of the gene categories, the over-represented functions and the over-represented groups obtained after running Mapman software. Data set 4 shows the count of reads for ASR1-binding sequences along all the peaks found by the Macs software. The Additional file 1: (pdf) contains 3 figures, 2 tables and 1 parameter list. Figures 1 and 2 show the results of ASR1 protein purification and anti-ASR1 antibody affinity chromatography. Figure 3 shows qRT-PCR experiments using a second housekeeping gene to express the relative amounts of mRNAs. Tables 1 and 2 list primers used for ChIP-qPCR and qRT-PCR, respectively. Finally, the Additional file 1: List 1 shows the parameters used for Macs and Gimmemotif softwares.
The scripts and in-house softwares developed for this paper are included in the Additional file 3: “tomatoProgramCode.zip” separate file.
Abbreviations
- MYA:
-
Million years ago
- LEA:
-
Late embryogenesis abundant
- ABA:
-
Abscisic acid
- ASR:
-
ABA/ Stress/Ripening (lower case: gene
- upper case:
-
protein)
- ChIP:
-
Chromatin immunoprecipitation
- DNA:
-
Deoxyribonucleic acid
- cDNA:
-
Complementary DNA
- TF:
-
Transcription factor
- ROC:
-
Receiver operating characteristic
- MIP:
-
Major intrinsic proteins
- AQP:
-
Aquaporins
- WT:
-
Wild type.
References
Cominelli E, Conti L, Tonelli C, Galbiati M: Challenges and perspectives to improve crop drought and salinity tolerance. N Biotechnol. 2013, 30 (4): 355-361. 10.1016/j.nbt.2012.11.001.
Qiu YL, Palmer JD: Phylogeny of early land plants: insights from genes and genomes. Trends Plant Sci. 1999, 4 (1): 26-30. 10.1016/S1360-1385(98)01361-2.
Rensing SA, Lang D, Zimmer AD, Terry A, Salamov A, Shapiro H, Nishiyama T, Perroud PF, Lindquist EA, Kamisugi Y, et al: The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science. 2008, 319 (5859): 64-69. 10.1126/science.1150646.
Dure L, Greenway SC, Galau GA: Developmental biochemistry of cottonseed embryogenesis and germination: changing messenger ribonucleic acid populations as shown by in vitro and in vivo protein synthesis. Biochemistry. 1981, 20 (14): 4162-4168. 10.1021/bi00517a033.
Tunnacliffe A, Wise MJ: The continuing conundrum of the LEA proteins. Naturwissenschaften. 2007, 94 (10): 791-812. 10.1007/s00114-007-0254-y.
Caramelo JJ, Iusem ND: When cells lose water: Lessons from biophysics and molecular biology. Prog Biophys Mol Biol. 2009, 99 (1): 1-6. 10.1016/j.pbiomolbio.2008.10.001.
Battaglia M, Olvera-Carrillo Y, Garciarrubio A, Campos F, Covarrubias AA: The enigmatic LEA proteins and other hydrophilins. Plant Physiol. 2008, 148 (1): 6-24. 10.1104/pp.108.120725.
Frankel N, Hasson E, Iusem ND, Rossi MS: Adaptive evolution of the water stress-induced gene Asr2 in Lycopersicon species dwelling in arid habitats. Mol Biol Evol. 2003, 20 (12): 1955-1962. 10.1093/molbev/msg214.
Giombini MI, Frankel N, Iusem ND, Hasson E: Nucleotide polymorphism in the drought responsive gene Asr2 in wild populations of tomato. Genetica. 2009, 136 (1): 13-25. 10.1007/s10709-008-9295-1.
Iusem ND, Bartholomew DM, Hitz WD, Scolnik PA: Tomato (Lycopersicon esculentum) transcript induced by water deficit and ripening. Plant Physiol. 1993, 102 (4): 1353-1354. 10.1104/pp.102.4.1353.
Konrad Z, Bar-Zvi D: Synergism between the chaperone-like activity of the stress regulated ASR1 protein and the osmolyte glycine-betaine. Planta. 2008, 227 (6): 1213-1219. 10.1007/s00425-008-0693-5.
Maskin L, Frankel N, Gudesblat G, Demergasso MJ, Pietrasanta LI, Iusem ND: Dimerization and DNA-binding of ASR1, a small hydrophilic protein abundant in plant tissues suffering from water loss. Biochem Biophys Res Commun. 2007, 352 (4): 831-835. 10.1016/j.bbrc.2006.11.115.
Cakir B, Agasse A, Gaillard C, Saumonneau A, Delrot S, Atanassova R: A grape ASR protein involved in sugar and abscisic acid signaling. Plant Cell. 2003, 15 (9): 2165-2180. 10.1105/tpc.013854.
Shkolnik D, Bar-Zvi D: Tomato ASR1 abrogates the response to abscisic acid and glucose in Arabidopsis by competing with ABI4 for DNA binding. Plant Biotechnol J. 2008, 6 (4): 368-378. 10.1111/j.1467-7652.2008.00328.x.
Seki M, Narusaka M, Ishida J, Nanjo T, Fujita M, Oono Y, Kamiya A, Nakajima M, Enju A, Sakurai T, et al: Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 2002, 31 (3): 279-292. 10.1046/j.1365-313X.2002.01359.x.
Gazanchian A, Hajheidari M, Sima NK, Salekdeh GH: Proteome response of Elymus elongatum to severe water stress and recovery. J Exp Bot. 2007, 58 (2): 291-300.
The-Tomato-Genome-Consortium: The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012, 485 (7400): 635-641. 10.1038/nature11119.
Infante CR, Park S, Mihala AG, Kingsley DM, Menke DB: Pitx1 broadly associates with limb enhancers and is enriched on hindlimb cis-regulatory elements. Dev Biol. 2013, 374 (1): 234-244. 10.1016/j.ydbio.2012.11.017.
Zhang JX, Zhang J, Yan W, Wang YY, Han L, Yue X, Liu N, You YP, Jiang T, Pu PY, et al: Unique genome-wide map of TCF4 and STAT3 targets using ChIP-seq reveals their association with new molecular subtypes of glioblastoma. Neuro Oncol. 2013, 15 (3): 279-289. 10.1093/neuonc/nos306.
Muino JM, Angenent GC, Kaufmann K: Visualizing and characterizing in vivo DNA-binding events and direct target genes of plant transcription factors. Methods Mol Biol. 2011, 754: 293-305. 10.1007/978-1-61779-154-3_17.
Feng J, Liu T, Qin B, Zhang Y, Liu XS: Identifying ChIP-seq enrichment using MACS. Nat Protoc. 2012, 7 (9): 1728-1740. 10.1038/nprot.2012.101.
Thorvaldsdottir H, Robinson JT, Mesirov JP: Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14 (2): 178-192. 10.1093/bib/bbs017.
Ji H, Jiang H, Ma W, Wong WH, et al: Using CisGenome to Analyze ChIP-Chip and ChIP-seq Data. Current Protocols in Bioinformatics. Edited by: Baxevanis AD. 2011, Hoboken, New Jersey, USA: Wiley Online Library, Unit 2.13, 2
Muino JM, Kaufmann K, van Ham RC, Angenent GC, Krajewski P: ChIP-seq Analysis in R (CSAR): an R package for the statistical detection of protein-bound genomic regions. Plant Methods. 2011, 7: 11-10.1186/1746-4811-7-11.
Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kruger P, Selbig J, Muller LA, Rhee SY, Stitt M: MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37 (6): 914-939. 10.1111/j.1365-313X.2004.02016.x.
van Heeringen SJ, Veenstra GJ: GimmeMotifs: a de novo motif prediction pipeline for ChIP-sequencing experiments. Bioinformatics. 2011, 27 (2): 270-271. 10.1093/bioinformatics/btq636.
Zhu W, Ouyang S, Iovene M, O’Brien K, Vuong H, Jiang J, Buell CR: Analysis of 90 Mb of the potato genome reveals conservation of gene structures and order with tomato but divergence in repetitive sequence composition. BMC Genomics. 2008, 9 (1): 286-10.1186/1471-2164-9-286.
Frankel N, Nunes-Nesi A, Balbo I, Mazuch J, Centeno D, Iusem ND, Fernie AR, Carrari F: ci21A/Asr1 expression influences glucose accumulation in potato tubers. Plant Mol Biol. 2007, 63 (5): 719-730. 10.1007/s11103-006-9120-0.
Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al: ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2011, 22 (9): 1813-1831.
Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH: An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol. 2008, 26 (11): 1293-1300. 10.1038/nbt.1505.
Aoki K, Yano K, Suzuki A, Kawamura S, Sakurai N, Suda K, Kurabayashi A, Suzuki T, Tsugane T, Watanabe M, et al: Large-scale analysis of full-length cDNAs from the tomato (Solanum lycopersicum) cultivar Micro-Tom, a reference system for the Solanaceae genomics. BMC Genomics. 2010, 11: 210-10.1186/1471-2164-11-210.
Ritter DI, Dong Z, Guo S, Chuang JH: Transcriptional enhancers in protein-coding exons of vertebrate developmental genes. PLoS One. 2012, 7 (5): e35202-10.1371/journal.pone.0035202.
Moore JP, Vicre-Gibouin M, Farrant JM, Driouich A: Adaptations of higher plant cell walls to water loss: drought vs desiccation. Physiol Plant. 2008, 134 (2): 237-245. 10.1111/j.1399-3054.2008.01134.x.
Maurel C, Verdoucq L, Luu DT, Santoni V: Plant aquaporins: membrane channels with multiple integrated functions. Annu Rev Plant Biol. 2008, 59: 595-624. 10.1146/annurev.arplant.59.032607.092734.
Chaumont F, Barrieu F, Wojcik E, Chrispeels MJ, Jung R: Aquaporins constitute a large and highly divergent protein family in maize. Plant Physiol. 2001, 125 (3): 1206-1215. 10.1104/pp.125.3.1206.
Johanson U, Karlsson M, Johansson I, Gustavsson S, Sjovall S, Fraysse L, Weig AR, Kjellbom P: The complete set of genes encoding major intrinsic proteins in Arabidopsis provides a framework for a new nomenclature for major intrinsic proteins in plants. Plant Physiol. 2001, 126 (4): 1358-1369. 10.1104/pp.126.4.1358.
Danielson JA, Johanson U: Unexpected complexity of the aquaporin gene family in the moss Physcomitrella patens. BMC Plant Biol. 2008, 8: 45-10.1186/1471-2229-8-45.
Jang JY, Kim DG, Kim YO, Kim JS, Kang H: An expression analysis of a gene family encoding plasma membrane aquaporins in response to abiotic stresses in Arabidopsis thaliana. Plant Mol Biol. 2004, 54 (5): 713-725.
Alexandersson E, Fraysse L, Sjovall-Larsen S, Gustavsson S, Fellert M, Karlsson M, Johanson U, Kjellbom P: Whole gene family expression and drought stress regulation of aquaporins. Plant Mol Biol. 2005, 59 (3): 469-484. 10.1007/s11103-005-0352-1.
Lian HL, Yu X, Lane D, Sun WN, Tang ZC, Su WA: Upland rice and lowland rice exhibited different PIP expression under water deficit and ABA treatment. Cell Res. 2006, 16 (7): 651-660. 10.1038/sj.cr.7310068.
Secchi F, Lovisolo C, Uehlein N, Kaldenhoff R, Schubert A: Isolation and functional characterization of three aquaporins from olive (Olea europaea L.). Planta. 2007, 225 (2): 381-392.
Li GW, Peng YH, Yu X, Zhang MH, Cai WM, Sun WN, Su WA: Transport functions and expression analysis of vacuolar membrane aquaporins in response to various stresses in rice. J Plant Physiol. 2008, 165 (18): 1879-1888. 10.1016/j.jplph.2008.05.002.
Lopez D, Bronner G, Brunel N, Auguin D, Bourgerie S, Brignolas F, Carpin S, Tournaire-Roux C, Maurel C, Fumanal B, et al: Insights into Populus XIP aquaporins: evolutionary expansion, protein functionality, and environmental regulation. J Exp Bot. 2012, 63 (5): 2217-2230. 10.1093/jxb/err404.
Sarda X, Tousch D, Ferrare K, Legrand E, Dupuis JM, Casse-Delbart F, Lamaze T: Two TIP-like genes encoding aquaporins are expressed in sunflower guard cells. Plant J. 1997, 12 (5): 1103-1111. 10.1046/j.1365-313X.1997.12051103.x.
Salma N, Xiao H, Imbalzano AN: Temporal recruitment of CCAAT/enhancer-binding proteins to early and late adipogenic promoters in vivo. J Mol Endocrinol. 2006, 36 (1): 139-151. 10.1677/jme.1.01918.
Geitmann A, Ortega JK: Mechanics and modeling of plant cell growth. Trends Plant Sci. 2009, 14 (9): 467-478. 10.1016/j.tplants.2009.07.006.
Wu Y, Cosgrove DJ: Adaptation of roots to low water potentials by changes in cell wall extensibility and cell wall proteins. J Exp Bot. 2000, 51 (350): 1543-1553. 10.1093/jexbot/51.350.1543.
Levy A, Erlanger M, Rosenthal M, Epel BL: A plasmodesmata-associated beta-1,3-glucanase in Arabidopsis. Plant J. 2007, 49 (4): 669-682. 10.1111/j.1365-313X.2006.02986.x.
Ueki S, Citovsky V: Identification of an interactor of cadmium ion-induced glycine-rich protein involved in regulation of callose levels in plant vasculature. Proc Natl Acad Sci USA. 2005, 102 (34): 12089-12094. 10.1073/pnas.0505927102.
Maeda H, Song W, Sage TL, DellaPenna D: Tocopherols play a crucial role in low-temperature adaptation and Phloem loading in Arabidopsis. Plant Cell. 2006, 18 (10): 2710-2732. 10.1105/tpc.105.039404.
Arenhart RA, Bai Y, de Oliveira LF V, Bucker Neto L, Schunemann M, Maraschin FD, Mariath J, Silverio A, Sachetto-Martins G, Margis R, et al: New insights into aluminum tolerance in rice: the ASR5 protein binds the STAR1 promoter and other aluminum-responsive genes. Mol Plant. 2013
Herve C, Dabos P, Bardet C, Jauneau A, Auriac MC, Ramboer A, Lacout F, Tremousaygue D: In vivo interference with AtTCP20 function induces severe plant growth alterations and deregulates the expression of many genes important for development. Plant Physiol. 2009, 149 (3): 1462-1477. 10.1104/pp.108.126136.
Welchen E, Gonzalez DH: Differential expression of the Arabidopsis cytochrome c genes Cytc-1 and Cytc-2. Evidence for the involvement of TCP-domain protein-binding elements in anther- and meristem-specific expression of the Cytc-1 gene. Plant Physiol. 2005, 139 (1): 88-100. 10.1104/pp.105.065920.
Zsigmond L, Rigo G, Szarka A, Szekely G, Otvos K, Darula Z, Medzihradszky KF, Koncz C, Koncz Z, Szabados L: Arabidopsis PPR40 connects abiotic stress responses to mitochondrial electron transport. Plant Physiol. 2008, 146 (4): 1721-1737. 10.1104/pp.107.111260.
Krochko JE, Abrams GD, Loewen MK, Abrams SR, Cutler AJ: (+)-Abscisic acid 8’-hydroxylase is a cytochrome P450 monooxygenase. Plant Physiol. 1998, 118 (3): 849-860. 10.1104/pp.118.3.849.
Mahony S, Benos PV: STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 2007, 35: W253-258. 10.1093/nar/gkm272.
Kalifa Y, Gilad A, Konrad Z, Zaccai M, Scolnik PA, Bar-Zvi D: The water- and salt-stress-regulated Asr1 (abscisic acid stress ripening) gene encodes a zinc-dependent DNA-binding protein. Biochem J. 2004, 381 (Pt 2): 373-378.
Vignault C, Vachaud M, Cakir B, Glissant D, Dedaldechamp F, Buttner M, Atanassova R, Fleurat-Lessard P, Lemoine R, Delrot S: VvHT1 encodes a monosaccharide transporter expressed in the conducting complex of the grape berry phloem. J Exp Bot. 2005, 56 (415): 1409-1418. 10.1093/jxb/eri142.
Carrari F, Nunes-Nesi A, Gibon Y, Lytovchenko A, Loureiro ME, Fernie AR: Reduced expression of aconitase results in an enhanced rate of photosynthesis and marked shifts in carbon partitioning in illuminated leaves of wild species tomato. Plant Physiol. 2003, 133 (3): 1322-1335. 10.1104/pp.103.026716.
Liu XJ, Prat S, Willmitzer L, Frommer WB: cis regulatory elements directing tuber-specific and sucrose-inducible expression of a chimeric class I patatin promoter/GUS-gene fusion. Mol Gen Genet. 1990, 223 (3): 401-406.
Ricardi M, Gonzalez R, Iusem N: Protocol: fine-tuning of a Chromatin Immunoprecipitation (ChIP) protocol in tomato. Plant Methods. 2010, 6 (1): 11-10.1186/1746-4811-6-11.
Langmead B, Trapnell C, Pop M, Salzberg SL: Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10 (3): R25-10.1186/gb-2009-10-3-r25.
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al: Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9 (9): R137-10.1186/gb-2008-9-9-r137.
R Development Core Team: R: A Language and Environment for Statistical Computing. 2011, Vienna, Austria: the R Foundation for Statistical Computing, Available online at http://www.R-project.org/, 3-900051-07-0
Livak KJ: ABI Prism 7700 Sequence Detection System. 1997, PE Applied Biosystems: Foster City, CA, vol. User Bulletin 2
Acknowledgments
The authors are indebted to the following Argentinian institutions: CONICET (Consejo Nacional de Investigaciones Científicas y Tecnológicas) for salaries, fellowships and grants, ANPCyT (Agencia Nacional de Promoción de Ciencia y Tecnología) for grants and UBA (the University of Buenos Aires) for salaries and grants.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
All the authors declare that they have no competing interests.
Authors’ contributions
MMR and RMG (equal contributions) performed all the experimental work on Asr1 purification, antibody making, ChIP for deep sequencing as well as subsequent ChIP-qPCR and qRT-PCR for validation of individual loci and transcripts; JJG and SZ provided the equipment and setups for library making and deep sequencing (SZ made the libraries); TD carried out the initial bioinformatic analyses; PGT developed in-house softwares for in-depth bioinformatic interpretation of the data; FC and PGD generated the ASR1-silenced plants used for functional validation; KA interpreted the results regarding aquaporin genes; JME interpreted the results regarding cell wall-related genes, JDSS made valuable suggestions throughout the work; NDI introduced the theoretical frame, coordinated the project and drafted the manuscript. All authors read and approved the final manuscript.
Martiniano M Ricardi, Rodrigo M González contributed equally to this work.
Electronic supplementary material
12870_2013_1759_MOESM1_ESM.pdf
Additional file 1: Figure S1: Purification of ASR1 by a Ni2+ affinity column chromatography. Figure S2. Purification of the anti-ASR1 antiboby. Figure S3. Testing another housekeeping gene for normalization of transcript levels. Table S1. Primers used for ChlP validation. Table S2. Primers used for expressions analysis (qRT-PCR). List 1. Software parameters used in this work. (PDF 124 KB)
12870_2013_1759_MOESM5_ESM.xlsx
Additional file 5: Data Set 3. List of the gene categories, the over-represented functions and the over-represented groups obtained after running the Mapman software. (XLSX 50 KB)
12870_2013_1759_MOESM6_ESM.xls
Additional file 6: Data set 4. Count of reads for ASR1-binding sequences along all the peaks found by the Macs software. (XLS 43 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Ricardi, M.M., González, R.M., Zhong, S. et al. Genome-wide data (ChIP-seq) enabled identification of cell wall-related and aquaporin genes as targets of tomato ASR1, a drought stress-responsive transcription factor. BMC Plant Biol 14, 29 (2014). https://doi.org/10.1186/1471-2229-14-29
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-14-29