
Abstract
Background
The medicinal plant Artemisia annua is covered with filamentous trichomes and glandular, artemisinin producing trichomes. A high artemisinin supply is needed at a reduced cost for treating malaria. Artemisinin production in bioreactors can be facilitated if a better insight is obtained in the biosynthesis of artemisinin and other metabolites. Therefore, metabolic activities of glandular and filamentous trichomes were investigated at the transcriptome level.
Results
By laser pressure catapulting, glandular and filamentous trichomes as well as apical and sub-apical cells from glandular trichomes were collected and their transcriptome was sequenced using Illumina RNA-Seq. A de novo transcriptome was assembled (Trinity) and studied with a differential expression analysis (edgeR).
A comparison of the transcriptome from glandular and filamentous trichomes shows that MEP, MVA, most terpene and lipid biosynthesis pathways are significantly upregulated in glandular trichomes. Conversely, some transcripts coding for specific sesquiterpenoid and triterpenoid enzymes such as 8-epi-cedrol synthase and an uncharacterized oxidosqualene cyclase were significantly upregulated in filamentous trichomes. All known artemisinin biosynthesis genes are upregulated in glandular trichomes and were detected in both the apical and sub-apical cells of the glandular trichomes. No significant differential expression could be observed between the apical and sub-apical cells.
Conclusions
Our results underscore the vast metabolic capacities of A. annua glandular trichomes but nonetheless point to the existence of specific terpene metabolic pathways in the filamentous trichomes. Candidate genes that might be involved in artemisinin biosynthesis are proposed based on their putative function and their differential expression level.
Similar content being viewed by others


Background
Artemisia annua L. (Sweet Wormwood) is a medicinal plant that produces artemisinin which is a sesquiterpene with anti-malarial properties. Since every year around 216 million people are infected with malaria [1], a high supply of artemisinin is needed at a reduced cost. Artemisinin production is being enhanced in A. annua by crossing high-producing plants [2]. Another strategy to increase production of artemisinin is synthesis of artemisinic-acid in engineered yeast and subsequent (photo)chemical conversion to artemisinin [3, 4]. A better insight in artemisinin biosynthesis could lead to a cheaper production method.
An important breakthrough unravelling artemisinin biosynthesis, was the localization of artemisinin production in the glandular trichomes [5]. Trichomes, named after the Greek word for hair are epidermal outgrowths covering plant organs. In A. annua, two types of trichomes are present: biseriate peltate glandular trichomes (Figure 1A) and filamentous trichomes composed of stalk cells and an elongated cell in a T-shape (Figure 1B) [6]. Duke et al. compared a normal biotype of A. annua with both filamentous and glandular trichomes to a biotype with only filamentous trichomes. Only in the presence of glandular trichomes, artemisinin was detected [5].

Glandular and filamentous trichomes of A. annua . A: Glandular trichome with on top a pair of white apical cells and two pairs of green sub-apical cells surrounded by a sub-cuticular cavity (cells at the basis of the trichome are partially removed); B: Filamentous trichomes (T-shape) with stalk cells and elongated cell; C: Glandular trichomes captured by laser pressure catapulting in the cap of a tube; D: Filamentous trichomes captured by laser pressure catapulting in the cap of a tube.
This information was used to discover candidate genes for artemisinin biosynthesis with an expressed sequence tag (EST) approach [7]. Three EST libraries were constructed: glandular trichome, flower bud and glandular trichome-minus-flower-bud subtracted library. Several genes that were preferentially expressed in glandular trichomes are involved in artemisinin biosynthesis such as CYP71AV1, ALDH1, DBR2 and ADH1[7–10].
Enzymes involved in artemisinin biosynthesis are known up to the formation of dihydroartemisinic acid. It is not yet clear whether the last step(s) from dihydroartemisinic acid to artemisinin involves a spontaneous auto-oxidation or is catalyzed by enzymes. Brown and Sy favour the theory of spontaneous chemistry because they see parallels between in vitro auto-oxidation and intermediates present in vivo[11]. Additionally, plants fed with labelled dihydroartemisinic acid and dried, contained the same proportion of labelled artemisinin as plants that were kept alive [11]. These are the main arguments for chemical conversion. On the other hand, while 70% of label incorporation was detected in the metabolites derived from dihydroartemisinic acid; artemisinin and arteannuin H had only 5-15% label incorporation [11]. Therefore, it cannot be excluded that there is another more important pathway leading to artemisinin which was not accessible by the labelled precursor and that enzymes might be involved to catalyze this process in A. annua.
To find candidate genes that catalyze the last step(s) to artemisinin, a more detailed analysis was needed of the transcriptome of glandular trichomes. Sequencing of the transcriptome from enriched glandular trichome preparations from A. annua was performed by Graham et al. on the Roche 454 platform to identify genes and markers for fast-track breeding [2]. In another study, Wang et al. performed a global transcriptome characterization of glandular trichomes. To confirm the expression of some genes in glandular trichomes a semi-quantitative RT-PCR analysis was performed on filamentous and glandular trichomes. Three genes involved in terpene biosynthesis were tested: amorpha-4,11-diene synthase, a sesquiterpene cyclase and (3R)-linalool synthase. They found that these genes were expressed in glandular trichomes as well as in filamentous trichomes [12] and this raised the question whether filamentous trichomes are involved in the production of secondary metabolites.
Trichomes are in general classified as non-glandular (e.g. filamentous trichomes in A. annua) or glandular, based on their secretory capacity. In plants, glandular trichomes are most often production sites for multiple secondary metabolites which form a first-line defence at the surface of the plant through their capacity to entrap, deter or poison pathogens and herbivores [13]. Because of their interesting metabolite content as illustrated with artemisinin, a large number of studies have been devoted to glandular trichomes.
In contrast to the extensive literature describing glandular trichomes, less attention has been paid to non-glandular trichomes. Non-glandular trichomes are assumed to form a physical barrier by steric hindrance of herbivores [14, 15]. Non-glandular trichomes are mainly described for taxonomic and phylogenetic purposes [16–20] but little is known about their production of secondary metabolites [21].
To detect potential candidate genes for artemisinin biosynthesis and to investigate if filamentous trichomes produce important secondary metabolites, it is interesting to compare filamentous and glandular trichomes. Therefore, we performed a comparative transcriptome analysis of filamentous and glandular trichomes on the Illumina HiSeq platform. Several cytochromes, peroxidases and dioxygenases that are potentially involved in the biosynthesis of artemisinin and/or other terpenes were upregulated in glandular trichomes. Our transcriptome analysis confirms the established metabolic capacities of A. annua glandular trichomes but also points to specific metabolic activities in A. annua filamentous trichomes.
Additionally, two other transcriptome experiments were set up to discover potential candidate genes. First, the effect of jasmonic acid (JA) elicitation on glandular and filamentous trichomes was investigated as e.g. Maes et al. showed that artemisinin production can be stimulated by JA [22].
In another experiment, the transcriptome of two cell types from glandular trichomes was investigated to examine in which cells the production of artemisinin, and/or other secondary metabolites occurs. Glandular trichomes contain morphologically distinct types of secretory cells: white apical cells and green-coloured sub-apical cells. In both cell types, Olofsson et al. detected expression of genes involved in artemisinin biosynthesis [23]. In our data set, artemisinin biosynthesis genes were detected in both cell-types and no significant conclusion could be drawn about differential expression.
Methods
Overview of collected samples
In total 6 sample-types have been collected from capitula (flower heads) of A. annua (Table 1). Three independent repeats of glandular (Figure 1C) and filamentous (Figure 1D) trichomes were collected from the same mock- and JA-treated plants. In addition to this, two repeats of separated apical and sub-apical cells of glandular trichomes were collected from mock-treated plants. The RNA from one of the 2 repeats for each cell type was split in two and separately amplified, creating 3 repeats for sequencing.
Plant preparation
Experiments were executed on Artemisia annua L. Anamed A3 cultivar (http://www.anamed.net). This cultivar contains up to 1,4% artemisinin (dry weight leaves) and is the result of cross breeding high-artemisinin producing plants by Mediplant Inc. (Conthey, Switzerland) [24]. Anamed A3 can grow well in tropical regions and is not as photosensitive as other breeds [25]. Under 8 hours day and 16 hours night, Anamed A3 starts flowering at the earliest after 6 months [see Additional file 1]. The length of this pre-flowering period is in line with observations under field conditions [26].
Seeds from A. annua Anamed were sterilized for 2 min in 70% EtOH (Merck, Darmstadt, Germany) and 10 min in a solution with 3.84 ml NaOCl (10-13% chlorine from Sigma-Aldrich, Steinheim, Germany), 5 μl Tween 20 (MP Biomedicals, Illkirch, France) and 6.16 ml sterile water. Subsequently, seeds were rinsed with sterile water and germinated on moist paper. After 1 or 2 weeks, shoots were transferred to soil and grown under a regime with 8 hours day, 16 hours night and a temperature of 20°C. After 6 months, flowers were appearing. For JA elicitation, plants were treated for 8 days before the start of the sampling procedure by spraying every 2 days with a solution of 100 μM JA (Duchefa, Haarlem, The Netherlands) containing 1.5 mM Tween20 (MP Biomedicals) and adding every 2 days 5 ml of 100 μM JA to the soil. During the sampling procedure, treatment was continued every 2 days. Control groups were treated with a mock (water) solution in a separate room.
Glandular and filamentous sample preparation
On the basal bracts and pedicel of the capitulum, filamentous trichomes are abundantly present [6]. Glandular trichomes are protruding on the corolla of the floret buds from the capitulum but are sunken in the capitulum bracts and in leaves [6]. An image of a flower head from A. annua Anamed was taken with Tabletop SEM (TM-1000, Hitachi, Tokyo, Japan) [see Additional file 2]. Sunken trichomes are difficult to collect with laser capture microdissection and therefore, capitula were used to collect glandular and filamentous trichomes. Trichomes were collected from mock and JA-treated plants in 3 independent biological repeats. For each repeat, trichomes were collected from a pool of 3 plants. The same plants were used for capturing 190 glandular and 670 filamentous trichomes.
On a RNA free microscope slide, a capitulum was cut in a drop of cold buffer with 25 mM MOPSO (pH6.3) (Sigma-Aldrich), 200 mM sorbitol (Alfa Aesar, Karlsruhe, Germany), 10 mM sucrose (Acros, Geel, Belgium), 5 mM thiourea (Sigma-Aldrich), 2 mM DTT (Sigma-Aldrich), 5 mM MgCl2 (Sigma-Aldrich) and 0.5 mM sodium-phosphate (Acros) [23]. Trichomes were captured, using the Palm MicroBeam system (P.A.L.M Microlaser Technologies, München, Germany) with a nitrogen UV-A laser (wavelength 355 nm). The laser-beam was focussed on the tissue for laser microdissection, to separate trichomes or cells from the surrounding tissue. Free trichomes were thereafter captured with laser pressure catapulting by focussing the laser beam just below the tissue. Samples were collected in 30 μl lysis buffer with ß-mercaptoethanol (Absolutely RNA Nanoprep Kit Stratagene, La Jolla, CA). An image of collected glandular and filamentous trichomes is shown in Figure 1C and D. RNA was extracted with the Absolutely RNA Nanoprep protocol with DNase treatment. Samples were eluted in 10 μl and half was used as starting material for RNA amplification.
Sample preparation of apical and sub-apical cells
Apical and sub-apical cells were collected from mock-treated plants in the same way as described for glandular and filamentous trichomes. In a first set of samples, 300 unfixated cells of each cell type were collected. In a second set, 500 fixated cells of each cell-type were collected. In this second set, the cells were fixated before laser microdissection and catapulting to make the separation of the cells easier. Fixation of flowers was carried out by subjecting the samples to a 4% formaldehyde phosphate buffered saline solution for 3 to 4 hours in vacuum. RNA was extracted with the Absolutely RNA Nanoprep protocol with DNase treatment. The RNA from the unfixated samples was split in two and each subsample was separately amplified to create a third sample set for sequencing.
RNA amplification and sequencing
Samples were amplified with a linear amplification system: the Ovation RNA-Seq System with 1 h 30 min Spia-amplification (NuGen, AC Bemmel, The Netherlands). In this amplification procedure, random primers and oligo dT primers are used during RNA amplification and consequently the 5’ end of the mRNA is better amplified compared to when only oligo dT primers are used. The cDNA priming reaction used in the NuGEN amplification kits are designed to avoid amplification of rRNA sequences. After RNA amplification, barcoded Illumina sequencing libraries were made using a post amplification ligation-mediated strategy [27]. The 18 samples were sequenced with a read-length of 100 bp in 3 lanes of an Illumina HiSeq 2000 flowcell.
Quantitative real time (qRT)-PCR
Nugen amplified DNA from 3 independent mock-treated glandular and filamentous trichome samples was analyzed with qRT-PCR. As template, 2 ng DNA was used in 10 μl reactions containing 5 μl iTaq SYBR Green Supermix with ROX (Bio-Rad, Watford, UK) and 400 nM primers. The qRT-PCR experiment was performed on a Light Cycler 480 (Roche) with hotstart at 95°C for 2 min. and 42 cycles 95°C (15 sec.), 52°C (1 min.), including melting curve analysis. Each qRT-PCR reaction was executed in duplo and these technical repeats were averaged prior to qbasePLUS (version 2.3) analysis with normalization to input DNA concentration [28]. To validate this normalization strategy, 3 genes were included that are expected to be similarly expressed in both trichome types: Actin 2 (homologue AT3G18780), protein phosphatase 2A subunit A3 (PP2AA3; homologue AT1G13320) and a pentatricopeptide repeat (PPR) superfamily protein (homologue AT5G55840). These genes were selected based on Arabidopsis data from Czechowski et al. [29] and expression of homologous transcripts in our RNASeq experiment. Other transcripts analyzed were artemisinin-synthesis and triterpene-synthesis related. Primers [see Additional file 3] were adopted from other manuscripts [22, 30] or designed with Primer-BLAST from NCBI [31].
Trinity and Blast2GO
A de novo transcriptome was assembled from all 18 samples. Trinity Release-2011-07-13 was used to perform an ALLPATHS error correction [32] on the reads prior to the de novo assembly which was made with Trinity Release-2011-08-20 [33]. The 150,288 Trinity contigs were annotated using BLAST and the NCBI non-redundant protein database. The best 5 BLAST hits were used to indicate a putative function. The Blast2GO suite [34] was used to generate gene ontology terms based on the BLAST output. Two sets of settings were sequentially used: strict and more permissive. In the more strict settings, a BLAST high-scoring segment pairs (hsp) length of 33 and a minimum hsp coverage of the query of 33 was required. The more permissive setting allowed for a shorter BLAST hsp length of 20 with no minimum hsp coverage of the query.
Bowtie and RSEM
Bowtie [35] and RSEM (RNASeq by Expectation Maximization) [36] were used for mapping the 289 million reads to the Trinity de novo assembly and counting the number of reads that matched to each contig. Standard options were used, but RSEM’s polyA tail option was disabled.
edgeR
To perform the differential expression analysis, an R script was developed that makes use of the Bioconductor edgeR package [37]. All glandular and filamentous samples were normalized together. Normalization was performed by trimmed mean of M values (TMM). TMM equates the overall expression levels of genes between samples under the assumption that the majority of them are not differentially expressed [38]. A p-value < 0.05, adjusted for multiple testing, was used to determine which contigs could be differentially expressed. The edgeR parameter prior.n was set to 1.
MapMan
MapMan is a program to visualize various pathways and to indicate genes or contigs that are up or down-regulated [39]. Prior to MapMan, functional bins need to be assigned to contigs of the de novo assembly. To assign the bins, Mercator was used (default options, Blast_cutoff: 50 and IS_DNA). Not all artemisinin biosynthesis genes were automatically assigned to a bin. For those genes, additional bins were added based on the definitions of the best 5 blast hits. The output of edgeR was visualized with MapMan to show which metabolic pathways are significantly differentially expressed.
Results and discussion
De novoassembly of the transcriptome
All 18 samples were sequenced in 3 lanes of an Illumina HiSeq 2000 flowcell. For each sample, on average 16 million single-end reads of 100 bp were generated. Around 60% of the reads had hits with a combined LSU and SSU rRNA reference database [40] and tRNA database with all known tRNAs [41]. The total of 289 million reads generated were processed using Trinity [33] to assemble a de novo transcriptome [see Additional file 4] containing 150,288 contigs in 108,400 homologous groups with an average contig-length of 412 bp and a minimum and maximum length of 201 and 7775 bp, respectively (Table 2).
To have a better estimate of the quality of the Trinity contigs, all 88 full-length A. annua mRNA sequences available in the NCBI non-redundant protein database were compared with the Trinity contigs. Of those, only 2 had no BLAST hits to the Trinity contigs. Each trinity contig that showed a hit with one of the NCBI sequences, covered on average 58% of the length of the NCBI sequence. A combination of contigs with the same BLAST-hit could together cover on average 84% of the NCBI sequence. If the set of NCBI sequences is assumed to be representational of the real transcripts, one can expect that more than 80% of the length of a random transcript is present in our assembly.
Annotation and characterization of de novotranscripts
Eighty-four% of the 150,288 contigs showed at least one BLAST hit with the NCBI non-redundant protein database. The first five BLAST hits served as an indication for the putative function of a contig. The contigs were also annotated with the Blast2GO suite [34], which links BLAST hits to gene ontology (GO) terms: 46,711 contigs had a good connection to GO terms, using strict settings for Blast2GO; 5,596 contigs still had a connection to GO terms with more permissive settings of Blast2GO; the remaining 73,389 contigs with BLAST hits did not point to any GO terms. As A. annua is not a model organism and does not belong to a family of a model organism it is logical that a lot of contigs with BLAST hits were not annotated with GO terms. It is mostly for genes that show strong homologies with genes of model organism that one can expect a successful annotation using this strategy. Moreover, a gene that performs a different function but has a homology with a gene of a model organism, can be annotated wrongly.
To further characterize our de novo Illumina transcripts, they were also compared to the 454 glandular trichome A. annua contigs of Wang et al. [12]. Of their 42,678 contigs, 79% had a BLAST hit within our Trinity contigs. Vice versa, only 20% of our contigs had a BLAST hit within their contig set. This was not due to contamination of rRNA and tRNA reads that could have resulted from our method of mRNA amplification since only 0.6% of all our contigs showed a hit with rRNA or tRNA. Therefore the fact that the majority of our contigs are differential from those of Wang et al. is most likely the result from a greater coverage.
Influence of JA treatment
The transcriptome of mock and JA-treated trichome samples was compared and no significant differences (adjusted p-value < 0.05) were detected. Accordingly, at the metabolite level, equal amounts of artemisinin, arteannuin B, dihydroartemisinic acid and artemisinic acid were measured with HPLC-MS/MS as shown in additional data [see Additional file 5 and Additional file 6]. This indicates that the JA-treatment did not have a major influence in our experimental set-up. This was not expected since Maes et al. measured higher artemisinin levels after JA treatment [22]. A possible explanation is the use of plants in different developmental stages: Maes et al. used young seedlings while in our RNASeq experiment 6-months old plants with closed capitula were used. It has been shown that in flowers of Arabidopsis thaliana, JA levels increase 6.7-fold just before flower bud opening [42, 43]. Therefore it is plausible that endogenous JA-signalling already reached a maximum effect in our samples and that exogenous JA treatment did not trigger an additional response.
Glandular versus filamentous trichomes
Transcript levels from glandular trichomes and filamentous trichomes were compared to obtain a list of significantly differentially expressed contigs (adjusted p-value < 0.05). To make a robust statistical comparison, 3 samples of glandular trichomes with JA and 3 samples with mock-treatment were compared to the 6 samples (mock and JA) of filamentous trichomes. Of 150,288 contigs; 631 were significantly differentially expressed and all these contigs are listed in additional data [see Additional file 7]. From these, 204 contigs were more expressed in filamentous trichomes whereas, 427 contigs were more expressed in glandular trichomes. An overview with contigs discussed in this article and their normalized counts for each sample, log2-fold changes and adjusted p-values are shown in additional data [see Additional file 8].
MVA and MEP pathway in glandular and filamentous trichomes
From the upregulated contigs, 5% in glandular and 0% in filamentous trichomes were involved in the mevalonate (MVA) or 2-C-methyl-D-erythritol 4-phosphate (MEP) pathways. The MEP and MVA pathways produce isopentenyl diphosphate and its isomer dimethylallyl diphosphate, which are precursors for the production of terpenes.
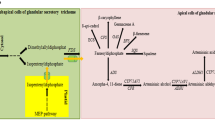
MEP and MVA pathways were detected in glandular and filamentous trichomes. All transcripts coding for enzymes of the MEP pathway were significantly upregulated in glandular trichomes (Figure 2). From the MVA pathway, only acetyl-CoA C-acetyltransferase (AACT) and 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) were significantly upregulated in glandular trichomes (Figure 2). Up-regulation of HMGR in glandular trichomes is important since it is shown that HMGR activity limits artemisinin biosynthesis [44, 45].

Differentially expressed MVA and MEP-pathway genes in glandular and filamentous trichomes. MapMan figure (adapted from [46]) with comparison of MVA and MEP pathways in glandular and filamentous trichomes. Significantly more expressed contigs in glandular trichomes are shown in red with colour scale to indicate the log2-fold changes. No contigs were significantly more expressed in filamentous trichomes. Grey represents contigs that were not significantly differentially expressed. MVA pathway: acetyl-CoA C-acetyltransferase (AACT), 3-hydroxy-3-methyl-glutaryl coenzyme A synthase (HMGS), 3-hydroxy-3-methyl-glutaryl coenzyme A reductase (HMGR), mevalonate kinase (MVK), phosphomevalonate kinase (PMK), diphosphomevalonate decarboxylase (PMD), isopentenyl diphosphate isomerase (IDI). MEP pathway: 1-deoxy-D-xylulose-5-phosphate synthase (DXS), 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR), 2-C-methyl-D-erythritol-4-phosphate cytidylyltransferase (MCT), 4-cytidine 5’-diphospho-2-C-methyl-D-erythritol kinase (CMK), 2-C-methyl-D-erythritol-2,4-cyclodiphosphate synthase (MCS), hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate synthase (HDS) and hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase (HDR).
Artemisinin biosynthesis in glandular and filamentous trichomes
Terpene synthesis genes were accounting for 6% of the upregulated contigs in glandular trichomes and only 1% in filamentous trichomes. Starting from the MVA and MEP pathway, farnesyl diphosphate is synthesised by farnesyl diphosphate synthase (FDS) whose transcripts were not significantly upregulated in glandular trichomes (Figure 3 and Additional file 8). Subsequently, farnesyl diphosphate is converted to amorpha-4,11-diene which is the starting product for artemisinin biosynthesis. This reaction is catalyzed by amorpha-4,11-diene synthase (ADS) [47] and transcripts coding for this enzyme were detected more in glandular trichomes . By CYP71AV1, amorpha-4,11-diene is converted to artemisinic alcohol [3, 7]. Thereafter, artemisinic alcohol is oxidized by CYP71AV1 [3, 7] and alcohol dehydrogenase 1 (ADH1) [10] to artemisinic aldehyde. Artemisinic aldehyde is further oxidized by aldehyde dehydrogenase 1 (ALDH1) [8] and CYP71AV1 [3, 7] to artemisinic acid or reduced by artemisinic aldehyde Δ11(13) double bond reductase (DBR2) [9] to form dihydroartemisinic aldehyde. A broad substrate oxidoreductase (RED1) can convert dihydroartemisinic aldehyde to dihydroartemisinic alcohol [48]. This reaction competes with ALDH1 using dihydroartemisinic aldehyde to form dihydroartemisinic acid [8]. Dihydroartemisinic acid is considered to be the precursor leading to artemisinin [9, 49, 50]. Transcripts corresponding to all these enzymes involved in the conversion of amorpha-4,11-diene to dihydroartemisinic acid were significantly upregulated in glandular trichomes, except for RED1 (Figure 3).

Differentially expressed artemisinin biosynthesis genes in glandular and filamentous trichomes. MapMan file with comparison of artemisinin biosynthesis pathways in glandular and filamentous trichomes. Significantly more expressed contigs in glandular trichomes are shown in red with colour scale to indicate the log2-fold change. There were no contigs with higher expression in filamentous trichomes. Grey represents contigs that were not significantly differentially expressed.
These results confirm previous data pointing to glandular trichomes as the major artemisinin production site. A short dip in chloroform causes the collapse of the sub-cuticular space of glandular trichomes and extracts almost all artemisinin [5, 51]. In addition to this, no artemisinin has been detected in a biotype of A. annua with only filamentous trichomes [5]. Nevertheless, in all samples of filamentous trichomes, transcripts corresponding to known artemisinin biosynthesis genes were detected, albeit at very low levels [see Additional file 8]. This is in agreement with previous reports, in which expression of ADS was also detected by RT-PCR in filamentous trichomes [12]. It should be noted though that in contrast, no staining of filamentous trichomes was observed in a promoter-GUS fusion study with the ADS promoter [52].
Candidate genes for artemisinin biosynthesis
Contigs that are upregulated in glandular trichomes are possibly linked to artemisinin biosynthesis genes. The subset of these contigs which were annotated as cytochromes, peroxidases and dioxygenases might be possible candidates for the endoperoxide ring formation in artemisinin. An overview of these contigs is given in Table 3, more results per repeat are given in additional data [see Additional file 8].
Comp69 was detected 6 log2-fold more in glandular compared to filamentous trichomes. The best BLAST-hit was with CYP81B1 [GenBank: CAA04117] from Helianthus tuberosus. This enzyme is functionally characterized to hydroxylate medium-chain saturated fatty acids [53]. In addition to this, CYP81B1 can be assumed to epoxidize these fatty acids [53, 54]. In glandular trichomes, comp548_c0_seq1 and comp548_c0_seq2 were detected 5 log2-fold more than in filamentous trichomes, these contigs were annotated as P450 mono-oxygenase and showed homology with predicted sequences from the CYP82 family and CYP82C9v3 [GenBank: XP_002327091] from Populus trichocarpa. This CYP82 family is relatively uncharacterized [55]. A characterized enzyme from this family is CYP82G1 [GenBank: Q9LSF8] from Arabidopsis thaliana which is involved in homoterpene biosynthesis in which epoxidation might occur [56, 57]. But this characterized protein was not present in the list of best-BLAST hits with comp548. Another cytochrome P450 significantly more expressed in glandular trichomes was comp2774. The full length of this transcript was determined by Misra et al. and called CIM_CYP03 (CYP72A) [GenBank: GU318227] [58]. The activity of this enzyme was tested in vitro with artemisinic acid, dihydroartemisinic acid, arteannuin B and artemisinin as substrates but no activity was detected [58]. Other contigs significantly more expressed in glandular trichomes were comp15043_c0_seq2 annotated as CYP76B1 and comp3673 as cytochrome P450. Comp586_c0_seq3 and comp586_c0_seq4 were annotated as a cytochrome c-type. Some other cytochromes were significantly more expressed in filamentous trichomes as shown in additional data [see Additional file 8].
Comp252 (seq1, seq3, seq4 and seq5), 2084 and 6217_c0_seq4_1 are peroxidases detected respectively 6 log2-fold, 4 log2-fold and 30 log2-fold more in glandular trichomes. Comp2084 was more specifically annotated as peroxidase 49 precursor and comp6217 was annotated as gluthatione peroxidase. In filamentous trichomes, two contigs annotated as peroxidases were significantly more expressed than in glandular trichomes. These contigs are comp3274 annotated as peroxidase 1 from A. annua and comp16324 as alkaline leaf peroxidase from Cyanara cardunculus.
Two dioxygenases were significantly more expressed in glandular trichomes: comp225_c0_seq1 and comp453 were both annotated as naringenin 2-oxoglutarate 3-dioxygenase (flavanone 3-hydroxylase).
Other terpene synthases in glandular and filamentous trichomes
Several contigs corresponding to enzymes involved in the biosynthesis of other sesquiterpenoids were significantly upregulated in glandular trichomes as well. These contigs are listed in the supplementary data [see Additional file 8]. Despite a higher germacrene-A synthase expression in glandular trichomes, germacrene-A was only detected in biotypes without glandular trichomes [59]. This can be the effect of up-regulation of germacrene-A oxidase which further oxidizes germacrene-A [60].
In filamentous trichomes, contig comp2645 corresponding to 8-epi-cedrol synthase, was significantly upregulated [see Additional file 8]. Low expression of 8-epi-cedrol synthase in glandular trichomes has been observed by qRT-PCR [23]. In vitro, recombinant 8-epi-cedrol synthase converts farnesyl diphosphate to 8-epicedrol, cedrol and minor amounts of α- cedrene and (E)-β-farnesene [61, 62]. Since a higher amount of 8-epi-cedrol synthase was detected in filamentous trichomes, these trichomes might synthesize the majority of these metabolites. Differences in (E)-β-farnesene concentration in glanded and glandless biotypes were estimated from Tellez et al. by correlating relative peak area to the oil content in fresh plant material [59]. Based on these estimates, the level of (E)-β-farnesene is approximately 1.8 times higher in glandless biotypes. This might correlate with the upregulated expression of 8-epi-cedrol synthase in filamentous trichomes. From α-cedrene, only trace amounts were measured in glanded and glandless biotypes [59] and the major product cedrol epimers were even not detected in extracts of A. annua[61, 62].
Regarding diterpenoid biosynthesis, only contigs annotated as momilactone A synthase were significantly more expressed in glandular trichomes [see Additional file 8]. Concerning monoterpenoid biosynthesis, many monoterpenoid synthases were significantly upregulated in glandular trichomes [see Additional file 8]. This is corroborated when comparing isoprenoid contents in glanded and glandless A. annua[59]. In oil from glanded biotypes, monoterpenes were predominant whereas in oil from glandless biotypes monoterpenes were almost absent.
β-amyrin synthase, an enzyme that converts 2,3-oxidosqualene to the triterpene saponin β-amyrin, has been characterized in A. annua[63]. This enzyme was represented in the de novo assembly by comp33386, comp59983, comp96251 and comp23239 and these contigs were not significantly differentially expressed. Contig comp7642_c0_seq2_1 shows homology with both dammarenediol synthase and (β-)amyrin synthase and is a yet uncharacterized oxidosqualene cyclase (U_OSC). This contig was detected significantly more in filamentous trichomes [see Additional file 8].
Lipid biosynthesis in glandular and filamentous trichomes
In glandular trichomes, 15% of the significantly upregulated contigs were annotated to lipid biosynthesis. Transcripts and their corresponding significantly differentially expressed contigs are listed in supplementary data [see Additional file 8]. Acetyl-CoA carboxylase converts acetyl-CoA to malonyl-CoA and was significantly upregulated in glandular trichomes as shown in Figure 4. Subsequently, malonyl CoA-acyl carrier protein transacylase converts malonyl-CoA to malonyl-ACP [64]. This transcript was not significantly upregulated. Fatty acid biosynthesis is initiated by the condensation of malonyl-ACP with acetyl-CoA by β-ketoacyl-ACP synthase III (KAS). β-ketoacyl-ACP is reduced by β-ketoacyl-ACP reductase, dehydrated by β-hydroxyacyl-ACP dehydratase and reduced by enoyl-ACP reductase to yield butyryl-ACP. The latter transcripts except enoyl-ACP reductase were significantly more expressed in glandular trichomes as shown in Figure 4. The acyl-ACP end product has two carbons more than the original acetyl molecule [64]. Similar elongation cycles are continued with condensation of malonyl-ACP and acyl-ACP and the removal of the β-ketogroup. Three types of KAS were present with different acyl chain length specificities: KASIII (C2 to C4), KASI (C4 to C16) and KASII (C16 to C18) [64]. All 3 types of KAS were upregulated in glandular trichomes.

Differentially expressed lipid biosynthesis genes in glandular and filamentous trichomes. MapMan file with comparison of lipid biosynthesis pathways in glandular and filamentous trichomes. Significantly more expressed contigs in glandular trichomes are shown in red with colour scale to indicate the log2-fold change. There were no contigs with higher expression in filamentous trichomes. Grey represents contigs that were not significantly differentially expressed.
Further extension of C16 and C18 to longer fatty acids requires their liberation from ACP by acyl-ACP thioesterase. Subsequently, fatty acids are exported out of the plastid to the endoplasmic reticulum [64]. The extension of fatty acids from long (C16,C18) to very long chains is catalyzed by β-ketoacyl-CoA synthase, β-ketoacyl-CoA reductase, β-hydroxyacyl-CoA dehydratase and enoyl-CoA reductase [65]. The rate-limiting step and specificity is determined by the β-ketoacyl-CoA synthase which was significantly more expressed in glandular trichomes [see Additional file 8].
Fatty acyl-CoA reductase 1 (TFAR1) [22] was significantly more expressed in glandular trichomes. The encoded enzyme catalyzes the formation from acyl-CoA to fatty alcohols and is potentially involved in wax formation [22]. For the formation of unsaturated fatty acids, omega-3 fatty acid desaturase is significantly upregulated in glandular trichomes. Contigs annotated as cyclopropane-fatty-acyl phospholipid synthase were highly expressed in glandular trichomes. This enzyme is forming a cyclopropane ring in unsaturated fatty acyl chains [66, 67]. Some contigs coding for lipid transfer proteins were significantly upregulated in glandular trichomes whereas other contigs coding for lipid transfer proteins were upregulated in filamentous trichomes [see Additional file 7].
The observed upregulation of lipid biosynthesis in glandular trichomes is in agreement with the results obtained by Tellez et al. [59], who measured that in glanded leaves 0.24% of fresh weight is oil and in glandless leaves only 0.06%.
qRT-PCR
Enrichment for artemisinin-synthesis and triterpene-synthesis related transcripts in glandular and filamentous trichomes, respectively, was verified by qRT-PCR [see Additional file 9]. In mock-treated filamentous and glandular trichomes, similar expression levels were detected for Actin2, PP2AA3 and PPR protein in both RNASeq and qRT-PCR. Similarly, with both techniques, no clear difference in FDS was observed between both trichome types. With qRT-PCR, the mean expression levels of CYP71AV1, DBR2, Aldh1, Fatty acyl-CoA reductase 1 (TFAR1) and 10 candidate genes for artemisinin biosynthesis were higher in glandular trichomes whereas the transcripts corresponding to uncharacterized oxidosqualene cyclase (U_OSC) and 8-epi-cedrol synthase were more abundant in filamentous trichomes. Hence, this qRT-PCR experiment confirmed the RNASeq data.
Apical versus sub-apical cells
The small number of microdissected cells yielded a suboptimal amount of RNA that could be used as input for the Nugen SPIA amplification. For this reason, the amplification was biased towards the high abundant transcripts and the counts of the low abundant transcripts showed a high variation. A comparison of apical and sub-apical cells revealed no significantly differentially expressed contigs (adjusted p-value < 0.05). Contigs coding for artemisinin biosynthesis genes were detected in both apical and sub-apical cells as shown in Figure 5, which is in agreement with the findings of Olofsson et al. [23].

Expression of artemisinin biosynthesis genes in apical and sub-apical cells from glandular trichomes. Normalized counts for artemisinin biosynthesis genes in apical (AP) and sub-apical (SA) cells. Error bars represent standard deviations.
Conclusions
On the transcript level, MEP and MVA pathways were significantly upregulated in glandular trichomes in comparison with filamentous trichomes. In addition to this, transcripts coding for the artemisinin biosynthesis pathway, other sesquiterpene biosynthesis and monoterpene pathways were predominantly expressed in glandular trichomes. Novel cytochrome-, peroxidase- and dioxygenases-encoding genes highly expressed in glandular trichomes were detected and these might be potential candidate genes for the formation of the endoperoxide bridge in artemisinin. Lipid biosynthesis pathways were highly expressed in glandular trichomes and less in filamentous trichomes. In filamentous trichomes, some specific genes from sesquiterpenoid and triterpenoid pathways such as 8-epi-cedrol synthase and oxidosqualene cyclase were detected significantly more than in glandular trichomes. Between the transcriptome of apical and sub-apical cells from glandular trichomes, no differences could be observed in the expression of artemisinin biosynthetic enzymes.
This transcriptome analysis underscores the vast metabolic capacities of A. annua glandular trichomes and simultaneously points to the existence of specific terpene metabolic pathways in the filamentous trichomes. Therefore, it would be interesting to examine metabolic activities in filamentous trichomes of other plant species. Besides this, it would also be interesting to characterize the potential candidate genes for artemisinin biosynthesis. If they are involved in the production of artemisinin, they can be used to produce artemisinin in yeast cells.
Abbreviations
- AACT:
-
Acetyl-CoA C-acetyltransferase
- ADS:
-
Amorpha-4,11-diene synthase
- ADH1:
-
Alcohol dehydrogenase
- ALDH1:
-
Aldehyde dehydrogenase 1
- CMK:
-
4-cytidine 5’-diphospho-2-C-methyl-D-erythritol kinase
- CYP71AV1:
-
Amorpha-4,11-diene monooxygenase
- DBR2:
-
Artemisinic aldehyde Δ11(13) double bond reductase
- DXS:
-
1-deoxy-D-xylulose-5-phosphate synthase
- DXR:
-
1-deoxy-D-xylulose-5-phosphate reductoisomerase
- EST:
-
Expressed sequence tags
- FDS:
-
Farnesyl diphosphate synthase
- GO:
-
Gene onthology
- HDS:
-
Hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate synthase
- HDR:
-
Hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase
- HMGR:
-
3-hydroxy-3-methyl-glutaryl coenzyme A reductase
- HMGS:
-
3-hydroxy-3-methyl-glutaryl coenzyme A synthase
- Hsp:
-
High-scoring segment pairs
- IDI:
-
Isopentenyl diphosphate isomerase
- LSU:
-
Large subunit
- JA:
-
Jasmonic acid
- KAS:
-
β-ketoacyl-ACP synthase
- MCS:
-
2-C-methyl-D-erythritol-2,4-cyclodiphosphate synthase
- MCT:
-
2-C-methyl-D-erythritol-4-phosphate cytidylyltransferase
- MEP:
-
2-C-methyl-D-erythritol 4-phosphate
- MVA:
-
Mevalonate pathway
- MVK:
-
Mevalonate kinase
- PMK:
-
Phosphomevalonate kinase
- PMD:
-
Diphosphomevalonate decarboxylase
- PP2AA3:
-
Protein phosphatase 2A subunit A3
- PPR:
-
Pentatricopeptide repeat superfamily protein
- RED1:
-
Dihydroartemisinic aldehyde reductase
- RSEM:
-
RNASeq by Expectation Maximization
- SSU:
-
Small subunit
- TFAR1:
-
Fatty acyl-CoA reductase 1
- TMM:
-
Trimmed mean of M values
- U_OSC:
-
Uncharacterized oxidosqualene cyclase.
References
WHO: World Malaria Report. Geneva: World Health Organization; 2011.
Graham IA, Besser K, Blumer S, Branigan CA, Czechowski T, Elias L, Guterman I, Harvey D, Isaac PG, Khan AM, et al: The genetic map of Artemisia annua L identifies Loci affecting yield of the antimalarial drug artemisinin. Science. 2010, 327 (5963): 328-331. 10.1126/science.1182612.
Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, et al: Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006, 440 (7086): 940-943. 10.1038/nature04640.
Levesque F, Seeberger PH: Continuous-flow synthesis of the anti-malaria drug artemisinin. Angew Chem-Int Edit. 2012, 51 (7): 1706-1709. 10.1002/anie.201107446.
Duke MV, Paul RN, Elsohly HN, Sturtz G, Duke SO: Localization of artemisinin and artemisitene in foliar tissues of glanded and glandless biotypes of Artemisia annua L. Int J Plant Sci. 1994, 155 (3): 365-372. 10.1086/297173.
Ferreira JFS, Janick J: Floral morphology of Artemisia annua with special reference to trichomes. Int J Plant Sci. 1995, 156 (6): 807-815. 10.1086/297304.
Teoh KH, Polichuk DR, Reed DW, Nowak G, Covello PS: Artemisia annua L. (Asteraceae) trichome-specific cDNAs reveal CYP71AV1, a cytochrome P450 with a key role in the biosynthesis of the antimalarial sesquiterpene lactone artemisinin. Febs Letters. 2006, 580 (5): 1411-1416. 10.1016/j.febslet.2006.01.065.
Teoh KH, Polichuk DR, Reed DW, Covello PS: Molecular cloning of an aldehyde dehydrogenase implicated in artemisinin biosynthesis in Artemisia annua. Botany. 2009, 87 (6): 635-642. 10.1139/B09-032.
Zhang Y, Teoh KH, Reed DW, Maes L, Goossens A, Olson DJH, Ross ARS, Covello PS: The molecular cloning of artemisinic aldehyde Delta 11(13) reductase and its role in glandular trichome-dependent biosynthesis of artemisinin in Artemisia annua. J Biol Chem. 2008, 283 (31): 21501-21508. 10.1074/jbc.M803090200.
Polichuk D, Teoh KH, Zhang Y, Ellens KW RD, Covello PS: Patent No. WO/2010/012074, February 4th. Nucleotide sequence encoding an alcohol dehydrogenease from Artemisia annua and uses thereof. 2010
Brown GD, Sy LK: In vivo transformations of dihydroartemisinic acid in Artemisia annua plants. Tetrahedron. 2004, 60 (5): 1139-1159. 10.1016/j.tet.2003.11.070.
Wang W, Wang YJ, Zhang Q, Qi Y, Guo DJ: Global characterization of Artemisia annua glandular trichome transcriptome using 454 pyrosequencing. Bmc Genomics. 2009, 10: 465. 10.1186/1471-2164-10-465.
Wagner GJ: Secreting glandular trichomes: more than just hairs. Plant Physiol. 1991, 96 (3): 675-679. 10.1104/pp.96.3.675.
Levin DA: The role of trichomes in plant defence. Q Rev Plant Biol. 1973, 48 (1): 3-15.
Mauricio R, Rausher MD: Experimental manipulation of putative selective agents provides evidence for the role of natural enemies in the evolution of plant defense. Evolution. 1997, 51 (5): 1435-1444. 10.2307/2411196.
Victorio CP, Moreira CB, Souza MD, Sato A, Arruda RDD: Secretory cavities and volatiles of Myrrhinium atropurpureum Schott var. atropurpureum (Myrtaceae): an endemic species collected in the Restingas of Rio de Janeiro, Brazil. Nat Prod Commun. 2011, 6 (7): 1045-1050.
Porto NM, De Figueiredo R, Oliveira AFM, Agra MD: Leaf epidermal characteristics of Cissampelos L. (Menispermaceae) species from Northeastern Brazil. Microsc Res Tech. 2011, 74 (4): 370-376.
Bhatt A, Naidoo Y, Nicholas A: The foliar trichomes of Hypoestes aristata (Vahl) Sol. ex Roem. & Schult var aristata (Acanthaceae) a widespread medicinal plant species in tropical sub-Saharan Africa: with comments on its possible phylogenetic significance. Biol Res. 2010, 43 (4): 403-409. 10.4067/S0716-97602010000400004.
Duarte MR, Lopes JF: Leaf and stem morphoanatomy of Petiveria alliacea. Fitoterapia. 2005, 76 (7–8): 599-607.
Bonzani NE, Barboza GE, Bugatti MA, Espinar LA: Morpho-histological studies in the aromatic species of Chenopodium from Argentina. Fitoterapia. 2003, 74 (3): 207-225. 10.1016/S0367-326X(03)00024-8.
Wagner GJ, Wang E, Shepherd RW: New approaches for studying and exploiting an old protuberance, the plant trichome. Ann Bot. 2004, 93 (1): 3-11. 10.1093/aob/mch011.
Maes L, Van Nieuwerburgh FCW, Zhang YS, Reed DW, Pollier J, Casteele S, Inze D, Covello PS, Deforce DLD, Goossens A: Dissection of the phytohormonal regulation of trichome formation and biosynthesis of the antimalarial compound artemisinin in Artemisia annua plants. New Phytol. 2011, 189 (1): 176-189. 10.1111/j.1469-8137.2010.03466.x.
Olofsson L, Lundgren A, Brodelius PE: Trichome isolation with and without fixation using laser microdissection and pressure catapulting followed by RNA amplification: expression of genes of terpene metabolism in apical and sub-apical trichome cells of Artemisia annua L. Plant Sci. 2012, 183: 9-13.
Delabays N, Simonnet X, Gaudin M: The genetics of artemisinin content in Artemisia annua L. and the breeding of high yielding cultivars. Curr Med Chem. 2001, 8 (15)): 1795-1801.
Ferreira JFS, Laughlin JC, Delabays N, Magalhaes PM: Cultivation and genetics of Artemisia annua L. for increased production of the antimalarial artemisinin. Plant Genet Resour-Characterization Util. 2005, 3 (2): 206-229. 10.1079/PGR200585.
WHO: Meeting on the production of artemisinin and artemisinin-based combination therapies; Tanzania. Geneva, Switzerland: World Health Organization; 2005.
Van Nieuwerburgh F, Soetaert S, Podshivalova K, Ay-Lin Wang E, Schaffer L, Deforce D, Salomon DR, Head SR, Ordoukhanian P: Quantitative bias in Illumina TruSeq and a novel post amplification barcoding strategy for multiplexed DNA and small RNA deep sequencing. PLoS One. 2011, 6 (10): e26969. 10.1371/journal.pone.0026969.
Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J: qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8 (2): R19. 10.1186/gb-2007-8-2-r19.
Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR: Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005, 139 (1): 5-17. 10.1104/pp.105.063743.
Olofsson L, Engstrom A, Lundgren A, Brodelius PE: Relative expression of genes of terpene metabolism in different tissues of Artemisia annua L. BMC Plant Biol. 2011, 11: 45. 10.1186/1471-2229-11-45.
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL: Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. Bmc Bioinformatics. 2012, 13: 13. 10.1186/1471-2105-13-13.
Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al: High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011, 108 (4): 1513-1518. 10.1073/pnas.1017351108.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al: Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011, 29 (7): 644-652. 10.1038/nbt.1883.
Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A: High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36 (10): 3420-3435. 10.1093/nar/gkn176.
Langmead B, Trapnell C, Pop M, Salzberg SL: Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10 (3): R25. 10.1186/gb-2009-10-3-r25.
Li B, Dewey CN: RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. Bmc Bioinformatics. 2011, 12: 323. 10.1186/1471-2105-12-323.
Robinson MD, McCarthy DJ, Smyth GK: edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010, 26 (1): 139-140. 10.1093/bioinformatics/btp616.
Robinson MD, Oshlack A: A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11 (3): R25. 10.1186/gb-2010-11-3-r25.
Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kruger P, Selbig J, Muller LA, Rhee SY, Stitt M: MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37 (6): 914-939. 10.1111/j.1365-313X.2004.02016.x.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig WG, Peplies J, Glockner FO: SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35 (21): 7188-7196. 10.1093/nar/gkm864.
Chan PP, Lowe TM: GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009, 37: D93-D97. 10.1093/nar/gkn787.
Reeves PH, Ellis CM, Ploense SE, Wu MF, Yadav V, Tholl D, Chetelat A, Haupt I, Kennerley BJ, Hodgens C, et al: A regulatory network for coordinated flower maturation. PLoS Genet. 2012, 8 (2): 17.
Nagpal P, Ellis CM, Weber H, Ploense SE, Barkawi LS, Guilfoyle TJ, Hagen G, Alonso JM, Cohen JD, Farmer EE, et al: Auxin response factors ARF6 and ARF8 promote jasmonic acid production and flower maturation. Development. 2005, 132 (18): 4107-4118. 10.1242/dev.01955.
Ram M, Khan MA, Jha P, Khan S, Kiran U, Ahmad MM, Javed S, Abdin MZ: HMG-CoA reductase limits artemisinin biosynthesis and accumulation in Artemisia annua L. plants. Acta Physiol Plant. 2010, 32 (5): 859-866. 10.1007/s11738-010-0470-5.
Aquil S, Husaini AM, Abdin MZ, Rather GM: Overexpression of the HMG-CoA reductase gene leads to enhanced artemisinin biosynthesis in transgenic Artemisia annua plants. Planta Med. 2009, 75 (13): 1453-1458. 10.1055/s-0029-1185775.
Usadel B, Nagel A, Thimm O, Redestig H, Blaesing OE, Palacios-Rojas N, Selbig J, Hannemann J, Piques MC, Steinhauser D, et al: Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of coresponding genes, and comparison with known responses. Plant Physiol. 2005, 138 (3): 1195-1204. 10.1104/pp.105.060459.
Bouwmeester HJ, Wallaart TE, Janssen MHA, van Loo B, Jansen BJM, Posthumus MA, Schmidt CO, De Kraker JW, Konig WA, Franssen MCR: Amorpha-4,11-diene synthase catalyses the first probable step in artemisinin biosynthesis. Phytochemistry. 1999, 52 (5): 843-854. 10.1016/S0031-9422(99)00206-X.
Ryden AM, Ruyter-Spira C, Quax WJ, Osada H, Muranaka T, Kayser O, Bouwmeester H: The molecular cloning of dihydroartemisinic aldehyde reductase and its implication in artemisinin biosynthesis in Artemisia annua. Planta Med. 2010, 76 (15): 1778-1783. 10.1055/s-0030-1249930.
Bertea CM, Freije JR, van der Woude H, Verstappen FWA, Perk L, Marquez V, De Kraker JW, Posthumus MA, Jansen BJM, de Groot A, et al: Identification of intermediates and enzymes involved in the early steps of artemisinin biosynthesis in Artemisia annua. Planta Med. 2005, 71 (1): 40-47. 10.1055/s-2005-837749.
Covello PS, Teoh KH, Polichuk DR, Reed DW, Nowak G: Annual Meeting of the Phytochemical-Society-of-North-American: 2006 2007. Functional genomics and the biosynthesis of artemisinin. 2007, Oxford, MS: Pergamon-Elsevier Science Ltd, 1864-1871.
Van Nieuwerburgh FCW, Casteele SRV, Maes L, Goossens A, Inze D, Van Bocxlaer J, Deforce DLD: Quantitation of artemisinin and its biosynthetic precursors in Artemisia annua L. by high performance liquid chromatography - electrospray quadrupole time-of-flight tandem mass spectrometry. J Chromatogr A. 2006, 1118 (2): 180-187. 10.1016/j.chroma.2006.03.121.
Wang H, Olofsson L, Lundgren A, Brodelius PE: Trichome-specific expression of amorpha-4,11-diene synthase, a Key Enzyme of Artemisinin Biosynthesis in Artemisia annua L., as reported by a promoter-GUS fusion. American. J Plant Sci. 2011, 2 (4): 619-628.
Cabello-Hurtado F, Batard Y, Salaun JP, Durst F, Pinot F, Werck-Reichhart D: Cloning, expression in yeast, and functional characterization of CYP81B1, a plant cytochrome P450 that catalyzes in-chain hydroxylation of fatty acids. J Biol Chem. 1998, 273 (13): 7260-7267. 10.1074/jbc.273.13.7260.
Salaun JP, Weissbart D, Durst F, Pflieger P, Mioskowski C: Epoxidation of cis-delta-9 and trans-delta-9-unsaturated lauric acids by a cytochrome P-450-dependent system from higher-plant microsomes. Febs Letters. 1989, 246 (1–2): 120-126.
Tholl D, Sohrabi R, Huh JH, Lee S: The biochemistry of homoterpenes - common constituents of floral and herbivore-induced plant volatile bouquets. Phytochemistry. 2011, 72 (13): 1635-1646. 10.1016/j.phytochem.2011.01.019.
Lee S, Badieyan S, Bevan DR, Herde M, Gatz C, Tholl D: Herbivore-induced and floral homoterpene volatiles are biosynthesized by a single P450 enzyme (CYP82G1) in Arabidopsis. Proc Natl Acad Sci U S A. 2010, 107 (49): 21205-21210. 10.1073/pnas.1009975107.
Donath J, Boland W: Biosynthesis of acyclic homoterpenes in higher-plants parallels steroid-hormone metabolism. J Plant Physiol. 1994, 143 (4–5): 473-478.
Misra A, Chanotiya CS, Gupta MM, Dwivedi UN, Shasany AK: Characterization of cytochrome P450 monooxygenases isolated from trichome enriched fraction of Artemisia annua L. leaf. Gene. 2012, 510 (2): 193-201. 10.1016/j.gene.2012.09.015.
Tellez MR, Canel C, Rimando AM, Duke SO: Differential accumulation of isoprenoids in glanded and glandless Artemisia annua L. Phytochemistry. 1999, 52 (6): 1035-1040. 10.1016/S0031-9422(99)00308-8.
Nguyen DT, Gopfert JC, Ikezawa N, MacNevin G, Kathiresan M, Conrad J, Spring O, Ro DK: Biochemical conservation and evolution of germacrene a oxidase in Asteraceae. J Biol Chem. 2010, 285 (22): 16588-16598. 10.1074/jbc.M110.111757.
Hua L, Matsuda SPT: The molecular cloning of 8-epicedrol synthase from Artemisia annua. Arch Biochem Biophys. 1999, 369 (2): 208-212. 10.1006/abbi.1999.1357.
Mercke P, Crock J, Croteau R, Brodelius PE: Cloning, expression, and characterization of epi-cedrol synthase, a sesquiterpene cyclase from Artemisia annua L. Arch Biochem Biophys. 1999, 369 (2): 213-222. 10.1006/abbi.1999.1358.
Kirby J, Romanini DW, Paradise EM, Keasling JD: Engineering triterpene production in Saccharomyces cerevisiae-beta-amyrin synthase from Artemisia annua. Febs J. 2008, 275 (8): 1852-1859. 10.1111/j.1742-4658.2008.06343.x.
Samuels L, Kunst L, Jetter R: Annual Review of Plant Biology. Sealing plant surfaces: Cuticular wax formation by epidermal cells. Palo Alto: Annual Reviews; 2008, 683-707.
Kunst L, Samuels L: Plant cuticles shine: advances in wax biosynthesis and export. Curr Opin Plant Biol. 2009, 12 (6): 721-727. 10.1016/j.pbi.2009.09.009.
Bao XM, Katz S, Pollard M, Ohlrogge J: Carbocyclic fatty acids in plants: Biochemical and molecular genetic characterization of cyclopropane fatty acid synthesis of Sterculia foetida. Proc Natl Acad Sci U S A. 2002, 99 (10): 7172-7177. 10.1073/pnas.092152999.
Yu XH, Rawat R, Shanklin J: Characterization and analysis of the cotton cyclopropane fatty acid synthase family and their contribution to cyclopropane fatty acid synthesis. BMC Plant Biol. 2011, 11: 97. 10.1186/1471-2229-11-97.
Acknowledgements
This research was supported by Flanders Fund for Scientific Research (FWO Flanders) and the Multidisciplinary Research Partnerships project (MRP) on bioinformatics of Ghent university. We would like to thank Sofie Vande Casteele for her excellent technical support on HPLC-MS/MS, Nico Smet and Miguel Riobello (VIB) who gardened the plants and Nicky Van Thuyne who gave assistance in informatics to this project. We are grateful to the ICT Department of Ghent University for assistance with our computations. Thanks to Mikael Olsson from the lab of Peter Brodelius (Linnaeus University Sweden) for demonstrating laser capture microdissection on trichomes. We are also thankful to the thesis students for their help during the laboratory work: Jill Deflou, Liselotte Van Den Eynde and Kim De Maré.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SS planned the experimental setup, performed all the laboratory work except for the sequencing, was involved in the bioinformatics analysis, interpreted the data and wrote the manuscript. CVN performed the bioinformatics analysis and wrote part of the manuscript. MV provided technical support on the laser microscope for collecting the samples. SH helped in optimizing the library preparation and performed the cluster generation as well as the sequencing. AG participated in designing the experiment, biological interpretation of the data and writing. FVN participated in designing the experiment, the bioinformatics analysis and the writing of the manuscript. DD participated in designing the experiment and writing. All authors reviewed and approved the final manuscript.
Electronic supplementary material
12870_2013_1414_MOESM1_ESM.jpeg
{kind=link}
Additional file 1: Artemisia annua Anamed grown in 8 h light, 16 h night. Pictures of 7 months old Artemisia annua Anamed plants (A, B) grown in a growth room under 8 h light, 16 h night photoperiod. C: detail of the flower bud stage used to collect trichomes. (JPEG 808 KB)
12870_2013_1414_MOESM2_ESM.tiff
Additional file 2: Trichomes on a flower head of A. annua. SEM picture of a flower head (capitulum) from A. annua Anamed with some bracts opened to show floret buds. On the floret buds, glandular trichomes are protruding whereas they are sunken in the capitulum bracts. Filamentous trichomes are abundantly present on the basal bracts. (TIFF 6 MB)
12870_2013_1414_MOESM4_ESM.zip
Additional file 4: Transcriptome assembly of trichomes from Artemisia annua. All 150,288 de novo assembled sequences and their contig-numbers generated with Trinity. (ZIP 16 MB)
12870_2013_1414_MOESM5_ESM.pdf
Additional file 5: Metabolite concentrations in flower heads of mock- and JA-treated plants. Graphs showing metabolite concentrations in flower heads of A. annua. Artemisinin, arteannuin B, dihydroartemisinic acid and artemisinic acid levels were measured with HPLC-MS/MS in mock- and JA-treated plants. (PDF 52 KB)
12870_2013_1414_MOESM6_ESM.pdf
Additional file 6: Metabolite concentrations in entire plant material of mock- and JA-treated plants. Graphs showing metabolite concentrations in entire plant material of A. annua. Artemisinin, arteannuin B, dihydroartemisinic acid and artemisinic acid levels were measured with HPLC-MS/MS in mock- and JA-treated plants. (PDF 52 KB)
12870_2013_1414_MOESM7_ESM.txt
Additional file 7: Significant differences in transcriptome expression in glandular and filamentous trichomes. List with all contigs that are significantly differentially expressed in glandular and filamentous trichomes. (TXT 601 KB)
12870_2013_1414_MOESM8_ESM.xlsx
Additional file 8: Overview from the commented differences between glandular and filamentous trichomes. Overview of all contigs discussed in this article including calculation of log2-fold changes and adjusted p-values. (XLSX 49 KB)
12870_2013_1414_MOESM9_ESM.pdf
Additional file 9: qRT-PCR analysis on filamentous and glandular trichomes. Bar chart showing qRT-PCR results on Nugen amplified material and on top the abundance of the corresponding transcripts in the RNASeq data (similar, decreased or enhanced abundance in filamentous and glandular trichomes). Of 3 biologic repeats, geometric averages from relative quantities were calculated with qbasePLUS and shown against a linear scale with as error bars the standard errors of the geometric mean. CF and CG represent mock-treated filamentous and glandular trichomes, respectively. (PDF 411 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Soetaert, S.S., Van Neste, C.M., Vandewoestyne, M.L. et al. Differential transcriptome analysis of glandular and filamentous trichomes in Artemisia annua. BMC Plant Biol 13, 220 (2013). https://doi.org/10.1186/1471-2229-13-220
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-13-220