
Abstract
Background
RT-qPCR is a preferred method for rapid and reliable quantification of gene expression studies. Appropriate application of RT-qPCR in such studies requires the use of reference gene(s) as an internal control to normalize mRNA levels between different samples for an exact comparison of gene expression level. However, recent studies have shown that no single reference gene is universal for all experiments. Thus, the identification of high quality reference gene(s) is of paramount importance for the interpretation of data generated by RT-qPCR. Only a few studies on reference genes have been done in plants and none in peach (Prunus persica L. Batsch). Therefore, the present study was conducted to identify suitable reference gene(s) for normalization of gene expression in peach.
Results
In this work, eleven reference genes were investigated in different peach samples using RT-qPCR with SYBR green. These genes are: actin 2/7 (ACT), cyclophilin (CYP2), RNA polymerase II (RP II), phospholipase A2 (PLA2), ribosomal protein L13 (RPL13), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 18S ribosomal RNA (18S rRNA), tubblin beta (TUB), tubblin alpha (TUA), translation elongation factor 2 (TEF2) and ubiquitin 10 (UBQ10). All eleven reference genes displayed a wide range of Cq values in all samples, indicating that they expressed variably. The stability of these genes except for RPL13 was determined by three different descriptive statistics, geNorm, NormFinder and BestKeeper, which produced highly comparable results.
Conclusion
Our study demonstrates that expression stability varied greatly between genes studied in peach. Based on the results from geNorm, NormFinder and BestKeeper analyses, for all the sample pools analyzed, TEF2, UBQ10 and RP II were found to be the most suitable reference genes with a very high statistical reliability, and TEF2 and RP II for the other sample series, while 18S rRNA, RPL13 and PLA2 were unsuitable as internal controls. GAPDH and ACT also performed poorly and were less stable in our analysis. To achieve accurate comparison of levels of gene expression, two or more reference genes must be used for data normalization. The combinations of TEF2/UBQ10/RP II and TEF2/RP II were suggested for use in all samples and subsets, respectively.
Similar content being viewed by others

Background
Reverse transcriptase quantitative real-time polymerase chain reaction (RT-qPCR) has become a very powerful technique for detection and quantification of mRNA transcription levels of a selected gene of interest [1, 2] due to its high sensitivity, specificity, reproducibility, no post-PCR processing and broad dynamic range [3], which allows a straightforward comparison between RNAs that differ widely in their abundance. To accurately quantify gene expression, many experimental variations should be taken into account, such as quality and amount of starting material, presence of inhibitors in different sample materials, primer design, and RNA extraction and retrotranscription efficiencies [1]. Therefore, selection of an appropriate normalization strategy is of crucial importance for the acquisition of biological meaningful data. Among several methods proposed so far [1, 4], reference genes are the most frequently used to normalize RT-qPCR data and to control the experimental possible errors generated in the quantification of gene expressions, since the reference genes are exposed to the same preparation steps as the gene of interest.
An ideal reference gene, known as an internal control gene or as reference gene, should be expressed at a constant level across various conditions, such as developmental stages or tissue types, and its expression is assumed to be unaffected by experimental parameters [5, 6]. Moreover, the reference gene and the target gene should have similar ranges of expression in the samples to be analyzed [7]. However, several recent studies have scrutinized the stability of commonly known reference genes like 18S ribosomal RNA (18S rRNA), β-actin (ACT), and glyceraldehydes-3-phosphate dehydrogenase (GAPDH) used for the quantification of mRNA expression, and have documented that these genes should be used with caution as internal controls, because they showed different behaviors under different experiment conditions [8–13]. The reason for these expressional variabilities may be that the reference genes not only participate in the basic cell metabolism but also take part in other cellular process [14, 15]. If the chosen reference gene has a large expression fluctuation, the normalization will lead to erroneous gene expression profiles of the target gene of interest [16, 17]. In addition, the choice of a suitable control gene will depend on the scope and nature of the experiment to be performed [6]. Therefore, the selection of the most stable gene or set of genes as internal controls is a critical step to control the variability between samples for quantitative gene expression studies with a sensitive RT-qPCR technique [8].
Recently, a growing number of published articles reflect the importance of reference genes and the need to validate them for each particular experimental model. Nevertheless, most of these studies mainly deal with human or animal tissues. However, only a few have concerned plants such as wheat [18], barley [19], rice [20–22], potato [13], soybean [23], grape [24], poplar [25], tomato [26, 27], coffee [28] and Arabidopsis thaliana [29, 30]. To the best of our knowledge, there have been no reports on the suitability of reference genes for RT-qPCR studies of differential expression of genes in peach (Prunus persica L. Batsch).
Peach fruit development and ripening are complex processes involving major changes in fruit metabolism [31]. Biochemical processes occur in a well-defined order under the control of a series of ripening-related genes leading to considerable changes in texture, pigmentation, taste and aroma [32]. The understanding of expression patterns of some key genes will help illuminate the mechanism involved in these processes in fleshy fruit and improve peach fruit quality and storage potential. Furthermore, studies of the molecular events associated with the ripening response of peach fruit to various exogenous regulators and melting and non-melting flesh genotypes may also help elucidate what contributes to fruit ripening.
The aim of this research was to select and evaluate the stability of 11 reference genes for the purpose of normalization in studying peach gene expression. Statistical methods implemented in geNorm [33], BestKeeper [34] and NormFinder [35] were used.
Results
To identify the best reference genes for studies of peach gene expression, a RT-qPCR assay, based on SYBR green detection, was designed for the transcription profiling of the eleven genes (18S rRNA, ACT, CYP2, TEF2, GAPDH, PLA2, RP II, RPL13, TUA, TUB and UBQ10, Table 1). The specificity of the amplifications was confirmed by the presence of a single band of expected size for each primer pairs in agarose gels following electrophoresis (data not shown) and by the single-peak melting curves of the PCR products. The melting temperatures of all PCR products were given in Table 2. No primer dimers or other products were resulted from non-specific amplification. No signals were detected in the minus RT and no-template controls. Efficiency of PCR reactions varied from 1.671 for RPL13 to 1.828 for ACT, and correlation coefficients ranged between 0.9952 and 0.9996 for RPL13 and RP II, respectively (Table 2).
Expression profiles of reference genes
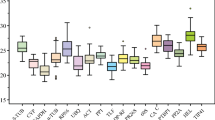
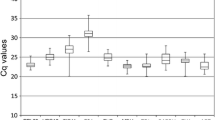
Analysis of the raw expression levels across all samples identified some variation amongst reference genes (Figure 1). Quantification cycle (Cq) [36] values for the 11 genes studied ranged from 8.2 to 30.9, while the majority of these values were between 18.6 and 24.6. The gene encoding 18S rRNA was highly expressed compared to the protein coding genes, reaching threshold fluorescence after only 8.2 amplification cycles, whereas the Cq average of all reference genes within the datasets was approximately 20.7 cycles. As a result, the 18S rRNA transcript levels were around 5700-fold more abundant than the dataset's average. The least abundant transcripts were PLA2 and RPL13, with Cq values of 27.6 and 30.9, respectively. The individual reference gene had different expression ranges across all studied RNA samples. ACT, TUA and TUB showed smaller gene expression variation (below 7 cycles) among studied reference genes, while PLA2, CYP2 and RPL13 had much higher expression variations (above 10 cycles). The wide expression ranges of the eleven tested reference genes confirmed that no single reference gene had a constant expression in different peach samples. Therefore, it is of utmost importance to select a reliable reference gene to normalize gene expression under a certain condition. Due to low expressed genes where Cqs were obtained around cycles 30–35 can lead to large variability 34, the candidate RPL13 was discarded from subsequent tests.

RT-qPCR C q values for reference genes. Expression data displayed as Cq values for each reference gene in all peach samples. A line across the box is depicted as the median. The box indicates the 25th and 75th percentiles. Whiskers represent the maximum and minimum values.
GeNorm analysis
Gene expression stability (M) of these ten reference genes studied was calculated using the software geNorm [33, 37]. The program is a Visual Basic application tool for Microsoft Excel and relies on the principle that the expression ratio of two perfect reference genes should be constant throughout the different experimental conditions or cell types. The M value is defined as the average pair-wise variation of a certain gene with all other tested reference genes, whereas the variation of this certain reference gene to another is determined as the standard deviation of the log2-transformed expression level ratios. The average expression M values of the 10 reference genes were plotted in Figure 2. The gene with the lowest M value is considered as the most stable expression, while the highest M value has the least stable expression. When all the samples were taken together, as shown in Figure 2A, the average expression stability value (M) of TEF2 and UBQ10 was the lowest, and that of CYP2 was the highest, indicating that TEF2 and UBQ10 had the most stable expression and that CYP2 was expressed most variably. TEF2 and UBQ10 were still the most stable genes, while 18S rRNA was the one with the highest M value, suggesting that it was variably expressed in fruit developmental stages (Figure 2B). In the different genotype samples, TEF2 and RP II were the most stable genes, while 18S rRNA was the least stable one (Figure 2C). The results remained very similar in the different storage time series, with the lowest M value for RP II and TEF2 and the highest M value for 18S rRNA (Figure 2D). TEF2 and RP II were still expressed much more stably than the other reference genes in different exogenous regulator treatments, while CYP2 was the least stable reference gene as in all samples (Figure 2E). CYP2 and TEF2 were two best genes among the ten tested reference genes, while 18S rRNA was the most variable one in different tissue samples of peach (Figure 2F).

Gene expression stability and ranking of the ten reference genes as calculated by geNorm. Expression stability and ranking of ten reference genes calculated with geNorm in all the samples (A), fruit developmental series (B), different genotypes (C), different storage time series (D), different exogenous regulator treatments (E), different tissues (F). A lower average expression stability M value indicates more stable expression.
Although most authors agree in using only one single gene as an internal control for normalization, it has been suggested that the use of two or more reference genes for RT-qPCR studies might generate more reliable results [33, 38]. To evaluate the optimal number of genes required for accurate normalization, pairwise variations Vn/Vn+1 between consecutively ranked normalization factors are calculated to determine the effect of adding the next reference gene in normalization. The normalization factors are defined by calculating the geometric mean of the 3 most stable gene relative quantities and stepwise inclusion of the other genes in the order of their expression stability. A large pairwise variation implies that the added gene has a significant effect on normalization and should be included for calculation of a reliable normalization factor [33]. As shown in Figure 3(B, C, D, E, F), the inclusion of the third reference gene did not contribute significantly to the variation of the normalization factor (V3/4 < 0.15). Based on the cut-off value of 0.15 proposed by geNorm program, below which the inclusion of an additional reference gene is not required, so the two most stable reference genes of each series subset would be sufficient for accurate normalization. When all the samples were taken together, the pairwise variation V2/3 was 0.213, higher than 0.15, while V3/4 was 0.146 (Figure 3A), indicating that the addition of the third reference gene was necessary to normalize gene expression. The 3 reference genes were TEF2, UBQ10 and TUB for this group samples. The recommended combinations of control genes of each sample series had mean stability values, M ≤ 1.0 and M ≤ 0.5, which are acceptable for heterogeneous and homogeneous sample panels, respectively [39]. Important to note is that the pairwise variation V and mean M values, calculated by the method of Exposito-Rodriguez et al. [27], for the combinations of TEF2/UBQ10/RP II and TEF2/RP II in all the sample pools and the series of fruit developmental and different tissue samples respectively, were all inside the ranges described as above (data not shown).

Determination of the optimal number of reference genes. Pairwise variation calculated by geNorm to determine the minimum number of reference genes for accurate normalization in all the samples (A), fruit developmental series (B), different genotypes (C), different storage time series (D), different exogenous regulator treatments (E), different tissues (F). Every bar represents change in normalization accuracy when stepwise adding more endogenous reference genes according to the ranking in Figure 2.
NormFinder analysis
NormFinder, another VBA applet, uses a model-based approach for identifying the optimal normalization gene(s) among a set of candidates. More stable gene expression is indicated by lower average expression stability values. In this mathematical model, estimation of both intra- and inter-group variation and a separate analysis of the sample subgroups in expression levels are included into the calculation of a gene expression stability value [35]. In this sense, five sample-subgroups were established as geNorm analysis. Moreover, expression data were also combined into "vegetative" (stems, roots, leaves, flowers and fruit at different developmental stages) and "mature" (including fruit obtained from different exogenous regulator treatments and two cultivars at different storage time) sample-subgroups. At the same time, all samples with no subgroups and the other five series were analyzed using this approach as well. The results of the NormFinder analysis were shown in Table 3 and Additional file 1. It is noteworthy that definition of sample-subgroups had a notable effect on NormFinder output. However, the NormFinder output with different sample-subgroups and no subgroups exhibited two common features: 1) TEF2, RP II and UBQ10 showed a remarkable stability of their expression levels and were always classified among the top four positions; 2) GAPDH, PLA2, ACT and 18S rRNA exhibited unstable expression profiles and were always included among the least stable reference genes. When it came to the other sample series, TEF2 was calculated to be the most stable single gene with a stability value of 0.007, while 18S rRNA was the most variable one in different exogenous regulator treatments. The results were broadly similar to the series of different tissues, with the highest stability value for TEF2 and lowest stability value for 18S rRNA. RP II was the most reliable gene in fruit developmental and different storage time series, while 18S rRNA remained the most variable one. In different genotype samples, TUB was identified as the most stable gene and PLA2 was the least one, with stability values of 0.002 and 0.053, respectively.
BestKeeper analysis
BestKeeper, an Excel-based tool, estimates inter-gene relations of possible reference gene pairs by performing numerous pairwise correlation analyses using raw Cq values of each gene. More important, all genes may be included in the calculation of the BestKeeper index, which can be used to rank the best reference genes because of stable reference genes showing a strong correlation with the BestKeeper index [34]. The results of the method analysis of the same data set were presented in Table 4 and Additional file 2. The 10 reference genes studied in our analysis correlated well one with another, if also compared with the BestKeeper index, except for 18S rRNA in different tissue samples. Particularly strong inter-gene correlations were found for the four most stable reference genes in all the sample pools (r > 0.89), especially in the other five series (r > 0.95). The high Pearson's coefficients of correlation indicated that these gene pairs had very similar overall expression patterns. When the complete data set was analyzed, TEF2, UBQ10, and RP II had strong correlation with the BestKeeper index (r > 0.95), and ranked among the top four genes, in accordance with the corresponding NormFinder output, thus identifying these three genes as the most reliable reference genes for normalization. The result was identical to the series of different tissues, different genotypes, different regulator treatments and fruit developmental stages, because of TEF2, UBQ10, and RP II were still included among the 4 top-ranked reference genes. In different storage time series, only TEF2 and RP II were classified among the top four genes. GAPDH, PLA2, and 18S rRNA consistently ranked poorly in the six series, and were identified as the least reliable reference genes.
Discussion
The reliability of RT-qPCR data will be greatly improved by inclusion of a reference gene whose transcription level should be invariable in the different experimental conditions [4]. The present study is the first detailed survey on the stability of a large number of genes used as internal controls for RT-qPCR studies of differential expression of genes in peach.
Several approaches have been proposed to identify stability of gene expression and select the best reference genes in the context of the relevant experimental conditions [33–35, 40–45], but to date, there is no consensus on which method should be used to examine reference gene expression stability. A comparison of different algorithms of reference gene selection allows a better evaluation of the most reliable controls and reduces the risk of artificial selection of co-regulated transcripts [46]. In order to select suitable reference gene(s) for accurate normalization, we compared three different statistical approaches, geNorm, NormFinder and BestKeeper to evaluate ten reference genes in peach.
The geNorm software is highly dependent on the assumption that none of the genes being analyzed are co-regulated as this would lead to an erroneous choice of optimum normaliser pair [35]. An obvious prediction about behavior of two co-regulated genes in the software is that they will occupy closed positions in the ranking [27]. In order to investigate whether the potential co-regulated genes TUA and TUB affected the outcome of our results, we removed one of them out of analysis and could not see any difference in the results, showing that in our data co-regulation did not affect the ranking of reference genes by stability. It should be worth mentioning that reference genes belonging to the same functional class that are not top-ranked by geNorm software in many previous studies [27]. Since it is very difficult to foresee common expression patterns, the stability of each reference gene expression was further assessed by NormFinder and BestKeeper that are less sensitive towards co-regulation of the reference genes.
The most prominent observation after completing the three analysis softwares was that each produced a different set of top ranked reference genes, and a fact that was not unexpected because the three programs based on different algorithms and analytical procedures. Generally, the analyses found that TEF2, UBQ10 and RP II were the most reliable internal controls for accurate normalization when looking at the expression data set as a whole, because these three genes were always classified among the 4 best performing reference genes except for RP II analyzed by geNorm in all the sample pools. For the other five series, TEF2 and RP II always ranked on top positions, exhibited stable expression patterns, and could serve as internal controls. On the other hand, 18S rRNA and PLA2 ranked poorly based on all the three software programs, indicating that these two genes were not consistently expressed and should be avoided as internal controls when doing gene expression studies in our experimental setup.
TEF2 and RP II were abundantly and constantly transcribed in all of the peach samples. Indeed, these two genes are known to be required for elongation and mRNA transcription in eukaryotes, respectively [43, 47]. So TEF2 and RP II remained continuously expressed over the different measured tissues and showed minimal changes in RNA transcription under different conditions. Regarding UBQ10, it was suggested to be an inappropriate internal control for RT-qPCR studies in different tissues at different developmental stages in rice [21] and soybean [23]. However, in an earlier study in Arabidopsis [29] and tomato [48], UBQ10 showed highly stable expression. But in the current study, results from all the three software analysis showed that UBQ10 underwent variation according to the experimental conditions. Consequently, it should be used with caution as an internal control. An ubiquitin tag is not only used to mark particular proteins for proteolytic elimination, but also has non-proteolytic functions [49] which may affect its level of expression in different plants. Based on the results from three software analysis, CYP2, stable in different peach tissue samples, was not the most stable in the other five series. Similarly, CYP2 was not among the best reference genes in any of the earlier analyses [13, 24]. The reason may be that CYP expression is significantly regulated by development or exposure to certain stress inducers, such as ethephon, salicylic acid in plants [50]. Other reference genes, like TUB, TUA, and PLA2 displayed unacceptably variable expression patterns, limiting their use as internal controls. Surprisingly, TUA showed highly stable expression in tested tissue samples of poplar among the 10 reference genes [25]. Taken together, these results suggested that a reference gene with stable expression under a certain condition may not be suitable to normalize gene expression under another condition, that is to say, reliable reference genes are highly specific for a particular experimental situation, thus requiring a careful evaluation for every individual experimental setup.
The most striking result was the poor performance of the most popular reference genes. GAPDH has been the one that is widely used in many areas of research [9] and is one of the best reference genes for measuring the gene expression in many tissues [24, 28, 51]. However, there have been also previous examples of this gene leading to wrong results due to its lack of stability in specific experimental conditions [9, 21]. In present analysis, GAPDH was not among the best reference genes between experimental groups. Reasons for those discrepancies may be that GAPDH not only acts as a component of the glycolytic pathway but also takes part in other processes as well. Thus, the expression profile of GAPDH might fluctuate according to the corresponding experimental conditions. Another most commonly used reference gene, 18S rRNA, performed worst and were not among the more stable genes in our tests. The poor stability of 18S rRNA in broomrape tissues was also found by Gonzalez-Verdejo et al. [52]. Previously, the 18S rRNA gene was considered to be an ideal internal control in RT-qPCR analysis [53]. However, there are several arguments against the use of 18S rRNA as an internal control. Its high abundance compared with target mRNA transcripts makes it difficult to subtract the baseline value in RT-qPCR data analysis accurately [33], and also makes it necessary to dilute the cDNA samples prior to real-time analysis, thus risking dilution errors [54]. Again, 18S rRNA can not be used as a reference gene when reverse transcription reaction is carried out using oligo-dT primers or only mRNA is used as template [21]. Furthermore, 18S rRNA synthesis is also regulated [55]. It is precisely for these reasons that 18S rRNA has failed to replace the use of other reference genes [56]. ACT, the third mainly used reference gene, has been widely used as reference gene in gene expression studies in many organisms. Nevertheless, recent studies revealed that ACT did not satisfy certain basic requirements for application as an internal control [13, 57]. Our analysis also showed that ACT was not the best reliable gene for comparative expression analysis. This may partly be explained by the fact that ACT, one of the major components of cytoplasmic microfilaments in eukaryotic cells, not only supports the cell and determines its shape but also participates in other cellular functions [56]. These results confirmed, once more, the need to evaluate reference genes in each experimental setting.
Earlier studies on the physiology of peach ripening have indicated that ethylene, abscisic acid (ABA), jasmonic acid (JA), 1-methylcyclopropene (1-MCP) and indole acetic acid (IAA) could modulate ripening [58–61]. However, the effects of those regulators on the expression of ripening-related genes, such as pectate lyase (PL), expansin (EXP) galactosidase (GAL), lipoxygenase (LOX), and so on, have not been elucidated in peach in detail. Moreover, the transcript levels of these genes in melting and non-melting flesh cultivars are quite variable. Studies of the molecular events associated with the ripening responses of fruit to various exogenous regulators and different genotypes will be beneficial in improving peach fruit quality and storage potential. In the present study, based on geNorm, NormFinder and BestKeeper methods, the most stable reference genes in the different cultivar and treatment samples were TEF2 and RP II. Analyses by geNorm applet suggested that the combination of the two genes was the optimal set of internal controls for studying differential gene expression in peach by RT-qPCR under the two conditions. Using the most reliable reference genes for normalization would be helpful to understand the molecular mechanisms involved in peach fruit ripening for different genotypes and regulator treatments.
Conclusion
Our data showed that expression stability varied considerably between genes in different tissue samples and under different experimental conditions in peach. Using the software applications BestKeeper, geNorm and NormFinder, TEF2, UBQ10 and RP II appeared to be the three most suitable reference genes for all the sample pools, and TEF2 and RP II for the other series, while 18S rRNA, RPL13 and PLA2 seemed to be unsuitable as internal controls. GAPDH and ACT also performed poorly and were less stable in our analysis. In order to get the most reliable results in peach gene profiling studies, more than one reference gene was recommended as internal controls for relative gene quantification. These results may provide a guideline for future works on gene expression in peach using RT-qPCR.
Methods
Plant materials and treatments
Tissues of Yuhua 1, a melting flesh peach genotype, were sampled from 8-year-old trees growing in national germplasm orchard of Institute of Horticulture of Jiangsu Academy of Agricultural Sciences, Nanjing, Jiangsu, China. Vegetative tissue samples, such as root, leaf and stem, were taken from young tissue; flowers were harvested at full bloom; and fruit at different developmental stages were taken at 2-week intervals after anthesis over the growing season. At each sampling time, plant materials except for fruits were frozen in dry ice after immediately harvesting transported to the laboratory at Nanjing Agricultural University and then stored at -70°C until total RNA was isolated.
Fruit of Jingyu peach, a non-melting flesh peach genotype, was from Institute of Forestry and Pomology, Beijing Academy of Agricultural and Forestry Sciences, China. At a stage equivalent to commercial ripeness, at about 9.56% and 12.16% soluble solids concentration (SSC) for fruits of Jingyu and Yuhua 1, respectively, free from visual symptoms of any disease or blemishes were chosen and directly stored at 25 ± 1°C for 6 days. For 1-methylcyclopropene (1-MCP) and ethylene treatments, Yuhua 1 fruits were sealed in two closed airtight containers, and 1 μl/L 1-MCP and 100 μl/L ethylene were injected into the two containers through a rubber septum, respectively. Fruits were incubated with 1-MCP or ethylene for 24 h at 25 ± 1°C and then containers were open to allow ripening in air in the same temperature conditions. Fruits of Yuhua 1 were also treated with ABA and IAA by dipping in 100 μM solutions in 0.2% Tween 80 for 20 min, respectively. In the JA treatment, Yuhua 1 fruits were dipped for 10 min in 50 mM methyl-jasmonate (MeJA) solution which also contained 0.2% Tween 80. After being dipped, fruits were dried and then allowed ripening at 25 ± 1°C in the air. Sampling from fruits of the two cultivars in normal room temperature was carried out daily for 6 days. Samples of Yuhua 1 fruit from different storage were used to evaluate the stability of 11 reference genes, while Jingyu fruit samples were only used to study the effect of different genotypes on stability values of 11 reference genes. Samples for different regulator treatments were taken for day 3, including one sample taken before treatment. All experiments were replicated three times with ten fruit as an experiment unit for gene expression studies. For all fruit samples epicarp and endocarp were excluded and the mesocarp was frozen in liquid nitrogen and stored at -70°C until further use.
Total RNA extraction
Total RNA was isolated according to the method described by Meisel et al. [62]. Genomic DNA was eliminated by treating each sample with RNase-free DNase I (TaKaRa, Japan) according to the instructions manual. The concentration of isolated total RNA was calculated from absorbance at 260 nm with BioPhotometer (Eppendorf, Hamburg, Germany), the purity was verified by optical density (OD) absorption ratio OD260 nm/OD280 nm between 1.80 and 2.05, and OD260 nm/OD230 nm ranging from 2.00 to 2.60 and the integrity was evaluated by electrophoresis on ethidium bromide-stained 1.0% agarose gels. Intact rRNA subunits of 18S and 28S were observed on the gel and absence of smears indicating minimal degradation of the RNA.
First strand cDNA synthesis
One microgram RNA was reverse-transcribed using the SYBR PrimeScript RT-PCR kit II (TaKaRa, Japan) for first-strand cDNA synthesis with 2.5 μM oligonucleotide dT primer and 5 μM random hexamer priming method according to the manufacturer's recommendations. Before transcription, RNA and primers were mixed and incubated at 70°C for 5 min followed by cooling on ice immediately. The first strand cDNA synthesis was started after adding transcription mixture at 37°C lasting 15 min for reverse transcriptase reaction. Finally, the PrimeScript Reverse Transcriptase was inactivated by heating the reaction mixture for 5 sec at 85°C. Each RNA sample was controlled for genomic DNA contamination without reverse transcriptase addition into cDNA synthesis mixture. All cDNA samples were stored at -20°C and diluted 1:10 with RNase-free water before being used as template in RT-qPCR analysis.
Selection of peach sequences and primer design
Eleven genes were selected for investigation to identify the most stably expressed reference gene(s) to be used in RT-qPCR studies. This group of genes comprised several classical reference genes which are the most commonly used as internal control for expression studies, such as GAPDH, 18S rRNA and ACT, the others based on previous reports [25, 43]. The peach EST database [63] was queried with Arabidopsis protein sequences using TBLASTN to select peach homologs of genes commonly used as internal controls for gene expression analysis. The chosen peach ESTs were then used to query the Arabidopsis protein database using BLASTX [64] to obtain the description of peach reference genes. The reference genes evaluated are listed on Table 1, as are the corresponding accession numbers, Arabidopsis homolog locus, Arabidopsis locus description and main functions.
Primer pairs for RT-qPCR amplification were designed based on selected sequences using Beacon Designer 7.0 software (Premier Biosoft International, Palo Alto, California, USA) with a melting temperature between 60–62°C, 20–26 bp and about 50% GC content. Amplicon lengths were optimized to 103–146 bp to ensure optimal polymerization efficiency and minimize the impact of RNA integrity on relative quantification of gene expression [65]. MFOLD software [66] was subsequently used to evaluate the target sequences amplified by the primer pairs to avoid the formation of secondary structures at the site of primer binding. The primers were further used to query peach EST database with BLASTN to confirm the identity of the genes. Before RT-qPCR, each primer pair was tested via standard RT-PCR to check for size specificity of the amplicon by 2.5% agarose gel electrophoresis and ethidium bromide staining. In addition, target amplicons were sequenced to confirm specificity of the PCR products. The primer sequences, amplicon sizes, and melting temperatures of all PCR products were indicated in Table 2.
RT-qPCR with SYBR green
RT-qPCR was performed using a Rotor-Gene 3000 (Corbett Robotics, Australia) and the SYBR Green Real-time Master Mix (TOYOBO, Japan). The PCR reaction volume was 20 μL containing 1.5 μL of diluted cDNA and 0.2 μM of each primer. Thermocycling conditions were set as an initial polymerase activation step for 2 min at 95°C, followed by 45 cycles of 15 sec at 94°C for template denaturation, 15 sec at 60°C for annealing and 20 sec at 72°C for extension and fluorescence measuration. Afterwards, a dissociation protocol with a gradient from 57°C to 95°C was used for each primer pair to verify the specificity of the RT-qPCR reaction and the absence of primer dimer. In addition, each PCR reaction included a reverse transcription negative control to check for potential genomic DNA contamination. Reagent contamination was also detected by a reaction mix without template. All samples were amplified in triplicates and the mean was used for RT-qPCR analysis.
Data analysis
Expression levels of the tested reference genes were determined by the number of amplification cycles (Cq) needed to reach a specific threshold level of detection. All amplification plots were analyzed with a threshold fluorescence value of 0.1 to obtain Cq values using the Rotor-Gene software version 6.1 (Corbett Research). The PCR efficiency showed in Table 2 was calculated for each gene with LinRegPCR program [67] from raw fluorescence data taken from the Rotor-Gene 3000 detection system. Results from the LinRegPCR and Rotor-Gene software were imported into Microsoft Excel and transformed to relative quantities using the comparative Cq method and specific efficiencies for each gene [68]. The data obtained were converted into correct input files, according to the requirements of the software, and analyzed using three different VBA applets, geNorm (version 3.4) [33, 37], NormFinder (version 0.953) [35, 69] and BestKeeper (version 1.0) [34, 70].
References
Ginzinger DG: Gene quantification using real-time quantitative PCR: An emerging technology hits the mainstream. Exp Hematol. 2002, 30 (6): 503-512.
Bustin SA: Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol. 2002, 29 (1): 23-39.
Garson JA, Grant PR, Ayliffe U, Ferns RB, Tedder RS: Real-time PCR quantitation of hepatitis B virus DNA using automated sample preparation and murine cytomegalovirus internal control. J Virol Methods. 2005, 126 (1–2): 207-213.
Huggett J, Dheda K, Bustin S, Zumla A: Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005, 6 (4): 279-284.
Schmittgen TD, Zakrajsek BA: Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods. 2000, 46 (1–2): 69-81.
Thellin O, Zorzi W, Lakaye B, De Borman B, Coumans B, Hennen G, Grisar T, Igout A, Heinen E: Housekeeping genes as internal standards: use and limits. J Biotechnol. 1999, 75 (2–3): 291-295.
Cappelli K, Felicetti M, Capomaccio S, Spinsanti G, Silvestrelli M, Supplizi AV: Exercise induced stress in horses: selection of the most stable reference genes for quantitative RT-PCR normalization. BMC Mol Biol. 2008, 9: 49-
Dheda K, Huggett J, Bustin SA, Johnson MA, Rook G, Zumla A: Validation of housekeeping genes for normalizing RNA expression in real-time PCR. BioTechniques. 2004, 37: 112-119.
Suzuki T, Higgins PJ, DR C: Control selection for RNA quantitation. BioTechniques. 2000, 29: 332-337.
Bas A, Forsberg G, Hammarstrom S, Hammarstrom ML: Utility of the housekeeping genes 18S rRNA, beta-actin and glyceraldehyde-3-phosphate-dehydrogenase for normalization in real-time quantitative reverse transcriptase-polymerase chain reaction analysis of gene expression in human T lymphocytes. Scand J Immunol. 2004, 59 (6): 566-573.
Yperman J, De Visscher G, Holvoet P, Flameng W: Beta-actin cannot be used as a control for gene expression in ovine interstitial cells derived from heart valves. J Heart Valve Dis. 2004, 13 (5): 848-853.
Barber RD, Harmer DW, Coleman RA, Clark BJ: GAPDH as a housekeeping gene: analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol Genomics. 2005, 21 (3): 389-395.
Nicot N, Hausman JF, Hoffmann L, Evers D: Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J Exp Bot. 2005, 56 (421): 2907-2914.
Ishitani R, Sunaga K, Hirano A, Saunders P, Katsube N, Chuang DM: Evidence that glyceraldehyde-3-phosphate dehydrogenase is involved in age-induced apoptosis in mature cerebellar neurons in culture. J Neurochem. 1996, 66 (3): 928-935.
Singh R, Green M: Sequence-specific binding of transfer RNA by glyceraldehyde-3-phosphate dehydrogenase. Science. 1993, 259 (5093): 365-368.
Dheda K, Huggett JF, Chang JS, Kim LU, Bustin SA, Johnson MA, Rook GAW, Zumla A: The implications of using an inappropriate reference gene for real-time reverse transcription PCR data normalization. Anal Biochem. 2005, 344 (1): 141-143.
Tatsumi K, Ohashi K, Taminishi S, Okano T, Yoshioka A, Shima M: Reference gene selection for real-time RT-PCR in regenerating mouse livers. Biochem Biophys Res Commun. 2008, 374 (1): 106-110.
Paolacci A, Tanzarella O, Porceddu E, Ciaffi M: Identification and validation of reference genes for quantitative RT-PCR normalization in wheat. BMC molecular biology. 2009, 10 (1): 11-
Burton RA, Shirley NJ, King BJ, Harvey AJ, Fincher GB: The CesA Gene Family of Barley. Quantitative Analysis of Transcripts Reveals Two Groups of Co-Expressed Genes. Plant Physiol. 2004, 134 (1): 224-236.
Ding J, Jia J, Yang L, Wen H, Zhang C, Liu W, Zhang D: Validation of a rice specific gene, sucrose phosphate synthase, used as the endogenous reference gene for qualitative and real-time quantitative PCR detection of transgenes. J Agric Food Chem. 2004, 52 (11): 3372-3377.
Jain M, Nijhawan A, Tyagi AK, Khurana JP: Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochem Biophys Res Commun. 2006, 345 (2): 646-651.
Kim BR, Nam HY, Kim SU, Kim SI, Chang YJ: Normalization of reverse transcription quantitative-PCR with housekeeping genes in rice. Biotechnol Lett. 2003, 25 (21): 1869-1872.
Jian B, Liu B, Bi Y, Hou W, Wu C, Han T: Validation of internal control for gene expression study in soybean by quantitative real-time PCR. BMC Mol Biol. 2008, 9 (1): 59-
Reid KE, Olsson N, Schlosser J, Peng F, Lund ST: An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT-PCR during berry development. BMC Plant Biol. 2006, 6 (1): 27-
Brunner AM, Yakovlev IA, Strauss SH: Validating internal controls for quantitative plant gene expression studies. BMC Plant Biol. 2004, 4 (1): 14-
Coker JS, Davies E: Selection of candidate housekeeping controls in tomato plants using EST data. BioTechniques. 2003, 35: 740-748.
Exposito-Rodriguez M, Borges A, Borges-Perez A, Perez J: Selection of internal control genes for quantitative real-time RT-PCR studies during tomato development process. BMC Plant Biology. 2008, 8 (1): 131-
Barsalobres-Cavallari C, Severino F, Maluf M, Maia I: Identification of suitable internal control genes for expression studies in Coffea arabica under different experimental conditions. BMC molecular biology. 2009, 10 (1): 1-
Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible W-R: Genome-wide identification and testing of superior reference genes for transcript normalization in arabidopsis. Plant Physiol. 2005, 139 (1): 5-17.
Remans T, Smeets K, Opdenakker K, Mathijsen D, Vangronsveld J, Cuypers A: Normalisation of real-time RT-PCR gene expression measurements in arabidopsis thaliana exposed to increased metal concentrations. Planta. 2008, 227 (6): 1343-1349.
Fischer RL, Bennett AB: Role of cell wall hydrolases in fruit ripening. Plant Mol Biol. 1991, 42: 675-703.
Fonseca S, Hackler L, Zvara Á, Ferreira S, Bald A, Dudits D, Pais MS, Puskás LG: Monitoring gene expression along pear fruit development, ripening and senescence using cDNA microarrays. Plant Sci. 2004, 167 (3): 457-469.
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F: Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3 (7): RESEARCH0034-
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP: Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper – Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004, 26 (6): 509-515.
Andersen CL, Jensen JL, Orntoft TF: Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64 (15): 5245-5250.
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT: The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009, 55 (4): 611-622.
geNorm Software. http://medgen.ugent.be/~jvdesomp/genorm/
Tricarico C, Pinzani P, Bianchi S, Paglierani M, Distante V, Pazzagli M, Bustin SA, Orlando C: Quantitative real-time reverse transcription polymerase chain reaction: normalization to rRNA or single housekeeping genes is inappropriate for human tissue biopsies. Anal Biochem. 2002, 309 (2): 293-300.
Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J: qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8: R19-
García-Vallejo JJ, Van het Hof B, Robben J, Van Wijk JAE, Van Die I, Joziasse DH, Van Dijk W: Approach for defining endogenous reference genes in gene expression experiments. Anal Biochem. 2004, 329 (2): 293-299.
Haller F, Kulle B, Schwager S, Gunawan B, Heydebreck Av, Sültmannd H, Füzesi L: Equivalence test in quantitative reverse transcription polymerase chain reaction: confirmation of reference genes suitable for normalization. Anal Biochem. 2004, 335 (1): 1-9.
Brunner AM, Yakovlev IA, Strauss SH: Validating internal controls for quantitative plant gene expression studies. BMC Plant Biology. 2004, 4 (1): 14-
Radonic A, Thulke S, Mackay IM, Landt O, Siegert W, Nitsche A: Guideline to reference gene selection for quantitative real-time PCR. Biochem Biophys Res Commun. 2004, 313 (4): 856-862.
de Kok JB, Roelofs RW, Giesendorf BA, Pennings JL, Waas ET, Feuth T, Swinkels DW, Span PN: Normalization of gene expression measurements in tumor tissues: comparison of 13 endogenous control genes. Lab Invest. 2005, 85 (1): 154-159.
Szabo A, Perou CM, Karaca M, Perreard L, Palais R, Quackenbush JF, Bernard PS: Statistical modeling for selecting housekeeper genes. Genome Biol. 2004, 5 (8): R59-
Ayers D, Clements D, Salway F, Day P: Expression stability of commonly used reference genes in canine articular connective tissues. BMC Veterinary Research. 2007, 3: 7-
Proud CG: Peptide-chain elongation in eukaryotes. Mol Biol Rep. 1994, 19 (3): 161-170.
Løvdal T, Lillo C: Reference gene selection for quantitative real-time PCR normalization in tomato subjected to nitrogen, cold, and light stress. Analytical Biochemistry. 2009, 387 (2): 238-242.
Hochstrasser M: Evolution and function of ubiquitin-like protein-conjugation systems. Nat Cell Biol. 2000, 2 (8): E153-157.
Marivet J, Frendo P, Burkard G: Effects of antibiotic stresses on cyclophilin in maize and bean and sequence analysis of bean cyclophilin cDNA. Plant Sci. 1992, 84: 171-178.
Iskandar H, Simpson R, Casu R, Bonnett G, Maclean D, Manners J: Comparison of reference genes for quantitative real-time polymerase chain reaction analysis of gene expression in sugarcane. Plant Mol Biol Rep. 2004, 22 (4): 325-337.
Gonzalez-Verdejo CI, Die JV, Nadal S, Jimenez-Marin A, Moreno MT, Roman B: Selection of housekeeping genes for normalization by real-time RT-PCR: analysis of Or-MYB1 gene expression in orobanche ramosa development. Anal Biochem. 2008, 379 (2): 176-181.
RT-PCR: The basic. http://www.ambion.com/techlib/basics/rtpcr/index.html
Takle GW, K TI, Brurberg MB: Evaluation of reference genes for real-time RT-PCR expression studies in the plant pathogen pectobacterium atrosepticum. BMC Plant Biol. 2007, 7: 50-
Vera MI, Norambuena L, Alvarez M, Figueroa J, Molina A, Leon G, Krauskopf M: Reprogramming of nucleolar gene expression during the acclimatization of the carp. Cell Mol Biol Res. 1993, 39 (7): 665-674.
Stürzenbaum SR, Kille P: Control genes in quantitative molecular biological techniques: the variability of invariance. Comp Biochem Phys B: Biochem Mol Biol. 2001, 130 (3): 281-289.
Gutierrez L, Mauriat M, Guenin S, Pelloux J, Lefebvre JF, Louvet R, Rusterucci C, Moritz T, Guerineau F, Bellini C, Van Wuytswinkel O: The lack of a systematic validation of reference genes: a serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnol J. 2008, 609-618. 6
Begheldo M, Ziliotto F, Rasori A, Bonghi C: The use of μPEACH 1.0 to investigate the role of ethylene in the initiation of peach fruit ripening. Advances in Plant Ethylene Research. 2007, 265-267.
Ziosi V, Bonghi C, Bregoli AM, Trainotti L, Biondi S, Sutthiwal S, Kondo S, Costa G, Torrigiani P: Jasmonate-induced transcriptional changes suggest a negative interference with the ripening syndrome in peach fruit. J Exp Bot. 2008, 59 (3): 563-573.
Liguori G, Weksler A, Zutahi Y, Lurie S, Kosto I: Effect of 1-methylcyclopropene on ripening of melting flesh peaches and nectarines. Postharvest Biol Tec. 2004, 31 (3): 263-268.
Trainotti L, Pavanello A, Zanin D: PpEG4 is a peach endo-beta-1, 4-glucanase gene whose expression in climacteric peaches does not follow a climacteric pattern. J Exp Bot. 2006, 57 (3): 589-598.
Meisel L, Fonseca B, González S, Baezayates R, Cambiazo V, Campos R, Gonzalez M, Orellana A, Retamales J, Silva H: A rapid and efficient method for purifying high quality total RNA from peaches (Prunus persica) for functional genomics analyses. Biol Res. 2005, 38: 83-88.
Peach EST database. http://compbio.dfci.harvard.edu/tgi/cgi-bin/tgi/gimain.pl?gudb=peach
TAIR BLAST. http://www.arabidopsis.org/Blast/index.jsp
Fleige S, MW P: RNA integrity and the effect on the realtime qRT-PCR performance. Mol Aspects Med. 2006, 27: 126-139.
Zuker M: Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31 (13): 3406-3415.
Ramakers C, Ruijter JM, Deprez RH, Moorman AF: Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett. 2003, 339 (1): 62-66.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001, 25 (4): 402-408.
NormFinder Software. http://www.mdl.dk/publicationsnormfinder.htm
BestKeeper Software. http://www.gene-quantification.de/bestkeeper.html
Acknowledgements
We gratefully acknowledge support of this work from the National Natural Science Foundation of China (303770987; 3067143). We also greatly appreciate Dr. (Max) Zong-Ming Cheng, Department of Plant Sciences, University of Tennessee for critical review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
ZT performed all the experimental procedures, data analysis, draft the manuscript and was the primary author of the manuscript. ZG and JZ assisted in manuscript revising and provided helpful discussions. FW performed the sample preparation and participated in tables and figures drawing. ZZ conceived and supervised the research, revised the manuscript and provided financial support. All authors read and approved the final manuscript.
Electronic supplementary material
12867_2008_439_MOESM1_ESM.pdf
Additional file 1: Stability values of reference genes calculated by NormFinder. File showing the stability values of the ten selected candidate reference genes calculated by NormFinder. (PDF 44 KB)
12867_2008_439_MOESM2_ESM.pdf
Additional file 2: Inter-gene relations and correlations between the reference genes and the BestKeeper index. The file shows pairwise correlation analyses were performed based on the Cq values of the ten reference genes. Pearson's correlation coefficients (r) are shown. All the correlations are over the significance threshold (p = 0.05). (PDF 91 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tong, Z., Gao, Z., Wang, F. et al. Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Molecular Biol 10, 71 (2009). https://doi.org/10.1186/1471-2199-10-71
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2199-10-71