
Abstract
Background
Transposable elements constitute a significant fraction of plant genomes. The PIF/Harbinger superfamily includes DNA transposons (class II elements) carrying terminal inverted repeats and producing a 3 bp target site duplication upon insertion. The presence of an ORF coding for the DDE/DDD transposase, required for transposition, is characteristic for the autonomous PIF/Harbinger-like elements. Based on the above features, PIF/Harbinger-like elements were identified in several plant genomes and divided into several evolutionary lineages. Availability of a significant portion of Medicago truncatula genomic sequence allowed for mining PIF/Harbinger-like elements, starting from a single previously described element MtMaster.
Results
Twenty two putative autonomous, i.e. carrying an ORF coding for TPase and complete terminal inverted repeats, and 67 non-autonomous PIF/Harbinger-like elements were found in the genome of M. truncatula. They were divided into five families, MtPH-A5, MtPH-A6, MtPH-D,MtPH-E, and MtPH-M, corresponding to three previously identified and two new lineages. The largest families, MtPH-A6 and MtPH-M were further divided into four and three subfamilies, respectively. Non-autonomous elements were usually direct deletion derivatives of the putative autonomous element, however other types of rearrangements, including inversions and nested insertions were also observed. An interesting structural characteristic – the presence of 60 bp tandem repeats – was observed in a group of elements of subfamily MtPH-A6-4. Some families could be related to miniature inverted repeat elements (MITEs). The presence of empty loci (RESites), paralogous to those flanking the identified transposable elements, both autonomous and non-autonomous, as well as the presence of transposon insertion related size polymorphisms, confirmed that some of the mined elements were capable for transposition.
Conclusion
The population of PIF/Harbinger-like elements in the genome of M. truncatula is diverse. A detailed intra-family comparison of the elements' structure proved that they proliferated in the genome generally following the model of abortive gap repair. However, the presence of tandem repeats facilitated more pronounced rearrangements of the element internal regions. The insertion polymorphism of the MtPH elements and related MITE families in different populations of M. truncatula, if further confirmed experimentally, could be used as a source of molecular markers complementary to other marker systems.
Similar content being viewed by others

Background
Transposable elements (TEs) are dispersed repetitive sequences constituting a major fraction of plant genomes, ranging from 10% of Arabidopsis thaliana genome [1], to an estimated value over 70% of maize genome [2]. Class I elements (retrotransposons), transposing via an RNA intermediate, form the most abundant fraction, while class II elements (DNA transposons), use a 'cut and paste' mechanism for transposition and are usually less numerous.
Advances in genome sequencing of model plant species enabled systematic, computer-based studies towards the identification of repetitive sequences, including those representing putative TEs. The presence of certain structural characteristics of particular groups of TEs allowed the development of a range of strategies for de novo or homology-based identification of novel elements. A number of methods for automatic mining of transposable elements were developed [3–6], To date, two model plant genomes, i.e. A. thaliana and Oryza sativa (rice) have been extensively studied [7–11].
Founder members of the PIF/Harbinger superfamily of class II TEs were identified in maize [12] and A. thaliana [7]. Other full-length elements were subsequently found in rice (Pong [13]), carrot, and M. truncatula (Master [14]). Autonomous PIF/Harbinger-like elements carry 14–25 bp long terminal inverted repeats (TIRs) flanked by 3 bp long (TTA/TAA) target site duplications (TSD), and a DDD/DDE transposase showing similarity to that of the bacterial IS5 insertion sequence. The group of PIF/Harbinger-like elements was shown to be widespread in the plant kingdom and composed of two easily distinguishable subgroups, i.e. PIF and Pong [15]. Elements representing both subgroups were related to certain miniature inverted repeat elements (MITEs), like Tourist in maize [12, 16] and mPING in rice [13].
Medicago truncatula (barrel medic) has been chosen as a model plant for the Fabaceae family, primarily to study relationships between plants and their symbiotic microbes. It has a relatively small genome of 500 to 600 Mbp [17], shows annual growth habit and self-fertility. The genome of M. truncatula has not been extensively analysed with respect to TE identification. A MITE element Bigfoot was reported in the genomes of M. truncatula and M. sativa [18], a set of Ty3/gypsy-like Ogre elements characteristic for legume species was described in M. truncatula [19], and several other M. truncatula elements were briefly characterized in Repbase Update database [20]. A recent study of another model legume, Lotus japonicus, identified a number of PIF- and Pong-like elements and a strong evidence for their recent amplification in the host genome [21].
In this paper we used the accumulated M. truncatula genomic sequence data to identify putative TEs belonging to the PIF/Harbinger superfamily and related to a previously characterized MtMaster element [14]. Therefore, our study was focused on identification and in-depth characterization of a strictly defined group of full-length (putative autonomous and non-autonomous) TEs carrying not only a PIF/Harbinger-specific transposase, but also a particular TIR motif characteristic of most of the PIF-like, but not of the Pong-like elements.
Results
Identification and phylogeny of PIF/Harbinger-like elements of M. truncatula
The initial search of the M. truncatula genome aimed at the identification of putative autonomous elements, i.e. those carrying an ORF showing homology to the predicted MtMaster TPase (transposase) protein sequence [14] and flanked with terminal inverted repeats of at least 14 bp, containing the G(N)5GTT motif, and followed by a 3 bp-long TSD (TAA or TTA). This resulted to 44 sequences showing significant homology (E-value < 10-20) to the TPase, after eliminating the redundancy coming from overlapping BACs. We obtained precisely the same hits using the whole TPase sequence and the DDE region, likely because of the very rigorous E-value threshold imposed during the search. Of the identified sequences, 22 were flanked by TIRs and TSDs characteristic for PIF/Harbinger-like elements and these were assumed to represent complete transposable elements. They ranged in length from 2,180 to 25,288 bp. In 11 of these elements, another coding region, similar to the MtMaster orf1 with E-value ranging from 10-4 to 10-99, could be found. The relative order of both ORFs was variable – five elements had orf1 upstream and six downstream the TPase (Table 1).
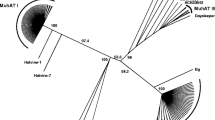
A phylogenetic analysis of the DDE domain region of the TPase revealed that the M. truncatula PIF/Harbinger-like elements could be divided into five lineages. Nine elements, including the previously described MtMaster, were grouped into lineage M, together with carrot DcMaster [14]. In seven of these elements the orf1 preceded the TPase as expected, while for the remaining two the orf1 was absent, most likely because of an internal deletion. Eight elements formed a new lineage designated as A6. Typically for the group A, the orf1 was located downstream the TPase in the five elements carrying both coding regions. Another new lineage, designated as E, was formed by two elements. In none of them could the orf1 be identified. Two other elements were included into lineage A5, together with maize ZmPIF [12] and one was placed into lineage D (Figure 1). However, in the latter case the orf1 was located downstream to TPase, contrary to previously described elements from that lineage [15].

Neighbor-joining tree representing the diversity of the M. truncatula PIF/Harbinger -like elements in relation with other previously identified TEs. Lineages are marked with color rectangles and letters, numbers show bootstrap values obtained using 1000 replicates.
Diversity and abundance of PIF/Harbinger-like elements in M. truncatula
In addition to the TPase-containing elements described above, using a strategy outlined in the Methods section, we identified additional 67 elements lacking any coding capacity and thus considered as non-autonomous. List of all identified elements and their coordinates are given in the Additional File 1. The grouping of the identified transposable elements was based on the full element sequence similarity or 5' and 3' terminal sequence similarity using two approaches: hierarchical clustering and multidimensional scaling (Additional File 2). This strategy allowed us to define families and subfamilies of PIF/Harbinger-like transposable elements in M. trunactula (Table 2), where families essentially reflected the lineages previously identified on the basis of the TPase phylogeny, and subfamilies grouped elements carrying homologous TIRs (Table 3) and showing a degree of overall DNA sequence similarity. For each but two subfamilies, one or two putatively autonomous core elements could be identified. The exception was a low copy number family MtPH-E, for which none of the elements contained a region homologous to the orf1.
The largest family, MtPH-A6, contained 54 elements, while family MtPH-D was represented only by a single element. The second most abundant family, containing 27 elements, was MtPH-M (Master), of which 18 was grouped into subfamily 3.
Detailed structure analysis of MtPH families
MtPH-A6 consisted of four subfamilies represented by putative autonomous elements sharing similar ORF organization, i.e. a TPase containing two introns, followed by orf1. MtPH-A6 TPases formed a well supported clade, containing four subclades with high bootstrap values, representing the corresponding subfamilies (Figure 1).
Subfamily MtPH-A6-1 contained nine elements ranging in length from 802 to 8,707 bp, the longest element carrying a nested insertion of the 7,555 bp long RAM12 gypsy-like retrotransposon.
Subfamily MtPH-A6-2 grouped six elements, 898 to 4,218 bp long, all being simple internal deletion derivatives of the core element MtPH-A6-2-Ia.
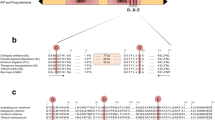
Sixteen elements belonged to subfamily MtPH-A6-3, ranging in length from 553 to 4,845 bp, except for a much larger, 23,892 bp long element MtPH-A6-3-IIIa, initially identified as being flanked by 15 bp TIRs unrelated to those of the MtPH-A6-3 subfamily. However, the element contained a 4.9 kb region 74% identical to the two elements mentioned above, but lacking the first 8 bases in the 5' TIR (Figure 2). Hence, the true boundaries of the elements could not be initially identified using our mining strategy. An interesting feature of that subfamily was the presence of a perfect microsatellite site in the first intron of the TPase. The three elements containing the region coding for the TPase, MtPH-A6-3-Ia, MtPH-A6-3-IIa, and MtPH-A6-3-IIIa had, respectively, 27, 8, and 21 repeats of the (TA)n core motif (Figure 2).

Structure of three elements representing family MtPH-A6-3. Arrows show terminal inverted repeats (TIRs), letters represent sequences of target site duplications (TSDs) and TIRs, solid lines show homologous regions with similarity rate written in italics, dotted lines show regions with no homology, numbers in bold show localization of nucleotide positions of important features, (TA) indicates presence of a microsatellite repeat, followed by the number of the core motif repeats.
MtPH-A6-4 subfamily members ranged in length from 431 to 25,288 bp. Among the 23 members of that subfamily, 18 were characterized by the presence of imperfect 60 bp long tandem repeats, variable in number, while in the remaining five elements the core repeat was entirely absent. Each repeat itself contained a triplicated AAACNNCTTATT motif. These elements contained from 2 to 35 repeats that in extreme cases covered almost the entire region between the TIRs (Figure 3A and 3B). In some elements, tandem repeats were present only in one subterminal region, while for the others they were present in both subterminal regions in opposite orientation. The 60 bp tandem repeats were identified in 27 other sites in the M. truncatula genome, initially not identified as occupied by MtPH-A6-4 elements. However, BLAST search with the terminal 214 bp + 3 bp TSD of the MtPH-A6-4-Ia and MtPH-A6-4-IIa elements indicated that in all instances at least one of the regions flanking the repeats showed residual homology to the TE terminus (E value < 1e-08). The presence of tandem repeats facilitated internal rearrangements resulting in inversions of the internal region (Figure 3C). Two nested insertions were identified in the longest element MtPH-A6-4-IIa, which showed three blocks of significant homology to the MtPH-A6-4-Ia core element, interrupted by an unidentified element of 2,191 bp carrying 15 bp TIRs and flanked by a 5 bp long TSD and a gypsy-like retrotransposon (Figure 3D).

VNTR regions, inversions, and nested insertions in elements belonging to family MtPH-A6-4. A. Consensus sequence of the 60 bp core VNTR motif, triplicated regions within the core motif are underlined, variable nucleotide positions within the triplicated motif are written in italics. B. Dot-plot and schematic representation of MtPH-A6-4-XXI, an example of TE carrying a large number of tandem repeats. Thick black arrowheads represent TIRs, gray arrows indicate localization and orientation of the VNTR region, number of repetitions is given below each arrow. C. Comparison of two elements containing an inversion of the internal region, thick black arrowheads show TIRs, gray arrows show localization of the VNTR, thin arrows indicate the orientation of the inverted region, solid lines represent homologous regions with similarity rates written in italics, dotted lines represent regions with no homology, numbers in bold show localization of nucleotide positions of important features. D. Organization of the long element MtPH-A6-4-IIa as compared to the core element MtPH-A6-4-Ia, thick black arrowheads show TIRs, solid lines represent homologous regions with percentages of similarity written in italics, dotted lines represent regions with no homology, numbers in bold show localization of nucleotide positions of important features, nested TEs are drawn above the MtPH-A6-4-IIa element.
MtPH-M family included three subfamilies with short (14 bp), similar TIRs and orf1 followed by TPase. Subfamily MtPH-M-1 contained only four elements, ranging in length from 812 to 5,140 bp. Two of them, MtPH-M-1-Ia (previously described as MtMaster [10]) and MtPH-M-1-IIa (showing 90% overall sequence identity to MtMaster) had both ORFs, and the remaining two were internally deleted derivatives.
Five elements were grouped into subfamily MtPH-M-2, three of them carrying both orf1 and TPase. The region containing element MtPH-M-2-IIa occurred to be a composite structure of two related TEs. The initially identified sequence flanked by TIRs and TSDs spanned over 21,696 bp. The 5,303 bp element MtPH-M-2-IIa occupied the 5' region of that sequence, however the downstream sequence also contained blocks of homology to the core element MtPH-M-2-Ia, and a nested insertion of a Gypsy-like retrotransposon (Figure 4). It indicates that an ancient copy of a TE related to those belonging to the subfamily MtPH-M-2 became a target for subsequent nested insertions. Other elements from that family ranged in length from 2,240 to 7,816 bp.

Mosaic structure of the MtPH-M-2-IIa element, as compared to the core element MtPH-M-2-Ia. Solid lines represent homologous regions with similarity rates written in italics, dotted lines represent regions with no homology, numbers in bold show the localization of nucleotide positions of important features, a nested retrotransposon is drawn above the AC149306 element.
Subfamily MtPH-M-3 was the largest within the family and contained 18 elements, of which two carried both ORFs. Their length varied from 442 to 4,048 bp, and interestingly, two 442 bp-long elements were 100% identical. As their length resembled that of miniature inverted repeat elements (MITEs), but unlike MITEs, their number in the M. truncatula genome was low, it would be tempting to speculate that these copies might become founders of a new MITE family. A slightly more advanced stage of proliferation of MITE-like elements could be observed with a group of 10 short (776–905 bp) elements from the same family. A more detailed comparison of the element sequences provided a further insight into the evolution of MtPH-M-3 subfamily. Internal deletions were accompanied by differentiation and rearrangement of variant sequences (blocks A, B, and C in Figure 5, Additional File 3) in the subterminal regions. Two lineages could be traced that originated from the core element MtPH-M-3-Ia, that included respectivley 5 and 11 elements. The element MtPH-M-3-VI showed apparently a mosaic structure, as it contained the 3' subterminal region from lineage I, while the major portion of the element contained sequence variants chracteristic for the lineage II (Figure 5).

Intra-family relationships among the MtPH-M-3 elements. Thick solid lines represent homologous regions, thick dotted lines represent regions with no homology, thin dashed lines represent internal deletions, blocks marked with orf1 and TPase show localization of the coding regions, blocks marked with A, B, and C show localization of sequence polymorphisms used to trace intra-family lineages, numbers show the length of the element.
Family MtPH-A5 was represented by four elements ranging in length from 1,182 to 6,770 bp. The two putative autonomous elements were 72% similar over the entire sequence, but within the coding region the nucleotide sequence similarity reached 95%. Two shorter elements were deletion derivatives of full-length elements. Interestingly, a recently reported MITRAV family of miniature elements of barrel medic [22] showed a high nucleotide sequence similarity of their termini to the MtPH-A5 elements, spanning over ca. 40 bp on both ends of the element.
Family MtPH-E consisted of three elements, none of which carried both ORFs. The elements ranged from 1,508 to 3,957 bp. The two largest elements were very similar, differing by one indel, while the similarity of the shortest element to the other two was restricted only to the 180 bp of the 5' terminus and 70 bp of the 3' terminus.
Family MtPH-D was represented by a single element of 3,160 bp, carrying both ORFs. However, their orientation was opposite to that of typical PIF/Harbinger-like elements representing the D lineage [15]. Its localization in the D lineage was not strongly supported by bootstrap analysis (Figure 1). The fact that no internally truncated elements were identified could suggest that the element might be capable of perfect excision, not triggering the process of abortive gap repair.
Documentation of the mobility of the mined elements
In order to find evidence for a possible mobility of identified elements we implemented a strategy proposed by Le et al. [8], i.e. we searched for regions, called RESites (Related to Empty Sites), paralogous to sequences flanking the insertion sites, but lacking the transposable element. We identified 11 RESites, of which five represented insertion sites of non-autonomous elements belonging to the MtPH-A6-4 subfamily, while two and one of them were related to non-autonomous elements of the MtPH-A6-3 and MtPH-A6-2 subfamilies, respectively. The remaining three RESites represented insertion sites of the putative autonomous (core) elements belonging to family MtPH-E and subfamilies MtPH-M-2, and MtPH-M-3 (Figure 6).

RESites corresponding to mined M. truncatula MtPH elements. For each group of sequences the upper one represents the insertion site and the lower one is the corresponding RESite. Numbers indicate the nucleotide position of the first and the last nucleotide of the presented sequence, related to the BAC clone from which it was extracted.
We identified several M. truncatula ESTs showing high similarity to putative expression products (orf1 and TPase) of the mined autonomous elements (Additional File 4). However, ESTs directly corresponding to the putative expression products, both to the orf1 (CX532696, 641 bp, 94% identity) and the TPase (AW686181, 304 bp, 99% identity), could be detected only in case of elements representing the MtPH-M-1 subfamily (Additional File 5). Interestingly, A number of ESTs similar to non-coding terminal regions of the TEs could also be identified (data not presented).
The PCR assay of MtPH insertion polymorphism was performed on eight M. truncatula populations selected to represent genetic diversity of the species, as proposed by Ronfort [31]. Fifty-six insertion sites identified in the reference genome of cv. Jemalong A17 were checked for presence of the TE. Thirty-seven primer pairs yielded products of the expected size for the reference sample, while 11 generated complex profiles, likely indicating that insertions were present in repetitive regions. The remaining eight primer pairs produced ambiguous results. Of the 37 successful amplifications, 20 occurred to be polymorphic. Usually, the size the shorter amplicon corresponded to the predicted size of the product amplified from the unoccupied site. However, amplicons slightly differing from the expected size were also observed, indicating a possible imperfect excision event (Figure 7).

Insertion related size polymorphisms of MtPH-A6-3 elements. A. Long PCR amplification of the region encompassing the MtPH-A6-3-IIa insertion site, B. PCR amplification of the region encompassing the MtPH-A6-3-VI insertion site, C. PCR amplification of the region encompassing the MtPH-A6-3-XVI insertion site. Lanes: M – 1 kB ladder (Fermentas), 1 – Jemalong A17, 2 – L163, 3 – L174, 4 – L368, 5 – L530, 6 – L544, 7 – L651, 8 – L734, 9 – negative control. Fragments representing occupied and unoccupied sites are marked by red and green arrows, respectively. Numbers in red indicate the expected length of products representing occupied sites, predicted from the original sequence.
Discussion
We developed a strategy for identification of transposable element families through in silico genome mining, based on initial assumptions on the type of transposase and the consensus sequences of terminal inverted repeats. It required several consecutive steps, i.e. (1) search for regions coding for the TPase, (2) identification of TIRs flanking the identified regions and matching a defined sequence motif, (3) identification of related elements with no coding capacity, and (4) grouping the identified elements into families on the basis of their sequence similarity. We applied this strategy to mine the genome of Medicago truncatula for PIF/Harbinger-like elements similar to the previously described MtMaster element [14]. In principle, the proposed strategy can be used to mine for any other type of class II TEs, provided that at least one 'seed' element is known.
Diversity of the identified PIF/Harbinger-like elements is high, although our search was limited by a specifically defined core TIR sequence. We focused on 22 ORFs coding for putative TPases, representing a half of all initially identified ORFs, as for the other half, TIRs flanking the ORF and containing the required motif could not be found. A recent broad analysis of the TE landscape in another legume, Lotus japonicus [21], revealed a presence of nine putative autonomous PIF-like elements (besides several more distantly related Pong-like elements) in ca. 32 Mb portion of the genome. This number is in agrrement with our results, as we found 22 full-length elements (2.5 times more) in ca. 200 Mb representing a certain level of redundancy. Interestingly, all PIF-like TEs from L. japonicus represented the A3 lineage, while no A3 members were identified in M. truncatula, which may indicate a strikingly different evolutionary fate of that group of TEs in each of the closely related species.
Detailed structure analysis of the mined element families indicates that their proliferation in the genome generally follows the model of abortive gap repair (AGR), as proposed for the Ac/Ds elements in maize [23]. Members of a particular family were usually direct deletion derivatives of the related, putative autonomous element. However, assuming that members of all PIF/Harbinger-like TE families in the genome of M. truncatula were mobilized with similar frequency, the efficiency of AGR seems to vary from one family to another. Two families, MtPH-A6 and MtPH-M, were the most numerous, while the remaining three were represented by a very small number of copies. Difference in the copy number may be a result of different transposition rates, but it may also indicate that some elements less efficiently trigger the process of AGR following excision, which would result in a higher frequency of perfect excision. The latter is further supported by two observations. Firstly, the members of subfamily MtPH-A6-4 contain a variable number of 60 bp tandem repeats in one or both subterminal regions, serving as targets for AGR and leading to increase of the TE copy number accompanied by changes in the number of VNTRs. The presence of 60 bp tandem repeats was inherently connected with MtPH-A6-4 elements throughout the M. truncatula genome, which implies that they likely evolved in the course of the proliferation of that subfamily. Probably, triggering the AGR from the VNTR region also led to an inversion of the internal region in MtPH-A6-4-XIV, as compared to MtPH-A6-4-Ia. Secondly, at least one member of the low copy number family MtPH-E was transpositionally active, as confirmed by the presence of the RESite, but despite the potential for mobility, the number of MtPH-E elements has remained low.
PIF/Harbinger-like elements are ancestors of certain groups of miniature transposons (MITEs), the relation of maize PIF element and MITEs belonging to the Tourist family has been well documented [12, 16]. Also, several other MITE families, e.g. Heartbreaker from maize [24], Kiddo from rice [25], and Krak from carrot [14] show TIR sequence similarities to those of PIF/Harbinger-like elements. We were able to directly link the previously identified MITRAV MITE family [22] to family MtPH-A5 of M. truncatula PIF/Harbinger-like elements. This suggests that both MtPH-A5 and MITRAV originated from a recent common ancestor and MtPH-A5 TPase might be the trans-acting factor for MITRAV mobilization, as experimentally proven for the Pong and mPing MITE in rice [13, 26, 27]. Also, two groups of two and ten TEs, all classified in the subfamily MtPH-M-3, might represent newly emerging MITE families. We performed an initial search for other MITEs showing a TIR homology to the consensus motif of the PIF/Harbinger TIRs leading to an identification of few other MITE families (data not presented). Altogether, it confirms that PIF/Harbinger-like elements and related MITEs are present in the genome of M. truncatula, similar to genomes of other plant species. However, the number of MITE copies is probably much lower than that present in the grass genomes.
A more detailed experimental evaluation of MtPH TEs diversity in a range of M. truncatula populations should be useful to further characterize the transpositional activity and the dynamics of particular families. Analysis of RESites and a high incidence of insertion related size polymorphisms shows that a significant fraction of the mined elements was mobile in the recent past. The presence of ESTs related to ORFs of the MtPH elements, including those directly derived from the MtPH-M-1 elements, suggests that they can still be mobile. As proven previously, one transcriptionally active autonomous element can cause trans-mobilization of a range of related, but not directly derived elements [13].
Polymorphic insertion sites could be used as a source of molecular markers, as shown previously for other species [28–30], to measure intraspecific diversity in relation to its geographic structure, complementing other molecular marker systems, e.g. these based on microsatellites [31].
Conclusion
Starting from a single previously described PIF/Harbinger-like TE of M. truncatula, we identified 89 elements representing the diversity of this superfamily in the host plant genome. They were divided into five families representing different evolutionary lineages, and further into subfamilies. Elements within each subfamily evolved essentially following the model of AGR, leading to the reconstruction of an internally deleted copy in the donor site following transposition. It is likely that different families vary in their potential to trigger the process of AGR. One peculiarity observed in a group of elements representing subfamily MtPH-A6-4 was the presence of 60 bp long VNTRs in one or both subterminal regions or even spanning over the entire internal region of the TE. Some of the identified elements are closely related to several MITE families, including a previously described MITRAV family. Also, some of the newly identified short elements can be viewed as in statu nascendi MITEs, provided that conditions for a rapid burst of their mobility would be met. Further investigation is necessary for a more detailed evaluation of the copy number, transpositional activity, and insertional polymorphism of the TEs, including MITEs, as they could be utilized as a source of molecular markers.
Methods
Semi-automated mining of PIF/Harbinger-like elements
The experiment was performed on the M. truncatula genomic DNA sequence database consisting of 1540 BACs, updated Aug 2005 [32]. As the size of the whole M. truncatula genome ranges from 500 to 600 Mbp [17] and the average non-overlapping coverage by each BAC was ca. 100 Kb [32], we estimated that the input sequence data amounted 26–30% of whole genome.
The predicted protein sequence of DDE domain and the whole TPase sequence of the previously identified MtMaster element [14] was used as the initial query for a TBLASTN search against the BAC sequence database, using the E-value threshold of 1e-20. The output file was then processed to eliminate redundancy coming from overlapping BACs, and significant hits were extracted, along with up to 30 kb flanking sequences. The extracted sequences were scanned for the presence TIRs and TSDs, using a newly developed tool named TIRfinder, identifying TIRs and TSDs and returning a file with a list of found elements fulfilling user-defined requirements. To provide fast computation on whole genome, the algorithm uses very efficient data structures, such as suffix trees. TIRfinder is an open source software accessible online [33]. The program was written in Java and can be run on Windows or Linux.
We allowed up to four mismatches inside 14 bp of the TIRs and no mismatch in TSDs. Another condition was the presence of the conserved G(N)5GTT motif at the 5' end of the TIR. In silico prediction of the presence of coding regions was performed for all identified sequences using FGENESH [34].
To identify internally deleted copies of elements related to those found previously, 217 bp-long (3 bp TSD + 14 bp TIR + 200 bp subterminal sequence) terminal regions were extracted from all putative autonomous elements. These sequences were used to scan the M. truncatula genomic DNA sequence database (BLASTN, E-value threshold – 1e-10), and regions showing homology to any of the terminal regions were identified. The output was automatically filtered to find sequences of length ranging from 400 to 30,000 bp, flanked with TIRs showing homology to the same autonomous element on both ends. All newly found sequences have been checked whether they contained a region coding for the TPase. All TEs were scanned using Censor [35], to identify the presence of nested elements.
Phylogenetic analyses, grouping, and visualization of TE sequence similarity
Multiple alignment of 48 transposase sequences of PIF/Harbinger-like transposable elements was obtained using T-Coffee [36]. Bootstrap analysis was performed with PHYLIP using seqboot, neighbor, protdist and consense programs [37]. The sequence similarity of 89 TEs was analyzed by the hierarchical clustering method and visualized with help of multidimensional scaling. For both tasks we used the R statistical environment [38]. As a measure of dissimilarity between sequences we used the E-value of BLAST. Hierarchical cluster analysis of a set of dissimilarities was done by hclust (complete linkage) method [39]. Multidimensional scaling [40] visualization is primarily dependent on the analogy of similarity and proximity (and hence of dissimilarity and distance). It re-scales a set of dissimilarity data into distances and produces the low-dimensional configuration that generated them. The visualization for our data was obtained with isoMDS R procedure.
TE structure analysis
Sequences were visually compared, aligned, edited, and analysed using BioEdit and the included accessory applications [41]. Pairwise sequence comparisons were performed using 'blast 2 sequences' [42] and Yass [43, 44]. Dot-plots were generated using Nucleic Acid Dot Plots [45] with a window size of 25 nucleotides and a mismatch limit of 5 positions. Tandem repeats identification was performed using 'mreps' software [46, 47].
Documentation of mobility
In order to find RESites (Related to Empty Sites) in the M. truncatula genome we performed a computer-based search, essentially as described by Le et al. [8]. Briefly, we extracted 1 Kb sequence flanking both sides of each of the mined elements, combined them into one sequence of 2 Kb, and used it as a query for a BLASTN search on the whole BAC sequence database. Hits spanning on both sides of the insertion were considered as those representing RESites.
EST search was performed using nucleotide sequences of the putative autonomous elements, using a BLAST tool run against the M. truncatula EST database [48].
PCR conditions
PCR assay was performed on plants representing cv. Jemalong A17 and seven populations from the core M. truncatula collection (CC8, as described by Ronfort et al. [31]). Primer pairs were anchored in the regions flanking the mined elements. They were designed using Primer3 [49] to obtain amplification of ca. 600 bp long fragment for the putative empty site. Two cycling protocols were employed. For TEs of length not exceeding 2 Kb a standard PCR was performed. The reaction was set up in the volume of 20 μl and contained 0.25 mM each dNTP, 2 mM MgCl2, 10 pmol of each primer, 1 unit of TAQ polymerase (Fermentas) and 2 μl of the PCR buffer supplied by the manufacturer. The thermal profile of the reaction was as followed: 94°C for 2 min., 35 cycles of: 94°C for 30 s, 53°C for 30 s, and 68°C for 90 s, and completed with 68°C for 5 min. For larger elements we used long PCR protocol. Amplification was performed in the volume of 20 μl containing 0.25 mM each dNTP, 10 pmol of each primer, 0,5 unit of long PCR enzyme mix (Fermentas) and 2 μl of the Long PCR buffer supplemented with MgCl2 (Fermentas), using the following thermal profile: 94°C for 2 min., 35 cycles of: 94°C for 15 s, 53°C for 30 s, and 68°C for 7 min., and completed with 68°C for 10 min. All reactions were carried out in the Mastercycler or Mastercycler Gradient (Eppendorf). Amplification products were separated on 1% agarose gels and visualized with ethidium bromide under UV.
References
Arabidopsis Genome Initiative: Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000, 408: 796-815. 10.1038/35048692.
Meyers BC, Tingey SV, Morgante M: Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res. 2001, 11: 1660-1676. 10.1101/gr.188201.
Price AL, Jones NC, Pevzner PA: De novo identification of repeat families in large genomes. Bioinformatics. 2005, 21: 351-358. 10.1093/bioinformatics/bti1018.
Yang G, Hall TC: MAK, a computational tool kit for automated MITE analysis. Nucleic Acids Res. 2003, 31: 3659-3665. 10.1093/nar/gkg531.
Bao Z, Eddy SR: Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12: 1269-1276. 10.1101/gr.88502.
Kurtz S, Schleiermacher C: REPuter: fast computation of maximal repeats in complete genomes. Bioinformatics. 1999, 15: 426-427. 10.1093/bioinformatics/15.5.426.
Kapitonov VV, Jurka J: Molecular paleontology of transposable elements from Arabidopsis thaliana. Genetica. 1999, 107: 27-37. 10.1023/A:1004030922447.
Le QH, Wright S, Yu Z, Bureau T: Transposon diversity in Arabidopsis thaliana. Proc Natl Acad Sci USA. 2000, 97: 7376-7381. 10.1073/pnas.97.13.7376.
Yu Z, Wright SI, Bureau TE: Mutator -like elements in Arabidopsis thaliana: Structure, diveristy and evolution. Genetics. 2000, 156: 2019-2031.
Mao L, Wood TC, Yu Y, Budiman MA, Tomkins J, Woo S, Sasinowski M, Presting G, Frisch D, Goff S, Dean RA, Wing RA: Rice transposable elements: A survey of 73,000 sequence-tagged-connectors. Genome Res. 2000, 10: 982-990. 10.1101/gr.10.7.982.
Tucrotte K, Srinivasan S, Bureau T: Survey of transposable elements from rice genomic sequences. Plant J. 2001, 25: 169-179. 10.1046/j.1365-313x.2001.00945.x.
Zhang X, Feschotte C, Zhang Q, Jiang N, Eggelston W, Wessler SR: P instability factor: an active maize transposon system associated with the amplification of Tourist-like MITEs and a new superfamily of transposases. Proc Natl Acad Sci USA. 2001, 98: 12572-12577. 10.1073/pnas.211442198.
Jiang N, Bao Z, Zhang X, Hirochika H, Eddy SR, McCouch SR, Wessler SR: An active DNA transposon family in rice. Nature. 2003, 421: 163-167. 10.1038/nature01214.
Grzebelus D, Yau YY, Simon PW: Master : a novel family of PIF/Harbinger-like transposable elements identified in carrot (Daucus carota L.). Mol Genet Genomics. 2006, 275 (5): 450-459. 10.1007/s00438-006-0102-3.
Zhang X, Jiang N, Feschotte C, Wessler SR: PIF- and Pong-like transposable elements: distribution, evolution and relationship with Tourist-like miniature inverted repeat transposable elements. Genetics. 2004, 166: 971-986. 10.1534/genetics.166.2.971.
Jurka J, Kapitonov VV: PIFs meet Tourists and Harbingers: a superfamily reunion. Proc Natl Acad Sci USA. 2001, 98: 12315-12316. 10.1073/pnas.231490598.
Blondon F, Marie D, Brown S, Kondorosi A: Genome size and base composition in Medicago sativa and M. truncatula species. Genome. 1994, 37: 264-270.
Charrier B, Foucher F, Kondorosi E, d'Aubenton-Carafa Y, Thermes C, Kondorosi A, Ratet P: Bigfoot : a new family of MITE elements characterized from the Medicago genus. Plant J. 1999, 18: 431-441.
Macas J, Neumann P: Ogre elements – A distinct group of plant Ty3/gypsy-like retrotransposons. Gene. 2007, 390: 108-116. 10.1016/j.gene.2006.08.007.
Jurka J: Repbase Update: a database and an electronic journal of repetitive elements. Trends Genet. 2000, 9: 418-420. 10.1016/S0168-9525(00)02093-X.
Holligan D, Zhang X, Jiang N, Pritham EJ, Wessler SR: The transposable element landscape of the model legume Lotus japonicus. Genetics. 2006, 174: 2215-2228. 10.1534/genetics.106.062752.
Shankar R, Jurka J: MITRAV: A miniature DNA transposon from barrel medic. Repbase Reports. 2007, 7: 38-
Rubin E, Levy AA: Abortive gap repair: underlying mechanism for Ds element formation. Mol Cell Biol. 1997, 17 (11): 6294-6302.
Casa AM, Brouwer C, Nagel A, Wang L, Zhang Q, Kresovich S, Wessler SR: The MITE family Heartbreaker (Hbr): molecular markers in maize. Proc Natl Acad Sci USA. 2000, 97: 10083-10090. 10.1073/pnas.97.18.10083.
Yang G, Dong J, Chandrasekharan MB, Hall TC: Kiddo, a new transposable element closely associated with rice genes. Mol Genet Genomics. 2001, 266: 417-424. 10.1007/s004380100530.
Kikuchi K, Terauchi K, Wada M, Hirano H-Y: The plant MITE mPing is mobilized in anther culture. Nature. 2003, 421: 167-170. 10.1038/nature01218.
Nakazaki T, Okumoto Y, Horibata A, Yamahira S, Teraishi M, Nishida H, Inoue H, Tanisaka T: Mobilization of a transposon in the rice genome. Nature. 2003, 421: 170-172. 10.1038/nature01219.
Casa AM, Mitchell SE, Smith OS, Register III JC, Wessler SR, Kresovich S: Evaluation of Hbr (MITE) markers for assessment of genetic relationships among maize (Zea mays L.) inbred lines. Theor Appl Genet. 2002, 104: 104-110. 10.1007/s001220200012.
Kwon SJ, Park KC, Kim JH, Lee JK, Kim NS: Rim2/Hipa CACTA transposon display; a new genetic marker technique in Oryza species. BMC Genetics. 2005, 6: 15-10.1186/1471-2156-6-15.
Grzebelus D, Jagosz B, Simon PW: The DcMaster Transposon Display maps polymorphic insertion sites in the carrot (Daucus carota L.) genome. Gene. 2007, 390: 67-74. 10.1016/j.gene.2006.07.041.
Ronfort J, Bataillon T, Santoni S, Delalande M, David JL, Prosperi J-M: Microsatellite diversity and broad scale geographic structure in a model legume: building a set of nested core collection for studying naturally occurring variation in Medicago truncatula. BMC Plant Biology. 2006, 6: 28-10.1186/1471-2229-6-28.
Medicago sequencing resources. [http://www.medicago.org/genome/]
TIRfinder. [http://www.sourceforge.net/projects/TIRfinder/]
Salamov AA, Solovyev VV: Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000, 10: 516-522. 10.1101/gr.10.4.516.
Kohany O, Gentles AJ, Hankus L, Jurka J: Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics. 2006, 7: 474-10.1186/1471-2105-7-474.
Notredame C, Higgins D, Heringa J: T-Coffee: A novel method for multiple sequence alignments. J Mol Biol. 2000, 302: 205-217. 10.1006/jmbi.2000.4042.
PHYLIP. [http://evolution.genetics.washington.edu/phylip.html]
Venables WN, Ripley BD: Modern Applied Statistics with S. Springer, New York. 2002
Defays D: An efficient algorithm for a complete link method. Comput J. 1977, 20: 364-366. 10.1093/comjnl/20.4.364.
Borg I, Groenen P: Modern Multidimensional Scaling: Theory and Applications. Springer-Verlag New York. 1997
Hall TA: BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999, 41: 95-98.
Tatusova TA, Madden TL: Blast 2 sequences – a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 1999, 174: 247-250. 10.1111/j.1574-6968.1999.tb13575.x.
Noe L, Kucherov G: YASS: enhancing the sensitivity of DNA similarity search. Nucleic Acids Res. 2005, 33: W540-W543. 10.1093/nar/gki478.
genomic DNA local alignment similarity search tool. [http://bioinfo.lifl.fr/yass/]
Nucleic Acid Dot Plots. [http://www.vivo.colostate.edu/molkit/dnadot/]
Kolpakov R, Bana G, Kucherov G: mreps: efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res. 2003, 31: 3672-3678. 10.1093/nar/gkg617.
mreps. [http://bioinfo.lifl.fr/mreps/]
Gene Indices – Blast Search. [http://compbio.dfci.harvard.edu/tgi/cgi-bin/tgi/Blast/index.cgi]
Rozen S, Skaletsky HJ: Primer3. 1998, [http://primer3.sourceforge.net]
Acknowledgements
The research project was funded by the Polish Ministry of Science and Higher Education grant no. N301 036 31/1203, for the years 2006–2008. SL, AG and GK were supported by the Polonium and ECO-NET programs of the French Ministry of Foreign Affairs. The authors wish to thank Dr. J-M Prosperi for donating seeds of M. truncatula populations used in the study, two anonymous reviewers for their helpful suggestions, and Mrs M Gladysz for her technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
DG developed strategy for the study, performed the fine-scale analysis of the TEs, performed the PCR, and prepared the final version of the manuscript, SL and TG developed algorithms for TE identification, TG edited HC and MDS graphs, GK analysed tandem repeats, and AG participated in the design of the study, performed HC and MDS analyses, and participated in drafting the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2007_1122_MOESM1_ESM.xls
Additional file 1: List of all PIF/Harbinger-like elements identified in the course of the study in the genome of Medicago truncatula. (XLS 22 KB)
12864_2007_1122_MOESM2_ESM.pdf
Additional file 2: Similarity-based grouping of M. truncatula PIF/Harbinger-like elements.. Results of multidimensional scaling (MDS): A. whole TE sequence, B. 5'end subterminal regions, C. 3'end subterminal regions, and hierarchical clustering (HC): D. whole TE sequence, E. 5'end subterminal regions, F. 3'end subterminal regions. (PDF 187 KB)
12864_2007_1122_MOESM3_ESM.ppt
Additional file 3: Sequence alignment of the A and B blocks differentiating individual elements belonging to the MtPH-M-3 family. (PPT 24 KB)
12864_2007_1122_MOESM4_ESM.pdf
Additional file 4: Identification of M. truncatula ESTs similar to putative expression products of orf1 and TPases coded by MtPH elements. Sequence of the whole element was used as query against M. truncatula EST database, hits in orf1 and TPase coding regions with E value lower than 1e-06 were scored. Nearly identical hits to orf1 and TPase of the MtPH-M-1 elements are marked red. (PDF 44 KB)
12864_2007_1122_MOESM5_ESM.pdf
Additional file 5: Alignment of MtPH-M-1-Ia and the ESTs corresponding to orf1 (CX532696) and TPase (AW686181). Predicted exons of the orf1 and TPase are highlighted yellow and green, respectively, TSDs of the element are marked gray. (PDF 58 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Grzebelus, D., Lasota, S., Gambin, T. et al. Diversity and structure of PIF/Harbinger-like elements in the genome of Medicago truncatula. BMC Genomics 8, 409 (2007). https://doi.org/10.1186/1471-2164-8-409
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-8-409