
Abstract
Background
At least three species of Borrelia burgdorferi sensu lato (Bbsl) cause tick-borne Lyme disease. Previous work including the genome analysis of B. burgdorferi B31 and B. garinii PBi suggested a highly variable plasmid part. The frequent occurrence of duplicated sequence stretches, the observed plasmid redundancy, as well as the mainly unknown function and variability of plasmid encoded genes rendered the relationships between plasmids within and between species largely unresolvable.
Results
To gain further insight into Borreliae genome properties we completed the plasmid sequences of B. garinii PBi, added the genome of a further species, B. afzelii PKo, to our analysis, and compared for both species the genomes of pathogenic and apathogenic strains.
The core of all Bbsl genomes consists of the chromosome and two plasmids collinear between all species. We also found additional groups of plasmids, which share large parts of their sequences. This makes it very likely that these plasmids are relatively stable and share common ancestors before the diversification of Borrelia species.
The analysis of the differences between B. garinii PBi and B. afzelii PKo genomes of low and high passages revealed that the loss of infectivity is accompanied in both species by a loss of similar genetic material. Whereas B. garinii PBi suffered only from the break-off of a plasmid end, B. afzelii PKo lost more material, probably an entire plasmid. In both cases the vls gene locus encoding for variable surface proteins is affected.
Conclusion
The complete genome sequences of a B. garinii and a B. afzelii strain facilitate further comparative studies within the genus Borrellia. Our study shows that loss of infectivity can be traced back to only one single event in B. garinii PBi: the loss of the vls cassettes possibly due to error prone gene conversion. Similar albeit extended losses in B. afzelii PKo support the hypothesis that infectivity of Borrelia species depends heavily on the evasion from the host response.
Similar content being viewed by others

Background
Infections with Borrelia species cause thousands of human disease cases per year [1]. The major causative agents of this disease are three species of the Borrelia burgdorferi sensu lato complex (Bbsl), B. burgdorferi sensu stricto, B. garinii, and B. afzelii [2–4]. As was shown previously using phylogenetic tree reconstructions [5, 6]B. garinii and B. afzelii are more closely related than either of them to B. burgdorferi sensu strictu, which branches at the basis of the three-species tree. Borreliae are obligatorily bound to host organisms for survival. During their life cycle they switch from the invertebrate host tick (Ixodes spec.) to various vertebrate hosts via a tick bite. The range of vertebrates and invertebrates used as hosts is thought to be mediated by factors encoded mainly on a large number of different plasmids found in Borreliae genomes. A further characteristic of the plasmids is their wealth of paralogous genes. During passage plasmids can be lost due to the lack of selection pressure as was shown recently [7]. This loss can be accompanied by the inability of Borreliae to prosper in the host. Furthermore, it is thought that Borreliae plasmids are not stable and are frequently rearranged leading to differing plasmid content within a species [8, 9]. To some extent the chromosome is also involved in fission/fusion events. It was shown that the right end of the chromosome of B. burgdorferi strains is variable due to its ability to catch plasmid material [10]. The high variability of Borreliae genomes has so far hindered a concise description of the genome properties of Borreliae based on plasmid size estimates and hybridizations alone.
Comparative genomics aims at the description of related organisms based on their common and discernible genetic material [11–13]. Furthermore, it enables the evaluation of the relationship between genotype and phenotype, if clear-cut phenotypic differences are described for the species in question. In case of prokaryotes a genome comparison can e.g. discern common genomic backbones from otherwise acquired genetic material [14].
To date the complete genome of B. burgdorferi sensu stricto (B31) is known [15]. It exhibits a wealth of circular as well as linear plasmids, some of which are nearly identical [8]. In a more recent study we reported on the genome of a B. garinii strain [16]. This comparative genomics approach showed clearly that three genomic elements, the chromosome and plasmids cp26 and lp54, are common to both Borrelia species and, more strikingly, collinear. At the same time we were able to show that some plasmids are confined to B. burgdorferi sensu stricto, since no trace of these plasmids including their coding capacity could be found in the whole genome shotgun sequencing (WGSS) data of B. garinii PBi. Yet, at this time it was not possible to give a clear picture of the plasmid content of B. garinii PBi, since plasmidal sequences were distributed over 36 sequence contigs that did not represent entire plasmids.
In this study we determine the complete sequences of the B. garinii PBi plasmids and the whole genomic sequence of a third species, B. afzelii PKo [GenBank: CP000397, GenBank: CP000398, GenBank: CP000399, GenBank: CP000400, GenBank: CP000401, GenBank: CP000402, GenBank: CP000403, GenBank: CP000404, GenBank: CP000405, GenBank: CP000406]. Furthermore, we wanted to shed light onto the requirements for pathogenicity in these species. For this purpose, sequences from pathogenic low passage and from high passage strains, which have lost their pathogenicity, were combined to yield the entire genome sequences of these two Borrelia strains.
Results
Chromosome and plasmid sequences
As we had previously shown by the genome analysis of an early (12th) passage of B. garinii PBi [16] a coverage as low as 3× is sufficient as a basis for genome analysis of Bbsl species. Extending this approach we applied low coverage WGSS to a late (300th) passage of B. garinii PBi as well as to early (8th) and late (70th) passages of B. afzelii PKo. The resulting sequences were assembled species-wise to give a coverage of around 6 for each genome (Table 1). Of the B. garinii genome we sequenced equal amounts of clones from high and low passage strains; of the B. afzelii genome only one third of all sequences were derived from the low passage strain. Employing additional gap closure procedures, we obtained nearly gap free genomic sequences. Contigs or parts thereof consisting only of low passage DNA derived clones were considered to have been lost during cultivation.
Collinearity and indels of the Bbsl chromosomes
Both B. garinii PBi and B. afzelii PKo genomes have collinear chromosomes of comparable sizes (Table 1) with only minor insertions and deletions. Pairwise alignments using stretcher revealed that most indel positions in B. garinii PBi and B. afzelii PKo are shared compared to B. burgdorferi B31 (Table 2) indicating a closer relationship between these two species than to B. burgdorferi B31. Most indels are small, but we also observed three regions exceeding 500 bp in B. afzelii. One of these differences between the chromosomes of Bbsl species is attributable to differing sequences at the origin of replication in the middle of the chromosome. The second affects the bmp locus at position 394,080 of the B. burgdorferi chromosome. We previously noticed that this locus is incompletely duplicated in B. garinii PBi compared to B. burgdorferi B31 [16]. Now, with a third sequence at hand we see that this locus seems to be instable in all species. In B. afzelii PKo this locus is incompletely triplicated. Most interestingly, the duplication events observed produce only functional copies of bmpA. All other duplicated bmp genes are rendered truncated during the duplication event (Fig 1). The third large indel affects a further outer membrane protein gene (lmp1), thus contributing to the diversity of the protein orthologs and possibly to the variations in the host pathogen interaction.

Chromosomal duplications of the bmp cluster genes in different Borrelia species. Grey boxes are functional genes, hatched boxes denote pseudogenes. Arrows indicate duplication events.
Striking differences in the plasmid content
The Bbsl plasmid fraction varies significantly between different species. Whereas the B. garinii PBi plasmids comprise only 29 % of the genome, the B. afzelii PKo plasmids add up to 36 % (Table 1). Yet, this is still well below the plasmid content of B. burgdorferii sensu stricto (47 %) [8]. Assemblies of the respective reads yielded 11 plasmids for the B. garinii PBi genome and at least 16 for the B. afzelii PKo genome (Table 3). Circular plasmids can be defined as finished once no gap remains in the sequence and the ring is closed. Using this criterion, we were able to define three circular plasmids in B. garinii PBi. In B. afzelii PKo we detected two circular plasmid types. On of these types comprises a group of eight very similar plasmids resembling the situation in B. burgdorferi B31. We were not able to completely resolve this group of eight plasmids since they are highly similar, if not identical, over long sequence stretches. This renders impossible the correct assignment of a considerable number of clones to a specific plasmid and PCR approaches to close gaps between plasmid segments. Only one plasmid with a size of over 30 kb could be entirely finished. Yet, the extraordinarily high identity of the group members to each other led us to the conclusion that all members of this group are circular.
We tested using PCR approaches, whether any of the remaining sequence contigs could be only part of another plasmid. Using this method, we closed 15 gaps between segments with unambiguous sequences. The remaining segments could not be linked together, since no or no unique PCR products could be produced with any of the combinations of ends possible. We sequenced several of the PCR products from primer combinations, which gave rise to more than one PCR product. Sequences derived from all these products were either of low sequence quality indicating PCR product mixtures or located elsewhere in the genome. Thus, since we were not able to find additional links between the sequence segments described here, we conclude that most of the described segments are indeed individual plasmids of these species and are supposed to be linear. The plasmid size range extends from 6 to over 60 kb (Table 3). Plasmid names were chosen according to size and to their circular (cp) or linear (lp) nature. The sequence reads of the small fragments in the range of 6 to 15 kb in both species were only found in low passage strains (see below).
The strain of B. garinii we present here has the fewest number of plasmids of all Borrelia species analysed so far. Including the indispensable cp26 and lp54 orthologs, there are three circular and eight linear plasmids. Conversely, the B. afzelii PKo genome comprises nine circular and seven linear plasmids.
Plasmid homologies between Borrelia species
We wondered whether we would be able to detect additional relationships between plasmids of different species besides the clearly homologous plasmids cp26 and lp54. In a first step we examined the relationships within a species. Most prominent in this respect are the B. burgdorferi B31 circular plasmids in the range of 30 kb, which are highly similar to each other with only minor sequence differences over large parts of the plasmids. We observed a homologous group of plasmids also in B. afzelii PKo, where we found 8 circular plasmids with a size of 30 kb. This group may even include further plasmids, since we were not able to assemble this group fully. Furthermore, despite the length differences between the two circular plasmids cp29 and cp31 the B. garinii PBi plasmids constitute a similarity group as shown by tupleplot analysis [see Additional file 1]. A three-species comparison revealed that all these circular plasmids belong to the same group and originated from a common ancestor. Most strikingly and despite their high sequence diversity, which renders an interspecies alignment difficult, these plasmids exhibit a conserved collinearity [see Additional file 1]. Intriguingly, these plasmids are amplified and diversified (i.e. they exhibit polymorphisms to each other) in all species albeit with differing copy numbers. The most remarkable distinguishing feature within this group of plasmids is the length difference observed in B. garinii PBi. An all-to-all comparison of the other plasmids showed that there are additional plasmid groups with a common ancestor although with slightly different lengths and minor unique sequence portions (Table 4 and Additional file 1).
To classify these plasmids we had to relax the criteria, according to which plasmids are grouped together. Thus, the minimal requirement for a group is to have detectable similarities (using tupleplot, see materials and methods) over a significant part of their sequence length irrespective of rearrangements. As is listed in Table 4 we can define six different groups according to their common presence in all Bbsl species. Group VI is the least well defined group since the similar sequence stretches are divided up in B. burgdorferi between three different plasmids. All other groups contain plasmids, of which the similar DNA sequences seem to be stably inherited within individual plasmids.
The relatedness between the plasmid content of B. afzelii PKo and B. garinii PBi is notable. Their plasmids within the same groups are more related to each other concerning size, shared similarity and extent of rearrangements than to the B. burgdorferi sensu stricto counterparts. Conversely, the otherwise highly amplified group III plasmids have undergone a reduction in the B. garinii PBi genome, whereas some other group specific plasmids were amplified (groups IV and VI).
Low passage versus high passage strains – genomic differences
As mentioned above we did not use a uniformly grown culture for the construction of whole genome shotgun libraries. Instead, we used low and high passage strains of the same isolate. The major difference between low and high passage is the loss of infectivity during repeated passages [9, 17, 18]. These strains should have accumulated only minor numbers of single base differences due to the short time of separation. Indeed, single nucleotide exchanges on the chromosomes were very rare. We found only 26 such events in B. garinii PBi and even less in B. afzelii PKo (4). We think that loss of infectivity is accompanied by larger changes in the genome of the strains, i.e. loss of whole parts of the genetic material.
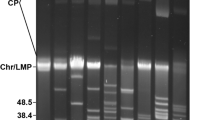
In case of B. garinii PBi we found all plasmids represented in both the low passage as well as the high passage culture. This is in good agreement with previous findings, where no plasmid differences could be detected in low and high passage strains of B. garinii PBi [9]. Yet, a small fragment of 6 kb was not detectable in the high passage strain shotgun data. An analysis of the gene content revealed, that nearly all of the 6 kb segment is occupied by cassettes of the vls gene cluster. This cluster was previously mapped to a 28 kb linear plasmid in B. garinii [19]. Yet, this fragment could not be assigned to one of the other plasmids despite testing all combinations possible using specific primers for all linear plasmid ends. It is very unlikely that we missed an entire plasmid with our shotgun cloning approach. Thus, this failure may be due to the repetitive nature of the vls locus and/or its frequent cassette switchings [20, 21], which makes the generation of matching unique primers impossible. Using different primers on this fragment we were able to amplify several DNA species of different lengths from the low passage strain (Fig. 2). Amplification in the high passage strain yielded a product only for the leftmost primer pair. All PCR products were sequenced and were confirmed to be derived from the correct genome position. This indicates that most of the vls cassettes and the vls gene itself was lost during passaging. A Southern blot analysis confirmed that the entire vls locus is missing in the high passage strain [see Additional file 1].

The vls cassette fragment in low and high passage strains of B. garinii. A: Designed primers and expected PCR products from the 6 kb low passage DNA fragment containing vls cassettes of B. garinii. B: Electrophoresis of PCR products obtained with various primer combinations. Roman letters indicate primer combinations from A. Low and high refer to DNA from low and high passage strains, respectively.
The genomic losses of B. afzelii PKo are more severe than those of B. garinii PBi. In B. afzelii, three segments with the cumulated size of 30 kb seem to be missing in the high passage strain. Previous results suggested the loss of a 31 kb and a 6 kb plasmid from the high passage strain of B. afzelii PKo [9]. Despite our inability to connect these segments via PCR it is very likely that these segments constitute two linear plasmids in B. afzelii PKo, The assembly of the plasmids consists almost entirely of low passage strain DNA material. Sequence reads of two clones derived from the high passage strain suggest that this loss is not achieved as a sudden event but progresses gradually within the culture. One of the segments (8 kb) contains a part of the vls gene cluster. Moreover, we found additional reads with similarities to vls gene and cassette fragments solely in the low passage shotgun data (adding up to 4 kb). Again, we were not able to connect all these reads to other plasmid contigs using PCR approaches. We therefore conclude that the vls cluster is missing from the high passage strain of B. afzelii PKo. Previous Pulsed Field Gel experiments indicated the loss of a 6 kb and a 31 kb linear plasmid from the high passage strain. Thus, the 8 kb and the 15 kb fragment together with the vls cluster should form this 31 kb linear plasmid, the 6 kb fragment is therefore one entire linear plasmid. Interestingly, the fragments of vls genes we sequenced do not properly align with the previously published sequence of the entire 8 kb vls locus of B. afzelii [21]. This finding demonstrates that this locus is highly polymorphic due to casette switching and homologous recombination.
Discussion
Borrelia burgdorferi sensu lato chromosome differences
The chromosomes of Bbsl species harbour only minor differences between them. This includes right arm extensions, which may be derived from linear plasmids and three major indels. Interestingly, two of these indels affect outer membrane proteins, thus contributing to the surface diversity between the species. This is most clearly seen at the bmp locus, where the copy number varies and the duplicated genes deviate slightly from the original sequence (Fig. 1). Since these differences are fixed in the respective genomes, it is likely that this diversity may contribute to subtle variations in the host range and initial success to invade the host.
Homologous plasmid groups in Borrelia species
The analysis of the B. burgdorferi plasmids [8] showed a wealth of similar regions of differing sizes residing on several plasmids. Our previous genome analysis of B. garinii PBi resulted in the assembly of the plasmid compartment in 37 segments, which in comparison to the B. burgdorferi plasmids revealed both common and unique sequences [16]. Yet, due the fragmented data set it was impossible to directly compare the entire plasmids. This work now presents the nearly gap-free sequence of the plasmids not only from B. garinii PBi but also from B. afzelii PKo.
All species contain three seemingly invariable and indispensable parts, the chromosome, a cp26 like circular plasmid, and an lp54 like linear plasmid. They are relatively well conserved and collinear. All other plasmids cannot directly be aligned between species due to a low sequence similarity. Furthermore, comparisons of genes on amino acid level can obscure real relationships between plasmids due to unclear orthology. Yet, tupleplot analysis revealed a striking overall similarity of long stretches of some plasmids with others within and between species. Therefore, despite high sequence and length variability, as well as broken collinearity, we can describe groups of plasmids apparently derived from a common ancestor (Table 4). The most prominent group consists of circular plasmids in the size range of 30 kb, which are highly amplified in B. burgdorferi B31 and B. afzelii PKo but not B. garinii PBi. The two B. garinii counterparts to this group are more diverse in size and sequence than in the other species indicating an accelerated evolution towards a higher diversification and possible secondary reduction of this group. The plasmids of this group encode outer membrane factors, erp proteins, which are thought to be involved in host range definition [22]. Since B. garinii PBi therefore is able to produce only two different erp proteins, the host range of this species is potentially reduced compared to the other two species. Groups IV and V share large parts of their sequences between species albeit with some intra-plasmid rearrangements. The weakest homology shows group VI where similar plasmid segments are dispersed over different plasmids in B. burgdorferi B31. Intriguingly, intra-plasmid divergence and shuffling seems to be favoured over inter- plasmid rearrangements. This finding indicates that the Borrelia genome is more stable and less prone to recombinations than previously thought [8]. Unique plasmids (i.e. with less than 50 % sequence similarity to any other plasmid) occur in all three species. These may contribute to the disease variations or adaptation to different environments. A previous study relying on Southern blots using probes derived from B. burgdorferi B31 plasmids showed relationships between the probes and plasmids of different strains and species [23]. Only one probe was used per plasmid restricting conclusions on homologies between plasmids to only the probe specific regions. Yet, in most cases this study is in agreement with our findings if possible cross-hybridisations with small similar regions are taken into account. The most prominent difference is within group VI. Our results indicate homology of lp25 to lp21-2 and lp22, whereas Palmer et al. detected homology to a 30 kb linear plasmid. We therefore conclude that the 6 kb fragment harbouring th vls gene cluster is most likely connected to one of these plasmids. This is also in good agreement to previous findings, where the vls cluster of B. garinii was located on a 28 kb linear plasmid [19].
From all sequence information, we can define three evolutionary trends: i) in B. garinii some plasmids (group IV and VI) were duplicated and diversified at the expense of plasmids from group III that are highly amplified in other species ii) unique plasmids in each species were either acquired by horizontal transfer [24] or diversification of existing plasmids. iii) plasmids of B. afzelii PKo and B. garinii PBi are more similar to each other than to those of B. burgdorferi B31 as it is especially pronounced in group VI. This is in good agreement with phylogenetic tree reconstructions using different genes [5, 6] and implies that at least these defined plasmid groups evolved without horizontal gene transfer.
Together, the three way comparison presented here provides a concise description of the plasmids of Bbsl and offers new insights into their evolutionary origin.
Loss of genetic material and infectivity during passage
Borrelia species can lose their infectivity, when passaged several times without selection pressure for infectivity. This loss is accompanied by loss of genetic material [18, 25]. It is not possible to estimate the extent of this loss based solely on PFGE or PCR, especially if the whole genomic sequence is not known. We therefore sequenced and analysed low and high passage strains of B. garinii as well as of B. afzelii together to be able to describe all sequence differences and to determine the cause of lost pathogenicity.
Surprisingly, not a whole plasmid but only a part of a plasmid was lost during passage in B. garinii PBi. The remaining part of the plasmid was either retained by chance or is essential. The lost part consists of vls gene cassettes whose frequent exchange is involved in the escape of B. burgdorferi from the host response [19, 20]. Thus, this selective loss may be caused by the failure to correctly repair the locus after recombination. Absence of selection pressure then allows this clone with the truncated plasmid to prosper in the culture. In order for the entire culture to lose the locus, cassette switching errors must be frequent or the mutated clone has a substantial growth advantage.
In B. afzelii PKo an entire plasmid is missing in the high passage strain. This plasmid also contained the vls genes as could be concluded from the presence of a few vls cassette derived reads. Loss of entire plasmids could occur if the plasmids are not correctly divided up between the daughter cells during division. Yet, we found that two sequences in the assembly of the fragments (Table 3) are derived from the high passage strain DNA indicating a two step process of plasmid loss. In this scenario the vls cluster is lost first as in B. garinii PBi, and in a second step the non essential remainder of the plasmid is lost successively.
Interestingly, in the tick vector the vls locus seems to be silent [26] and thus is stable in this host. This can explain, why the accidental loss does not occur in nature.
Thus, loss of infectivity is accompanied in both species by the loss of the vls locus. While this loss seems to be the sole reason for the loss of infectivity in B. garinii PBi, the influence of the additional material lost in B. afzelii PKo on infectivity remains to be determined. Yet, we believe that this additional loss occurred only by coincidence due to the non essential nature of the remaining parts of the plasmid. This leaves the vls locus as the prominent but fragile cause for the successful thriving of Borrelia species in their respective vertebrate hosts.
Conclusion
Comparative sequencing is a valuable tool to discern species specific and common genetic material. Our analysis of genomes from B. garinii and B. afzelii strains together with the previously described B. burgdorferi B31 genome shows that more plasmids are indispensable for B. burgdorferi sensu lato than previously thought. In Borreliae lack of selection can lead to the loss of genetic material accompanied by a phenotype change. A base by base analysis of these losses as we made it here can not only reveal genes important for the phenotype change upon their loss but also gives clues about underlying mechanisms for the losses. Further studies on additional low and high passage strains should reveal whether the loss of the vls cassette is the major or even single causative event which leads to loss of infectivity.
Methods
The B. garinii strain PBi (OspA serotype 4) is a CSF isolate from a German patient with neuroborreliosis. Passage 12 is still infectious for gerbils, but infectivity was lost between passage 30 and 60 [27]. B. afzelii strain PKo was originally isolated from human skin [28], the early passage is still infectious as can be demonstrated with gerbils. Early (8th subculture) and late (70th subculture) passages of B. afzelii PKo and a late passage of B. garinii PBi (300th) were grown in MVP-medium as described [29].
DNA was extracted using the Genomic DNA Bufferset (Quiagen GmbH, Hilden) and Genomic tip 500/6 and 100/6 (Quiagen, GmbH, Hilden). Genomic libraries with a target insert size of 1.5 kb using total DNA were constructed as described previously [16]. From all libraries sufficient clones were sequenced from both ends to give a sequence coverage of at least 5× per species (see Table 1).
All sequences were checked for similarity to chromosomal or cp26 and lp54 sequences of B. burgdorferi using BLAST [30] and assembled separately as described previously [16]. All other plasmid sequences were assembled together and manually checked for assembly errors. In the case of the multiple copies of cp30 in B. afzelii we used polymorphism and read pair information to generate unambiguously assembled contigs. This enabled us to define at least 8 different plasmids according to the placement of the contigs on a common backbone. Since we did not know, which contigs related to linear plasmids form a natural unit, we performed a PCR based analysis of possible connections between all segments. We tested all possible combinations of segment ends to determine combinations that produced PCR products when using whole genomic DNA as a template. Resulting PCR products were sequenced and used to close gaps in the plasmid assemblies. No further unambiguous PCR products were obtained linking the ends of the contigs. Thus, the obtained sequences represent either a full set of linear plasmids or, as may be the case for the smaller segments, missing links due to the inability to design unique primer pairs.
Initial gene finding was performed using GeneMarkS [31, 32]. Annotation was done using the GenColors system [33].
The alignments between the Borreliae collinear chromosomes [see Additional file 1] were generated using the program stretcher, which is part of the EMBOSS package [34]. The similarity plot program tupleplot [35] with default settings was used for a systematic comparison of plasmids of all species to each other.
All data are available from the Spirochetes Genome Browser [36] and were submitted to GenBank, as well as to NCBI Trace and Assembly Archives.
References
Lyme Disease. [http://textbookofbacteriology.net/Lyme.html]
Marti Ras N, Postic D, Foretz M, Baranton G: Borrelia burgdorferi sensu stricto, a bacterial species "made in the U.S.A."?. Int J Syst Bacteriol. 1997, 47 (4): 1112-1117.
Saint Girons I, Gern L, Gray JS, Guy EC, Korenberg E, Nuttall PA, Rijpkema SG, Schonberg A, Stanek G, Postic D: Identification of Borrelia burgdorferi sensu lato species in Europe. Zentralbl Bakteriol. 1998, 287 (3): 190-195.
Wilske B, Busch U, Eiffert H, Fingerle V, Pfister HW, Rossler D, Preac-Mursic V: Diversity of OspA and OspC among cerebrospinal fluid isolates of Borrelia burgdorferi sensu lato from patients with neuroborreliosis in Germany. Med Microbiol Immunol (Berl). 1996, 184 (4): 195-201.
Fukunaga M, Okada K, Nakao M, Konishi T, Sato Y: Phylogenetic analysis of Borrelia species based on flagellin gene sequences and its application for molecular typing of Lyme disease borreliae. Int J Syst Bacteriol. 1996, 46 (4): 898-905.
Wang G, van Dam AP, Schwartz I, Dankert J: Molecular typing of Borrelia burgdorferi sensu lato: taxonomic, epidemiological, and clinical implications. Clin Microbiol Rev. 1999, 12 (4): 633-653.
Strother KO, Broadwater A, De Silva A: Plasmid requirements for infection of ticks by Borrelia burgdorferi. Vector Borne Zoonotic Dis. 2005, 5 (3): 237-245. 10.1089/vbz.2005.5.237.
Casjens S, Palmer N, van Vugt R, Huang WM, Stevenson B, Rosa P, Lathigra R, Sutton G, Peterson J, Dodson RJ, Haft D, Hickey E, Gwinn M, White O, Fraser CM: A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol Microbiol. 2000, 35 (3): 490-516. 10.1046/j.1365-2958.2000.01698.x.
Busch U, Will G, Hizo-Teufel C, Wilske B, Preac-Mursic V: Long-term in vitro cultivation of Borrelia burgdorferi sensu lato strains: influence on plasmid patterns, genome stability and expression of proteins. Res Microbiol. 1997, 148 (2): 109-118. 10.1016/S0923-2508(97)87642-5.
Huang WM, Robertson M, Aron J, Casjens S: Telomere exchange between linear replicons of Borrelia burgdorferi. J Bacteriol. 2004, 186 (13): 4134-4141. 10.1128/JB.186.13.4134-4141.2004.
Hardison RC: Comparative genomics. PLoS Biol. 2003, 1 (2): E58. Epub 2003 Nov 17.-10.1371/journal.pbio.0000058.
Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES: Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003, 423 (6937): 241-254. 10.1038/nature01644.
Seshadri R, Myers GS, Tettelin H, Eisen JA, Heidelberg JF, Dodson RJ, Davidsen TM, DeBoy RT, Fouts DE, Haft DH, Selengut J, Ren Q, Brinkac LM, Madupu R, Kolonay J, Durkin SA, Daugherty SC, Shetty J, Shvartsbeyn A, Gebregeorgis E, Geer K, Tsegaye G, Malek J, Ayodeji B, Shatsman S, McLeod MP, Smajs D, Howell JK, Pal S, Amin A, Vashisth P, McNeill TZ, Xiang Q, Sodergren E, Baca E, Weinstock GM, Norris SJ, Fraser CM, Paulsen IT: Comparison of the genome of the oral pathogen Treponema denticola with other spirochete genomes. Proc Natl Acad Sci U S A. 2004, 101 (15): 5646-51. Epub 2004 Apr 2.. 10.1073/pnas.0307639101.
Anjum MF, Lucchini S, Thompson A, Hinton JC, Woodward MJ: Comparative genomic indexing reveals the phylogenomics of Escherichia coli pathogens. Infect Immun. 2003, 71 (8): 4674-4683. 10.1128/IAI.71.8.4674-4683.2003.
Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, Gwinn M, Dougherty B, Tomb JF, Fleischmann RD, Richardson D, Peterson J, Kerlavage AR, Quackenbush J, Salzberg S, Hanson M, van Vugt R, Palmer N, Adams MD, Gocayne J, Weidman J, Utterback T, Watthey L, McDonald L, Artiach P, Bowman C, Garland S, Fuji C, Cotton MD, Horst K, Roberts K, Hatch B, Smith HO, Venter JC: Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997, 390 (6660): 580-586. 10.1038/37551.
Glöckner G, Lehmann R, Romualdi A, Pradella S, Schulte-Spechtel U, Schilhabel M, Wilske B, Suhnel J, Platzer M: Comparative analysis of the Borrelia garinii genome. Nucleic Acids Res. 2004, 32 (20): 6038-6046. 10.1093/nar/gkh953.
Grimm D, Elias AF, Tilly K, Rosa PA: Plasmid stability during in vitro propagation of Borrelia burgdorferi assessed at a clonal level. Infect Immun. 2003, 71 (6): 3138-3145. 10.1128/IAI.71.6.3138-3145.2003.
Schwan TG, Burgdorfer W, Garon CF: Changes in infectivity and plasmid profile of the Lyme disease spirochete, Borrelia burgdorferi, as a result of in vitro cultivation. Infect Immun. 1988, 56 (8): 1831-1836.
Zhang JR, Hardham JM, Barbour AG, Norris SJ: Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell. 1997, 89 (2): 275-285. 10.1016/S0092-8674(00)80206-8.
Ohnishi J, Schneider B, Messer WB, Piesman J, de Silva AM: Genetic variation at the vlsE locus of Borrelia burgdorferi within ticks and mice over the course of a single transmission cycle. J Bacteriol. 2003, 185 (15): 4432-4441. 10.1128/JB.185.15.4432-4441.2003.
Wang D, Botkin DJ, Norris SJ: Characterization of the vls antigenic variation loci of the Lyme disease spirochaetes Borrelia garinii Ip90 and Borrelia afzelii ACAI. Mol Microbiol. 2003, 47 (5): 1407-1417. 10.1046/j.1365-2958.2003.03386.x.
Stevenson B, Miller JC: Intra- and interbacterial genetic exchange of Lyme disease spirochete erp genes generates sequence identity amidst diversity. J Mol Evol. 2003, 57 (3): 309-324. 10.1007/s00239-003-2482-x.
Palmer N, Fraser C, Casjens S: Distribution of twelve linear extrachromosomal DNAs in natural isolates of Lyme disease spirochetes. J Bacteriol. 2000, 182 (9): 2476-2480. 10.1128/JB.182.9.2476-2480.2000.
Qiu WG, Schutzer SE, Bruno JF, Attie O, Xu Y, Dunn JJ, Fraser CM, Casjens SR, Luft BJ: Genetic exchange and plasmid transfers in Borrelia burgdorferi sensu stricto revealed by three-way genome comparisons and multilocus sequence typing. Proc Natl Acad Sci U S A. 2004, 101 (39): 14150-14155. 10.1073/pnas.0402745101.
Xu Y, Kodner C, Coleman L, Johnson RC: Correlation of plasmids with infectivity of Borrelia burgdorferi sensu stricto type strain B31. Infect Immun. 1996, 64 (9): 3870-3876.
Indest KJ, Howell JK, Jacobs MB, Scholl-Meeker D, Norris SJ, Philipp MT: Analysis of Borrelia burgdorferi vlsE gene expression and recombination in the tick vector. Infect Immun. 2001, 69 (11): 7083-7090. 10.1128/IAI.69.11.7083-7090.2001.
Wilske B, Preac-Mursic V, Gobel UB, Graf B, Jauris S, Soutschek E, Schwab E, Zumstein G: An OspA serotyping system for Borrelia burgdorferi based on reactivity with monoclonal antibodies and OspA sequence analysis. J Clin Microbiol. 1993, 31 (2): 340-350.
Wilske B, Fingerle V, Preac-Mursic V, Jauris-Heipke S, Hofmann A, Loy H, Pfister HW, Rossler D, Soutschek E: Immunoblot using recombinant antigens derived from different genospecies of Borrelia burgdorferi sensu lato. Med Microbiol Immunol (Berl). 1994, 183 (1): 43-59.
Preac-Mursic V, Wilske B, Reinhardt S: Culture of Borrelia burgdorferi on six solid media. Eur J Clin Microbiol Infect Dis. 1991, 10 (12): 1076-1079. 10.1007/BF01984935.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25 (17): 3389-3402. 10.1093/nar/25.17.3389.
Genemark Gene Prediction. [http://opal.biology.gatech.edu/GeneMark]
Besemer J, Lomsadze A, Borodovsky M: GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29 (12): 2607-2618. 10.1093/nar/29.12.2607.
Romualdi A, Siddiqui R, Glöckner G, Lehmann R, Suhnel J: GenColors: accelerated comparative analysis and annotation of prokaryotic genomes at various stages of completeness. Bioinformatics. 2005, 21 (18): 3669-3671. 10.1093/bioinformatics/bti606.
EMBOSS package. [http://www.emboss.org]
Tupleplot Software. [http://genome.fli-leibniz.de/software.html]
Spirochetes Genome Browser. [http://sgb.fli-leibniz.de/]
Acknowledgements
We are grateful to A. Romualdi, the main developer of the GenColors system, for his support. We also thank A. Heidel for suggestions on the style of the manuscript. MF and MS were funded by a grant from the BMBF
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
GG, BW, and MP conceived the study, US-S and BW provided material and input on the manuscript, MS, MF, and JS performed the bioinformatics. GG wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2006_594_MOESM1_ESM.doc
Additional File 1: Analysis of plasmid relationships and losses. The additional file contains tuple plot analysis for all plasmids named in the text and a Southern analysis of the B. garinii PBi low and high passage genome using the vls cassette as a probe. (DOC 440 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Glöckner, G., Schulte-Spechtel, U., Schilhabel, M. et al. Comparative genome analysis: selection pressure on the Borrelia vls cassettes is essential for infectivity. BMC Genomics 7, 211 (2006). https://doi.org/10.1186/1471-2164-7-211
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-7-211