
Abstract
Background
Aureobasidium pullulans is a black-yeast-like fungus used for production of the polysaccharide pullulan and the antimycotic aureobasidin A, and as a biocontrol agent in agriculture. It can cause opportunistic human infections, and it inhabits various extreme environments. To promote the understanding of these traits, we performed de-novo genome sequencing of the four varieties of A. pullulans.
Results
The 25.43-29.62 Mb genomes of these four varieties of A. pullulans encode between 10266 and 11866 predicted proteins. Their genomes encode most of the enzyme families involved in degradation of plant material and many sugar transporters, and they have genes possibly associated with degradation of plastic and aromatic compounds. Proteins believed to be involved in the synthesis of pullulan and siderophores, but not of aureobasidin A, are predicted. Putative stress-tolerance genes include several aquaporins and aquaglyceroporins, large numbers of alkali-metal cation transporters, genes for the synthesis of compatible solutes and melanin, all of the components of the high-osmolarity glycerol pathway, and bacteriorhodopsin-like proteins. All of these genomes contain a homothallic mating-type locus.
Conclusions
The differences between these four varieties of A. pullulans are large enough to justify their redefinition as separate species: A. pullulans, A. melanogenum, A. subglaciale and A. namibiae. The redundancy observed in several gene families can be linked to the nutritional versatility of these species and their particular stress tolerance. The availability of the genome sequences of the four Aureobasidium species should improve their biotechnological exploitation and promote our understanding of their stress-tolerance mechanisms, diverse lifestyles, and pathogenic potential.
Similar content being viewed by others

Background
Aureobasidium pullulans (de Bary) G. Arnaud is a polyextremotolerant black yeast of considerable biotechnological importance, and it has exceptional stress tolerance and increasing medical relevance [1]. A. pullulans is well known for its production of pullulan, a neutral polysaccharide of repeating maltotriose units, which has numerous applications in medicine, pharmacy, the food industry, and other fields [2, 3]. A. pullulans also produces a β-glucan that shows high reactivity to human IgG antibodies [4] and possible beneficial immunomodulatory effects [5]. A. pullulans has an unusually large spectrum of extracellular enzymatic activities [6, 7]. Several of these are of biotechnological interest, and include: amylases, cellulases, lipases, proteases, xylanases, β-fructofuranosidases, maltosyltransferases, mannanases, and laccases (for review, see [8]). At least some of these appear to have interesting traits that are different from their homologues in other species [8, 9].
A strain of A. pullulans is used for the production of a cyclic peptide that has specific antifungal activity: aureobasidin A [10]. Due to its strong antagonistic activity against other microorganisms, A. pullulans is used as a biocontrol agent in agriculture [11]. Additionally, a recent study reported that some strains of A. pullulans can produce an antibacterial compound, exophilin A, as well as high yields of liamocins, and heavy oils with previously unknown acylated mannitol structures, which have possible industrial applications as surfactants [12].
The occurrence of A. pullulans is widespread in tropical, temperate and polar areas. It is frequently found in association with diverse plants, such as in the phyllosphere [13, 14], as an epiphyte or endophyte, on stored barley grain [15], and in coconut water [16]. A. pullulans has also been found in numerous other habitats, some of which are particularly unusual, such as coastal hypersaline water [17, 18], glacial ice [19, 20], other polar environments [21, 22], polluted water [23], refrigerated, frozen, salt-preserved and dried foods [24, 25], various indoor habitats (e.g., bathroom surfaces [26], house dust [27], dishwashers [28], tap water [29]), the surface of human skin [30], aviation fuel tanks [31], and the surface of synthetic polymers [32] and of degrading polyurethane and PVC plastics [33]. A. pullulans has been reported to cause a variety of localised infections in humans, and even systemic infections, although very rarely (for review, see [34, 35]). It has been suggested that this infection potential is at least partially supported by the production of extracellular enzymes [36], and by the pronounced stress tolerance of A. pullulans[1].
Aureobasidium pullulans has evolved an exceptional tolerance for a broad range of ecological conditions. It is considered a polyextremotolerant organism [1, 37], and it can survive hypersaline [17], acidic, basic [38, 39], cold and oligotrophic [40] conditions. Some of the A. pullulans adaptations to stress (and especially to elevated salt concentrations) are associated with rigorous management of intracellular concentrations of alkali-metal cations [41], synthesis of compatible solutes [42] and of mycosporines [43], and adaptation at the level of the membrane-lipid composition [44, 45]. A. pullulans can also mitigate environmental stress by rapid dimorphic switching from small colourless yeast cells to thick-walled, heavily melanised, meristematic forms [46].
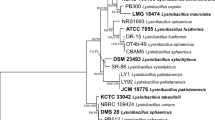
The genus Aureobasidium is a member of the order Dothideales (Ascomycota, Dothideomycetes; Figure 1), and it comprises 27 taxa (species and varieties), with A. pullulans being by far the most studied of these. Although the distinction of varieties and forms of A. pullulans has been suggested, the name A. pullulans is mainly used in research databases. The most recently described species of Aureobasidium are A. leucospermi Crous [47], A. proteae (Joanne E. Taylor & Crous) Joanne E. Taylor & Crous 2011 [47], and A. thailandense S.W. Peterson, Manitchotpisit & Leathers [48].

The Aureobasidium pullulans varieties. A. Phylogram showing the phylogenetic relationships of the four A. pullulans varieties and their phylogenetic position, inferred from super alignment of selected fungal proteomes. Chi2-based branch supports are shown, calculated according to the approximate Likelihood-Ratio Test, as implemented in Phyml 3.0. B. Representative images of one-month-old cultures of the four A. pullulans varieties (as indicated) on malt extract agar and microscopy images of cultures after one week of growth on malt extract agar blocks.
Various loci have been sequenced in the past to infer the taxonomy and phylogeny of the taxa in Aureobasidium, such as internal transcribed spacer (ITS) rDNA, intergenic spacer 1, translation elongation factor-1α, β-tubulin, and RNA polymerase II [19, 47–49]. Based on a multilocus analysis of a worldwide selection of A. pullulans-like isolates, it was confirmed that the earlier described variety A. pullulans var. melanogenum Hermanides-Nijhov is distinct from A. pullulans (de Bary) G. Arnaud 1918. In addition, two new varieties have been described: A. pullulans var. subglaciale Zalar, de Hoog & Gunde-Cimerman, and A. pullulans var. namibiae Zalar, de Hoog & Gunde-Cimerman. A. pullulans var. aubasidani Yurlova (Yurlova and de Hoog), which had been previously described due to its structurally unique polysaccharide, aubasidan, was synonymised with A. pullulans var. pullulans[19].
In 2008, the infraspecies classification of the varieties was retained in the redefinition of the species to prevent confusion linked to the use of new or additional epithets in applied fields [19]. Nevertheless, substantial phenotypic differences in growth-temperature range, melanisation, and tolerance to NaCl had already been observed among the varieties. A. pullulans var. pullulans is found mostly in mildly osmotic environments, it is frequently associated with plants, and it can tolerate up to 17% NaCl (w/v), the highest salt concentration of all four of the A. pullulans varieties [19]. A. pullulans var. melanogenum has been isolated mainly from oligotrophic, aqueous environments, and it grows at 37°C, while the other varieties cannot. On the other hand, A. pullulans var. subglaciale is unique for its psychrotolerant nature (it grows at 4°C) and its occurrence in glacial habitats in Svalbard (Norway). Finally, A. pullulans var. namibiae was named based on a single isolate from Namib Desert marble [19]. Polymorphism is evident within A. pullulans, even within the varieties (Figure 1), and the species has been proposed as a model for the investigation of fungal phenotypic plasticity, because of the unique colony morphologies, the frequent changes in appearance and growth-temperature regimes, and the different nutrient requirements and morphological responses to light among its strains and varieties [50].
The genome sequence of one strain of each of the above-described varieties of A. pullulans was obtained: A. pullulans var. pullulans, A. pullulans var. melanogenum, A. pullulans var. subglaciale (type strain), and A. pullulans var. namibiae (type strain). The results of the genome analyses performed are presented and discussed in the following sections.
Results and discussion
The sequencing of the genomes of the four varieties of A. pullulans was aimed to uncover the genetic basis of their many interesting and useful traits. This included three aspects in particular: (i) their biotechnological potential (e.g., genes for extracellular enzymes, and for production of pullulan, aureobasidin A, and other compounds of interest); (ii) their exceptional stress tolerance (mainly focusing on genes associated with the synthesis of compatible solutes and water management, transport of alkali-metal ions and water, the high osmolarity glycerol [HOG] pathway, and melanin synthesis); and (iii) evidence in favour of describing these varieties as separate species.
Genome properties: sequencing and assembly of the genomes
The genome assembly sizes of the four sequenced varieties of A. pullulans were 29.62 Mb (var. pullulans), 25.80 Mb (var. subglaciale), 25.43 Mb (var. namibiae), and 26.20 Mb (var. melanogenum) (Table 1). These are smaller than the sizes of all but one of the 18 Dothideomycete fungi that were compared by Ohm et al. [51], and that had an average genome size of almost 39 Mb.
Aureobasidium pullulans var. pullulans contains more repetitive sequences than the other A. pullulans varieties. However, the level of the repetitive sequences is low across all four of these varieties (0.78%-1.45%), and cannot explain the differences in their genome sizes. A. pullulans var. pullulans is the most halotolerant of the four investigated varieties [19], and it was isolated from hypersaline water. Therefore, it is interesting to note that investigations of Saccharomyces cerevisiae have revealed a tendency towards increased genome size as a response to high concentrations of salt [52, 53]. An increased genome size was also observed for the extremely halotolerant black yeast Hortaea werneckii, which has experienced recent whole-genome duplication [54]. However, the increase in the genome size of A. pullulans var. pullulans is less extensive, as its genome is only 14.8% larger than the genome of its closest relative, A. pullulans var. subglaciale.
The numbers of predicted proteins (from 10266 in A. pullulans var. namibiae, to 11866 in A. pullulans var. pullulans; Table 1) do not differ substantially from those observed in related fungi, where the average number of the predicted proteins in 18 related species that were analysed in a recent study was 11955 [51]. The median number of introns in the predicted genes of A. pullulans varieties is two introns per gene, with a median length of 57 bp (Table 1). Between 80% and 90% of the predicted proteins are in all four of the A. pullulans varieties, while 16% to 19% of the proteins are duplicated (Figure 2).

The shared, unique and duplicated proteins of the Aureobasidium pullulans varieties. A. Unique and shared protein families, as determined by the Markov clustering algorithm. B. Proteins shared between individual varieties, as determined by all-against-all blastp. C. Proteins present in at least two copies in the proteome of each of the four A. pullulans varieties. P, A. pullulans var. pullulans; S, A. pullulans var. subglaciale; N, A. pullulans var. namibiae; M, A. pullulans var. melanogenum.
(i) Biotechnological potential of the Aureobasidium pullulansvarieties
Synthesis of pullulan
Pullulan is a linear α-D-glucan that is made of maltotriose units connected with α-1,6 linkages, while the glucose units within the maltotriose are connected with α-1,4-glycosidic bonds. This alternation of the bonds gives the molecule its distinctive physical properties: flexibility, solubility, adhesive ability, biodegradability, and the ability to form viscous solutions, and oxygen-impermeable and transparent fibres and films. Pullulan thus has broad application value [8]. Despite its economic importance, relatively little is known about the biosynthesis of pullulan. Duan et al. [55] proposed a biosynthetic pathway in which the key enzymes for converting glucose units into pullulan were α-phosphoglucose mutase, uridine diphosphoglucose pyrophosphorylase, and glucosyltransferase.
There are single-copy genes for phosphoglucose mutase and uridine diphosphoglucose pyrophosphorylase in all four A. pullulans varieties. This is expected, as both of these enzymes catalyse important metabolic reactions, and they have been evolutionarily conserved. The enzyme phosphoglucose mutase catalyses a key step in hexose metabolism: the interconversion of glucose 1-phosphate and glucose 6-phosphate. The enzyme uridine diphosphoglucose pyrophosphorylase catalyses the reversible formation of uridine diphosphoglucose from glucose 1-phosphate and UTP. As described below, the predicted A. pullulans secretomes contain representatives of several Carbohydrate-Active enZYme (CAZy) database families [56], which include members with glucosyltransferase activities. Thus all four A. pullulans varieties contain all of the putative enzymes that were proposed to be involved in the biosynthesis of pullulan by Duan et al. [55].
Kang et al. [57] showed that disruption of the putative pullulan synthetase gene (pul) of A. pullulans [GenBank:AF470619] reduces its exopolysaccharide production to a pure β-glucan. Each of the predicted proteomes of the four A. pullulans varieties contains one similar protein (JGI Protein IDs: 349889, 3372, 53761, 64747, in A. pullulans var. pullulans, subglaciale, namibiae, and melanogenum, respectively; e-values between 10-22 and 10-27). All of these are predicted as being secreted. The low similarity scores can be attributed to the differences in annotation, as the pul gene prediction in GenBank contains an atypically long intron (almost 600 bp) and its second exon lies in a poorly conserved region (Additional file 1). When the annotation of the pul gene [GenBank:AF470619] was corrected to correspond to our annotation, it contained a one-nucleotide frameshift–insertion at nucleotide position 62 (Additional file 1, blue). After this was removed, the resulting protein-coding sequences and the predicted protein sequences were more than 70% identical. It is possible that the insertion in AF470619 was an artefact of the sequencing and that it led to the differences in the above-described gene annotation. If the pul gene is indeed involved in pullulan production, this observation is of significant importance.
Synthesis of aureobasidin A and siderophores
Strains of A. pullulans are used efficiently as biocontrol agents of post-harvest diseases [11]. Not only numerous hydrolytic enzymes [58], but also various antimicrobial compounds, such as exophilin A [12], siderophores [59] and aureobasidin A [10], might have roles in the strong antagonistic effects of A. pullulans towards other species.
The antibiotic aureobasidin A is a cyclic nonadepsipeptide [10] that shows strong fungicidal activity, including against Candida species, Cryptococcus neoformans, and some Aspergillus species [60]. Aureobasidin A has been shown to inhibit the phosphatidylinositol:ceramide phosphoinositol transferase that is involved in sphingolipid synthesis [60]. The synthesis of aureobasidin A is catalysed by the 11659-amino-acid-long biosynthesis complex that is encoded by the intronless gene aba1 of the A. pullulans strain R106 [61].
The four sequenced A. pullulans varieties have not been tested previously for aureobasidin A production. However, their genomes do not include any homologues of the aba1 gene. This is not entirely surprising, as the production of aureobasidin A is not a universal trait of A. pullulans.
However, in a search for aureobasidin A synthase, a group of similar non-ribosomal peptide synthases was identified, although these proteins are substantially shorter than aureobasidin A synthase (1123–2920 amino acids) and are most similar to synthases for siderophores. A. pullulans var. melanogenum and A. pullulans var. namibiae have one putative synthase for siderophores, whereas A. pullulans var. subglaciale has two copies, and A. pullulans var. pullulans has three.
Siderophores are iron-chelating compounds with substantial biotechnological potential that are known to be produced by A. pullulans[62]. Siderophores have been reported to act as antimicrobials [59]. Their presence is also in line with the oligotrophic nature of A. pullulans, as they are beneficial under conditions of iron-depletion. Additionally, in other fungi, siderophores have roles in virulence, fungal–host interactions, and resistance to oxidative stress [63].
Secondary metabolite biosynthesis clusters
Fungi produce a multitude of low-molecular-mass compounds, known as secondary metabolites, which have roles in a range of cellular processes, such as transcription, development, and intercellular communication [64]. Many of these compounds have important applications as antibiotics or immunosuppressants [64], and they are used in medicine or for plant protection [65]. Genome mining investigations have indicated that the ability of fungi to produce secondary metabolites has been substantially underestimated, because many of the fungal secondary metabolite biosynthesis gene clusters are not expressed under standard cultivation conditions [64].
In A. pullulans var. pullulans, only nine secondary metabolite biosynthetic clusters were identified with Antibiotics and Secondary Metabolites Analysis Shell (antiSMASH 2.0; http://antismash.secondarymetabolites.org/). However, there are 32 clusters in A. pullulans var. namibiae, and 37 in each of A. pullulans var. melanogenum and A. pullulans var. subglaciale (Table 2; Additional file 2). These cluster numbers are surprisingly high, and although many cannot be assigned to production of a certain metabolite, their abundance suggests that the genomes of these A. pullulans varieties represent rich resources for secondary metabolite biosynthesis. In comparison, antiSMASH predicted 45 T1 polyketide synthase/non-ribosomal peptide synthase/dmat gene clusters in Penicillium chrysogenum, 29 clusters in Aspergillus fumigatus[66], and a total of 35 putative gene clusters in a draft genome sequence of Streptomyces ansochromogenes (Streptomycetes are known for their complex secondary metabolism) [67]. For example, lantipeptide biosynthetic clusters from A. pullulans var. pullulans, var. namibiae and var. melanogenum might be of particular interest. Lantipeptides are post-translationally modified peptides that have antimicrobial properties. Lantipeptides were initially believed to be produced only by Gram-positive bacteria, but later studies revealed a much wider diversity of lantipeptide producers than previously appreciated (and also of their activity, structure, and biosynthetic machinery) [68]. Although dozens of new lantipeptides have been isolated in recent years, bioinformatic analyses indicate that many hundreds more await discovery, owing to the widespread frequency of the lantipeptide biosynthetic machinery in the bacterial genome [68].
Secreted proteins
An unusually large spectrum of extracellular enzymatic activities has been described for A. pullulans, many of which are of considerable biotechnological interest [6–8]. Some of these, such as alkaline serine proteases, glucanases and chitinases, are also believed to have roles in the above-described antagonistic effects of A. pullulans against phytopathogenic fungi [58].
The in-silico predicted set of secreted proteins comprises 869 proteins for A. pullulans var. pullulans (7.3% of the predicted proteome), 813 for A. pullulans var subglaciale (7.5%), 734 for A. pullulans var. namibiae (7.1%), and 725 for A. pullulans var. melanogenum (6.8%) (Table 3 and Additional file 3). The proportion of the proteins that contain signalP (predictor for secretory signal peptides) and TMHMM (predictor for transmembrane helices) sequences is comparable to that in other fungi [69]. The largest group of the secretory proteins are those similar to enzymes that are active against carbohydrates (41.1% to 45.8% of total secreted proteins; Table 3). Most of these are glycoside hydrolases (GHs; approximately a quarter of the secretome, and over half of the enzymes that are active against carbohydrates).
All four of these A. pullulans varieties also contain a large number of enzyme families, the members of which are known to be involved in the degradation of plant material, as designated by [70]. These are discussed below.
The enzymes involved in the degradation of pectin are represented by three families of polysaccharide lyases (PL1, PL3, PL4), and the families of GH28 (polygalacturonases), GH78 (rhamnosidases), GH105 (rhamnogalacturonyl hydrolases) and the carbohydrate esterases CE8 (pectinesterases). The GH88 family (d-4,5-unsaturated β-glucuronyl hydrolases) is only present in A. pullulans var. namibiae, although it is not recognised as secreted. No representatives of the polysaccharide lyases from the PL9 and PL11 families were identified.
In all four of these A. pullulans varieties we identified five out of seven protein families with members that are involved in (among other activities) the degradation of cellulose (they contain the GH6, GH7, GH12, GH45, and AA9 families, but not the GH74 and GH94 families). A. pullulans var. namibiae contains all of the 15 families that contain hemicellulases (GH10, GH11, GH27, GH29, GH35, GH36, GH39, GH43, GH51, GH53, GH54, GH62, GH67, GH93, GH115), while the other three A. pullulans varieties lack the GH39 family (Additional file 3). This diversity is comparable to, and in some cases even larger than, other plant pathogenic fungi [70], including those from Dothideomycetes (Additional file 4) [51], and it is larger than in most of the saprophytic fungi, especially in the case of the hemicellulases (Additional file 4) [70].
Although the sequence-based families of carbohydrate-active enzymes frequently group together enzymes of varying substrate specificities, they are nevertheless considered to be good reporters of fungal lifestyles, especially when considering broad substrate categories, such as cellulose, hemicellulose or pectin [51]. The richness of the enzyme families that are involved in the degradation of plant material is thus interesting in light of the epiphytic lifestyle of A. pullulans. When comparing individual varieties, proteins containing PFAM protein family domains (http://pfam.sanger.ac.uk/), which are characteristic for the GH3 (PF00933, PF01915), GH5 (PF00150), GH16 (PF00722), GH28 (PF01915), and GH43 (PF04616) families, are significantly enriched in A. pullulans var. pullulans compared to the other varieties (using the Computational Analysis of Gene Family Evolution [CAFE] software [71]), while the GH3 and GH5 families are depleted in A. pullulans var. melanogenum. This corresponds to the plant-related ecology of A. pullulans var. pullulans and to the largest phylogenetic distance between A. pullulans var. pullulans and A. pullulans var. melanogenum.
The numbers of genes and families of secreted proteases in the genomes of A. pullulans are also comparable to those found in the dothideomycete plant pathogens [51]. All of the subfamilies of the MEROPS database of proteolytic enzymes [72], which are expected to efficiently digest proteins and/or to work in inhospitable environments of the extracellular matrix (A01, C13, G01, M35, M20, S08, S09, S10; [51]), are represented in these predicted secretomes. The only exception is C13 protease, which is recognised as being secreted only in A. pullulans var. melanogenum, whereas the homologues from the other varieties are not. The family of scytalidoglutamic peptidases (G01) contains even more representatives than in the analysed plant pathogens. Aspartic proteases (PF00026) and the A4 family (PF01828) are enriched in A. pullulans var. pullulans.
The number of lipases in the predicted secretomes is relatively low, although the number of cutinases, which are enzymes that are important for the degradation of the plant cuticle, is in the range observed in related plant pathogens [51] (Table 3). However, some of the putative cutinases are not designated as secreted with the criteria used in the present analysis, so the actual number of these enzymes might be even higher.
Tannases are another group of enzymes that are significantly enriched within the genome of A. pullulans var. pullulans. Tannases, or tannin acyl hydrolases (EC, 3.1.1.20), catalyse the hydrolysis of ester bonds in the hydrolysable tannins and gallic acid esters. These enzymes are used industrially as catalysts in the manufacture of gallic acid, and they also have potential use in beverage and food processing [73].
Given the previously recognised substantial biotechnological potential of the large spectrum of the extracellular enzymatic activities of this fungus [6–8], which is confirmed here by the predicted secretome, their application should be brought to a new level with the availability of these genomic data. The wide array of enzymatic activities should also be considered when investigating the pathogenicity potential of A. pullulans. As already noted by Chan et al. [36], lipases, phospholipases, proteases and β-lactamases are important virulence factors that might have roles in A. pullulans as an emerging opportunistic human pathogen.
Biodegradation of plastic and aromatic compounds
Black yeast, including A. pullulans, are known for their ability to degrade aromatic pollutants [74, 75] and plastic [76, 77]. In the A. pullulans AY4 strain, Chan et al. [36] reported the presence of genes coding for 2-monooxygenases and catechol dioxygenases (biodegradation of aromatic substances) and depolymerase (biodegradation of plastic). There are two families of 2-monooxygenases in these four A. pullulans varieties. The larger of these has six representatives in each of A. pullulans var. pullulans and A. pullulans var. subglaciale, five in A. pullulans var. melanogenum, and four in A. pullulans var. namibiae. The second family contains only one representative in A. pullulans var. pullulans and A. pullulans var. subglaciale. A family of proteins with catechol 1,2-dioxygenase activity is also present, with four representatives in A. pullulans var. melanogenum and five in each of the other three A. pullulans varieties. There are single-copy genes for poly-β-hydroxybutyrate depolymerase, an enzyme that can degrade polyhydroxyalkanoates [78], in all four of these A. pullulans varieties. The presence of these genes underlines the considerable potential of A. pullulans for use in bioremediation or biodegradation processes.
Major facilitator superfamily sugar transporters
The major facilitator superfamily (MFS) of sugar transporters is the largest group of secondary transmembrane carriers. This consists of 74 protein families, each of which is specialised for the transport of a certain type of substrate. They have different transport modes, from uniport, to solute:cation (H+ or Na+) symport and/or solute:proton or solute:solute antiport. Most of these have 400 to 600 amino acyl residues, with two six-transmembrane segment repeat units [79]. According to the Transporter Classification (TC) database [80], two representatives in this group, the MFS family (TC no. 2.1) and the glycoside-pentoside-hexuronide:cation symporter family (TC no. 2.2), contain sugar-specific families of secondary active transporters. In Eukarya, only two families of MFS sugar transport substrates are known, of which the sugar porter family (TC no. 2.1.1) is the largest of all of the MFS families, with several hundred known members. The sugar porter family is diverse with respect to their substrate specificity, and they can work either as uniporters or as cation symporters. The other familiy of sugar transporters is the sialate:H1 symporter family (TC no. 2.1.12) [81].
According to CAFE analysis, proteins that contain the PFAM domain, which is characteristic for sugar-transport proteins (PF00083), are significantly enriched in the genomes of all four A. pullulans varieties. When comparing these varieties individually, the sugar-transport proteins are also significantly enriched in A. pullulans var. pullulans, while they are depleted in A. pullulans var. melanogenum. Detailed analysis has revealed great diversity of these transporters (Table 4). The sugar porter family contains from 73 (A. pullulans var. melanogenum) to 92 (A. pullulans var. pullulans) predicted proteins, which is much higher compared to the 32 transporters of Neurospora crassa[82], or the 31 of S. cerevisiae[82, 83] (Table 4). However, it is lower than the number of MFS sugar transporters in Aspergillus nidulans (100) and Aspergillus oryzae (119) (http://www.membranetransport.org, 1. 8. 2013). More than three quarters of the transporters in these four A. pullulans varieties are classified as sugar: H+ symporters (Table 4), while in S. cerevisiae, more than half are uniporters [83].
The high numbers of sugar transporters correspond to the nutritional diversity of A. pullulans, while the differences between the four A. pullulans varieties might reflect their ecological preferences. A. pullulans var. pullulans is most frequently associated with plants, and it thus has access to various sugars of plant origin. On the other hand, A. pullulans var. melanogenum has significantly fewer sugar transporters, and it is associated more with freshwater habitats, where simple sugars are not as readily available. It is also separated from A. pullulans var. pullulans by the largest evolutionary distance of all of these A. pullulans varieties. The most notable difference between these varieties is in the number of the putative maltose or α-glucoside: H+ symporters (TC no. 2.A.1.1.10 or 2.A.1.1.11), as A. pullulans var. melanogenum has almost half the number of copies compared to the other varieties.
(ii) Polyextremotolerance
Components of the high-osmolarity glycerol pathway
The mitogen-activated protein kinases (MAPKs) are involved in many cellular processes, such as stress responses and regulation of differentiation and proliferation, and they are highly conserved in eukaryotes. This system consists of three kinases that phosphorylate one another in a signalling cascade. One of the best-characterised is the high-osmolarity glycerol (HOG) pathway, which is a branched MAPK signal-transduction system, the physiological role for which in fungi is primarily the mediation of cellular adaptation to increased osmolarity of the surrounding medium. The HOG pathway is also required for adaptation to other stress conditions, such as oxidative, heavy-metal, and hot or cold stress [84] and in the virulence of pathogenic fungi [85].
The HOG pathway in fungi consists of two major signalling modules. One is the MAPK module that comprises MAPK, MAPK kinase (MAPKK), and MAPKK kinase (MAPKKK). The other module is a two-component phospho-relay system that is composed of hybrid sensor kinases, a histidine-containing phosphotransfer protein, and response regulators; this system senses and relays environmental signals, and subsequently activates the HOG pathway [84, 85].
As expected, the main components [86] in the four A. pullulans varieties are very similar to those in other fungi. These genomes contain one homologue of each kinase in the MAPK module (Hog1 MAPK, Pbs2 MAPKK, and two MAPKKKs, Ssk2/22 and Ste11), one Ste50 kinase homologue, and one homologue of all of the components in two sensory branches Sln1 and Sho1 (Sln1, Ypd1, Ssk1, Sho1). Interestingly, A. pullulans var. pullulans and A. pullulans var. namibiae have two predicted homologues of Ste20 or Cla4 kinases (activators of Ste11 MAPKKK), while the other two varieties have only one. One of the duplicated homologues is missing the pleckstrin-homology domain (PF00169). This domain has roles in the recruitment of proteins to different cellular membranes, and thus enables the proteins to interact with other components of the signal-transduction pathways [87]. Whether the observed difference has a role in the polyextremotolerance of A. pullulans remains a question for future studies.
Genes involved in the biosynthesis of compatible solutes
Stress tolerance is frequently associated with accumulation of small organic molecules that can act as protective compounds against different stress factors (for review, see [88]). In A. pullulans, exposure to high temperatures or high salt concentrations, or a combination thereof, leads to increased intracellular concentrations of the polyhydroxy compounds trehalose, mannitol and glycerol [42]. The concentrations of trehalose in A. pullulans increase in heat-stressed cells, and in simultaneously heat-stressed and salt-stressed cells, but not in cells subjected to salt stress alone. Mannitol increases under all of the stress conditions that have been examined, while an increase in intracellular glycerol has only been detected in salt-stressed cells [42]. Indeed, in response to a saline stress, glycerol is the most abundant compatible solute (Kogej, unpublished data). In other microbes, glycerol, trehalose and other polyols are also very important for life in extremely cold environments [89]. This might also be the case in the psychrotolerant A. pullulans, however, the role of compatible solutes at low temperatures has not yet been studied in this species.
Glycerol is synthesised from dihydroxyacetone phosphate, a glycolytic intermediate, via two reaction steps that are catalysed by nicotinamide adenine dinucleotide (NAD)-dependent glycerol-3-phosphate dehydrogenase (Gpd) and glycerol-3-phosphatase (Gpp) [90]. Both of the genes that encode these enzymes required for glycerol biosynthesis are present in the genomes of all four of these A. pullulans varieties, as single-copy genes. The yeast S. cerevisiae and the halophilic basidiomycete Wallemia ichthyophaga each have two copies of Gpd [91], while the extremely halotolerant H. werneckii has four, two of which can be attributed to a whole-genome duplication event [54]. The predicted Gpd proteins from A. pullulans lack the N-terminal PTS2 sequence that is important for peroxisome localisation, a trait also noted in other halotolerant fungi [92]. It was proposed that the constant cytosolic localisation of the Gpd1 homologues is advantageous for the organisms that live in extremely saline environments [92], as the osmoprotective role of Gpd is dependent on the cytosol and nuclear fractions of this protein [93].
The biosythesis of trehalose is a two-step process in which glucose 6-phosphate and UDP-glucose are first converted to α,α-trehalose 6-phosphate by trehalose-6-phosphate synthase (Tps), and then to trehalose by trehalose-6-phosphate phosphatase (Tpp) [94]. In Ascomycota, mannitol is also synthesised in two steps: fructose 6-phosphate is first reduced to mannitol 1-phosphate by NAD-dependent mannitol-1-phosphate dehydrogenase (Mpd), and then dephosphorylated to mannitol, by mannitol-1-phosphate phosphatase (Mpp) [95]. One copy of trehalose-phosphatase was found in each of the four investigated A. pullulans varieties, and two relatively dissimilar (~30% identical amino acids) trehalose synthases were identified. Two proteins in each variety have predicted mannitol dehydrogenase activities. One is similar to the homologues that are crucial for mannitol biosynthesis in other fungi, while the other is probably involved in the first step in the catabolism of mannitol [95]. Interestingly, no homologues of known fungal mannitol-1-phosphate phosphatases were found in any of these four A. pullulans genomes.
In the genomes of all four of the A. pullulans varieties, we identified a putative D-xylose reductase (cluster 19, 48900/38638, 70467, 36428 and 36838 for A. pullulans var. pullulans, var. subglaciale, var. namibiae and var. melanogenum, respectively). D-xylose reductase converts xylose to xylitol, a sweetener compound. The production of xylitol is a commercially interesting process [96, 97], which has rarely been contemplated in A. pullulans[98].
Melanin biosynthesis genes
The cells of A. pullulans produce a black pigment that has long been known to be 1,8-dihydroxynaphthalene (DHN)-melanin [99]. Melanin is a high-molecular-weight, dark brown or black pigment that can be produced by numerous fungi [100], and it has a known protective role under various stress conditions (for review, see [88]), and also under hypersaline conditions [101]. Melanin is located in the fungal cell wall, either enmeshed within the structure of the cell wall, or as its outermost layer [102]. Microbes predominantly produce melanin pigment via tyrosinases, laccases, catecholases, and the polyketide synthase pathway [103].
Dothideomycetes mostly produce DHN-melanin via the polyketide synthase pathway [104, 105]. DHN-melanin biosynthesis starts with polyketide synthase, with acetyl coenzyme A or malonyl coenzyme A as a precursor. The polyketide synthase produces 1,3,6,8-tetrahydroxynaphthalene, which is reduced by hydroxynaphthalene reductase to form scytalone. Dehydration of scytalone by scytalone dehydratase forms 1,3,8-trihydroxynaphthalene. In turn, 1,3,8-trihydroxynaphthalene reductase converts 1,3,8-trihydroxynaphthalene to vermelone, which is further dehydrated by scytalone dehydratase, to 1,8-DHN. Subsequent steps are believed to involve dimerisation of the 1,8-DHN molecules, followed by polymerisation that is catalysed by p-diphenol oxidase [106].
One melanin polyketide synthase gene was identified in each of these four A. pullulans varieties. This is in agreement with findings in the other DHN-melanin–producing fungi, which also have one melanin polyketide synthase [107–109]. Additionally, there are one of each of the trihydroxynaphthalene reductase-like gene and tetrahydroxynaphthalene reductase-like gene. Each of the two reductases from these four A. pullulans varieties falls within either a trihydroxynaphthalene-like or a tetrahydroxynaphthalene-like group, as has been observed in other melanised fungi. One of the traits not expected is the presence of two scytalone dehydratase-like genes in all of the investigated varieties except A. pullulans var. melanogenum. These duplicated genes form two distinct phylogenetic groups. Furthermore, our phylogenetic analysis of the scytalone dehydratases from these four A. pullulans varieties and the scytalone dehydratase sequences collected from the NCBI-NR protein database show the presence of two scytalone dehydratases in several other fungal species as well (Additional file 5). These duplicated genes form two distinct phylogenetic groups: the larger one (cluster I) contains scytalone dehydratases from known DHN-melanin–producing fungi, and proteins 67660, 42009, 345205 and 70470 (from A. pullulans var. melanogenum, var. subglaciale, var. pullulans, and var. namibiae, respectively). The second cluster has two subclusters: one (IIa) contains the three duplicate proteins from A. pullulans var. subglaciale, A. pullulans var. pullulans and A. pullulans var. namibiae (676013, 282919, 48527), together with both scytalone dehydratase proteins from Fusarium, Botryotinia and Metarhizium. The other subcluster (IIb) contains proteins that produce bluish-green pigment through the DHN-melanin pathway [110], and it has no homologues in the four A. pullulans varieties (Additional file 5). This is the first report of several scytalone dehydratase genes and proteins in a single fungal species. Thus, we have identified here genes that possibly encode essential components of the DHN-melanin biosynthesis pathway in all four of these A. pullulans varieties.
Aquaporins
A large family of major intrisic proteins (MIPs), which are membrane-channel proteins that are selective for the transport of water (aquaporins) or water plus glycerol (aquaglyceroporins), has been found in diverse life forms [111]. All of the aquaporins are transmembrane proteins with six transmembrane domains, and with their N-terminus and C-terminus in the cytosol. These are water channels that are 109-fold faster compared to ion channels and transporters. The presence of solute-permeable aquaporins represents a new concept in terms of absorption, not only because of the rate of the process, but also because of its osmotic implications [112].
Orthodox aquaporins mediate rapid and selective fluxes of water across biological membranes, and hence they have important roles in the osmoregulation of cells and organisms. Aquaglyceroporins, on the other hand, facilitate transmembrane transport of small uncharged molecules, like polyols, urea, arsenite, and many more, thereby having roles in nutrient uptake, osmoregulation, and probably other processes [113, 114]. Petterson et al. [115] divided the fungal aquaporins into four groups: one group of orthodox aquaporins, and three groups of aquaglyceroporins (Fps1-like, Yfl054-like, and aquaglyceroporins that do not fit into these groups). A later phylogenetic analysis of 229 major intrisic fungal proteins by Xu et al. [116] also classified the aquaporin-like genes into four clusters, delineated by functionally characterised major intrisic fungal proteins: orthodox aquaporins, aquaglyceroporins, facultative fungal aquaporins, and X intrinsic proteins.
Altogether, 11 aquaporin-like and aquaglyceroporin-like genes were identified in A. pullulans var. pullulans, A. pullulans var. melanogenum and A. pullulans var. subglaciale, and 12 in A. pullulans var. namibiae. These numbers are higher than those in other species: e.g., Laccaria bicolor has six or seven aquaporins, and the numbers in other investigated fungal species have been reported to be even lower [116] (Figure 3). Therefore, the high number of aquaporin-like and aquaglyceroporin-like genes in the genomes of these four polyextremotolerant A. pullulans varieties is striking. Such an abundance is typical for plants and mammals [116], but not for fungi. Our phylogenetic analysis of these A. pullulans aquaporin-like sequences and the sequences analysed by Xu et al. [116] shows that the major intrisic proteins from Aureobasidium varieties cluster with orthodox aquaporins, or true water channels (cluster I: five proteins in A. pullulans var. namibiae, four in each of the other varieties), with fungal aquaglyceroporins (cluster II: one protein in each of these four A. pullulans varieties), and with facultative aquaporins (cluster III: six proteins in each of these four A. pullulans varieties) (Figure 3, Additional file 6). As these water channels and osmoregulators enable sufficient water and/or compatible solute fluxes into and out of the cells under various osmotic conditions, the high abundance of aquaporins in these four A. pullulans varieties might be vital for survival and adaptability of these polyextremotolerant species in water-challenged environments. However, the possible beneficial effects of increased copy numbers of these proteins for survival in extreme environments have not been indicated previously. Furthermore, as only the MIPs in model yeast species and mycorrhizal fungal species have been studied so far, the aquaporins from the fungal species that inhabit various extreme habitats (e.g., drought-resistant and salt-tolerant species, like A. pullulans varieties) might have either high efficiency or unique mechanisms of aquaporin regulation [116] that are still awaiting discovery.

Aquaporin genes in Aureobasidium pullulans and other fungi. A. Protein tree of aquaporin-like genes from the four A. pullulans varieties and other fungi. The tree with GenBank accession numbers is available as Additional file 6. Colours correspond to previously recognised phylogenetic groups [116], and aquaporin-like proteins of the A. pullulans varieties are marked in red. B. Histogram showing the number of aquaporins in the four A. pullulans varieties, as compared to the other fungal species reported by Xu et al. [116].
Alkali-metal cation transporters
As a halotolerant species, A. pullulans has to maintain its intracellular cation homeostasis during changing and occasionally high environmental salinities. Under such conditions, the maintenance of a high and stable K+ content and the elimination of toxic Na+ ions is crucial for survival [117, 118]. However, it is not clear if and how the number of alkali-metal cation transporter genes is correlated with the halotolerance of an organism. The genome sequencing of the extremely halotolerant black yeast H. werneckii revealed an extensive enrichment of genes that encode transporters of alkali-metal cations [54]. These appear to allow H. werneckii to maintain the very low internal Na+ concentrations that are observed even at high external NaCl concentrations [41]. On the other hand, genome sequencing of the most halophilic fungus known, the basidomycete W. ichthyophaga, uncovered only modest numbers of ion-transporter genes, and additionally showed that their transcription is relatively low and non-responsive to different salt concentrations [91].
The analysis of these four A. pullulans genomes revealed several duplications of plasma-membrane transporters, but not of those located on the intracellular organelles (Table 5, Additional file 7). This is not entirely surprising, as the organelle membranes are protected by the relatively stable intracellular environment, rather than being exposed to the high extracellular fluctuations of inorganic salt ions [41]. From an additional analysis, which included the homologues from other Dothideomycetes along with the sequenced genomes of the four A. pullulans varieties, it is clear that most of the duplications observed occurred in the relatively distant evolutionary past (Figure 4). Three such events can be observed for the Nha Na+/H+ antiporter and the Trk K+ importer, two for the Pho Na+/Pi importer, and one for the Ena Na+ exporter, the Tok K+ exporter and the Acu K+ importer. Cochliobolus heterostrophus still has representatives in almost all of the resulting subgroups (15 of 17), followed by A. pullulans var. melanogenum (14), A. pullulans var. pullulans (13), A. pullulans var. subglaciale (13), H. werneckii (13), Cladosporium fulvum (13), and A. pullulans var. namibiae (12) (Additional file 8).

Protein trees of the various membrane transporters of Na+and K+. The protein trees are labelled according to the names of homologues from S. cerevisiae (except Acu transporters, which have no S. cerevisiae homologues). The trees (except for Acu) were rooted with homologous proteins from C. neoformans (Trk: [GenBank:XP_570017], [GenBank:XP_569339]; Tok: [GenBank:XP_568987], [GenBank:XP_568988]; Nha: [GenBank:XP_569560]; Ena: [GenBank:XP_572412], [GenBank:XP_568029], [GenBank:XP_570160]; Pho: [GenBank:XP_568082]) and root locations marked with an arrow. In addition to genes from A. pullulans, homologues from related fungi were used, as labelled with the fungal species name and GenBank accession number (H. werneckii, M. graminicola, S. cerevisiae), or the Joint Genome Institute Genome Portal protein ID (all of the rest). Putative gene duplications leading to the present diversity of these genes in A. pullulans are indicated by double arrows. ApP, A. pullulans var. pullulans; ApS, A. pullulans var. subglaciale; ApN, A. pullulans var. namibiae; ApM, A. pullulans var. melanogenum.
S. cerevisiae contains two typical K+-transport systems: Trk1 and Trk2, which are high-affinity channels for K+ uptake [119], while the Tok1 membrane-depolarisation-activated channel is for K+ efflux [120]. Ena1, Ena2 and Ena5 P-type ATPases [121–123] and the Nha1 antiporter, which is driven by a proton gradient, can also contribute to K+ efflux, although they are primarily known as exporters of Na+. In A. pullulans, both Trk and Tok channels are duplicated (Figure 4). A. pullulans var. pullulans contains another homologue of Trk that belongs to cluster 2, which is still present in H. werneckii and C. heterostrophus, but not in other fungi analysed. The same is true for A. pullulans var. subglaciale, although its homologue is uncharacteristically short (it contains only 137 amino acids, compared to the average of 808 for the other A. pullulans Trk proteins).
Unexpectedly, in addition to Trk, all of these four A. pullulans varieties contain two less-common K+ uptake systems (Table 5): the Hak H+/K+ symporters, which under some conditions appear to act as Na+/K+ symporters [124]; and the Acu P-type ATPases [124], which are encoded by not one, but two relatively divergent genes in each of these genomes (Figure 4). Both of these types of transporters are present in only a few other Dothideomycetes. According to the predicted phylogeny, it is parsimonious to assume that both of these K+ uptake systems were present in the ancestor of H. werneckii and were lost later in the evolution of this by-far-the-most halotolerant of all of the species analysed. Therefore, the primary purpose of the redundancy of these K+ uptake systems might not be adaptation to salt.
In other groups of transporters two duplications were observed in the above-mentioned Nha Na+/H+ antiporter, and one duplication in the Ena Na+ exporters (Figure 4). The smaller number of Ena genes compared to Nha (Table 5) has already been noted for H. werneckii[54]. Two duplication events and the subsequent loss of genes (Figure 4) led to different numbers of Pho Na+/Pi symporters in these individual A. pullulans varieties (Table 5). In salty environments, the Na+ gradient possibly represents an alternative energy source for the influx of Pi[54].
The most halotolerant of all of the species that have been analysed, H. werneckii, contains the highest number of transporter genes (30 genes), although it should be noted that the number is doubled due to the recent whole-genome duplication of H. werneckii[54]. The second largest number of transporter genes is present in C. heterostrophus (19 genes), which is not a halotolerant species (although, of note, it has not been studied in this respect), and is not found in hypersaline habitats. The high number of plasma-membrane K+ transporters of A. pullulans (seven or eight transporters across the four varieties) is in even starker contrast with the most halophilic fungus known, the basidiomycete W. ichthyophaga. The latter has only one Trk-encoding gene, and has no homologues of the Tok, Hak or Acu transporters [91].
When comparing only these four A. pullulans varieties, the most halotolerant A. pullulans var. pullulans has neither the highest number nor the largest diversity of transporters. Furthermore, although in S. cerevisiae the Ena pumps are the major determinant of salt tolerance (for review, see [117]), A. pullulans var. pullulans even has one Ena gene homologue less than the other A. pullulans varieties. Although the number of alkali-metal cation-transporter genes in A. pullulans is higher than in many related fungi, this difference is not particularly outstanding. It appears that the diversity and the total number of alkali-metal cation-transporter genes are (at best) only approximate predictors of halotolerance.
Only one gene for the plasma-membrane proton-exporting ATPase Pma is present in each of these four A. pullulans varieties. In contrast, H. werneckii has four copies of this gene, and their expression is salt responsive [54]. Pma pumps are responsible for establishing the electrochemical gradient of protons across the plasma membrane, and through this, they supply the energy to the secondary active symporters and antiporters [125, 126], among which there are also those responsible for maintaining ion homeostasis under hypersaline conditions.
In addition to the Pma pumps, other transporters might be responsible for the building of the electrochemical gradient of protons. Type 1 microbial rhodopsins can function as proton pumps to establish the electrochemical gradient for ATP production. This process has already been demonstrated in another dothideomycetous fungus, Leptosphaeria maculans[127]. An alternative way to build this proton-motive force would not only be useful in hypersaline conditions, as has already been proposed for the extremely halotolerant H. werneckii[128], but it might also be of importance in the light-exposed, oligotrophic biology of A. pullulans; e.g., to rapidly resume metabolism after cryptobiosis. The search for bacteriorhodopsins revealed at least two gene copies in the genomes of all four of the A. pullulans varieties (with three copies in A. pullulans var. melanogenum). The copies within these genomes differ in terms of their spliceosomal intron numbers and positions. According to our reconstructed phylogeny, these bacteriorhodopsin copies with one and two introns formed monophyletic groups (orthologues; see Additional file 9), and appear to represent old duplications before the split of the lineages within A. pullulans. However, A. pullulans var. melanogenum contains an additional third copy, suggesting either a recent duplication in this A. pullulans variety or a loss of a paralogue in the other A. pullulans varieties.
Although bacteriorhodopsins from A. pullulans have not yet been used for biotechnological purposes, they can be considered for the many different applications that this family of proteins has; e.g., mainly for optical appliances, but also for therapeutic/medical applications and research, including in optogenetics and bioelectronics [129, 130].
(iii) Description of new Aureobasidiumspecies
The genus Aureobasidium (Dothideales, Ascomycota, Dothideomycetes) is a conglomerate of cosmopolitian taxa. The correct taxonomic distinction of the phylogenetic clusters of this genus is of great importance for their industrial applications and investigations of their medical relevance. The analysis of the Kr distances between the genome pairs and the number of amino-acid substitutions per site of the aligned proteomes shows that the differences between these four A. pullulans varieties are comparable or larger than those observed between S. cerevisiae and three of its relatives, and especially its closest relative, Saccharomyces paradoxus (Figure 5, Additional file 10). Substantial differences between the genomes of these four A. pullulans varieties were also observed when their scaffolds were aligned with Mummer 3.23 (Additional file 11). Thus, in combination with previous knowledge, this suggested an elevation of these four A. pullulans varieties to the species level.

Kr distances between the genomes of Aureobasidium pullulans and selected pairs of genomes of Saccharomyces cerevisiae, Saccharomyces mikatae , and Saccharomyces kudriavzevii . Distances are represented by the lengths of the horizontal bars.
Elevation of these four A. pullulans varieties to the species level
A revised concept for the species A. pullulans is presented here. These four varieties of A. pullulans are elevated to the species level based primarily on the phylogenetic analysis of their whole genome sequences, as well as on their phylogenetically relevant genes.
Aureobasidium melanogenum (Hermanides-Nijhof) Zalar, Gostincar, Gunde-Cimerman, stat. nov. MycoBank MB 807698.
Basionym: Aureobasidium pullulans var. melanogenum Hermanides-Nijhof, Stud. Mycol. 15: 161, 1977. MycoBank MB352628.
Aureobasidium subglaciale (Zalar, de Hoog & Gunde-Cimerman) Zalar, Gostincar, Gunde-Cimerman, stat. nov. MycoBank MB 807700.
Basionym: Aureobasidium pullulans var. subglaciale Zalar, de Hoog & Gunde-Cimerman, Stud. Mycol. 61: 33, 2008. Mycobank MB512380.
Aureobasidium namibiae (Zalar, de Hoog & Gunde-Cimerman) Zalar, Gostincar, Gunde-Cimerman, stat. nov. MycoBank MB807701.
Basionym: Aureobasidium pullulans var. namibiae Zalar, de Hoog & Gunde-Cimerman, Stud. Mycol. 61: 34, 2008. Mycobank MB512381.
For the synonyms, see the previously published taxonomic study of Zalar et al. [19].
Taxonomic markers for distinguishing the new Aureobasidium species
Species/varieties within the genus Aureobasidium have most often been distinguished based on multilocus analyses of different regions of the rDNA gene clusters. The ITS region has been most commonly used [19, 47–49], although it is recognised as insufficient in certain genera [131]. Based on the ITS rDNA, the distinction of Aureobasidium pullulans was possible, but the resolution based on other household genes was better [19]. Therefore, the different household genes used earlier in the phylogenetic analyses of Aureobasidium and of other genera were compared here. The selected genes encode for the following proteins: actin (ACT), β-tubulin (BTUB), calmodulin (CAL), chytin synthase (CHS), NAD-dependent glycerol-3-phosphate dehydrogenase (GPD), mini-chromosome maintenance proteins essential for initiation of eukaryotic genome replication (MCM7), RNA polymerase 2, the largest subunit (RPB1), RNA polymerase 2, the second-largest subunit (RPB2), and translation elongation factor 1α (TEF1α). CAL and CHS are present in several different copies in these four Aureobasidium taxa and are thus less appropriate for use in phylogenetic analyses. Other markers were identified as single-copy genes in the genomes.
The overview of these genes – their length, number of exons/introns, possible candidate regions for phylogenetic markers in Aureobasidium species, and markers/regions already used in the phylogenetic analyses of Aureobasidium species are provided in Table 6.
The sequences of ACT, GPD and MCM7 have not yet been used in phylogenetic studies of Aureobasidium, although they represent appropriate candidate regions for such studies [135]. The positions of the introns in these genes are conserved in all four of the investigated genomes. To evaluate the usefulness of the listed gene exons and introns as barcodes for species identification or as phylogenetic marker loci, further analyses based on a larger set of Aureobasidium strains are required.
Comparisons of these loci show substantial differences among the sequenced Aureobasidium species. The largest variation is observed in the GPD gene. The lowest pairwise distance is between A. melanogenum and A. namibiae (0.158), and the highest between A. subglaciale and A. melanogenum (0.214). The smallest variation among these studied genes is in the ACT gene, where the pairwise distances range from 0.071 in A. melanogenum versus A. namibiae, to 0.092 in A. pullulans versus A. melanogenum (Table 7).
The genome sequences generated here also allowed the correct identification of two other ‘A. pullulans’ strains for which the genomes had been sequenced in the past and are publicly available. Strain ATCC 62921 (http://genome.fungalgenomics.ca/), which is of an unknown origin, but it has been used in numerous industrial applications, is confirmed as A. pullulans. The sequenced strain AY4, which was isolated from a human patient [36], is identified as A. melanogenum according to the above-presented species concept. The new species A. melanogenum also includes all other infection-causing ‘A. pullulans’ strains. Of the other commonly used ‘A. pullulans’ strains, strain NRRL Y-6220 is one of the commonly used strains for pullulan production, and it is related to A. melanogenum. On the other hand, the strain R106, which is used for aureobasidin A production, could not be reliably classified with the publicly available sequences, since in the phylogenetic analyses it was placed in a lineage separate from the here-described four species. The two Aureobasidium strains that are sold as commercial agricultural biocontrol products (DSM14940, DSM14941) remain in the species A. pullulans – for this purpose it is important to note that the species A. pullulans does not contain any potentially opportunistic pathogens, as these are placed in the newly described A. melanogenum.
Homothallic MAT loci and evidence for sexuality
A possible teleomorph of A. pullulans, Columnosphaeria fagi (H.J. Huds.) M.E. Barr, was suggested on the basis of the ITS sequence similarity [136], although this was not further confirmed either by a multilocus analysis or by a single-spore re-isolation of the fungus. Due to the lack of the recombination between the four previous A. pullulans varieties, it was concluded that A. pullulans is strictly clonal, although recombination within the four phylogenetic clusters of A. pullulans strains (at that time recognised as varieties) was not definitively ruled out [19]. However, the redefinition of these varieties as the four Aureobasidium species (see above) implies that the lack of the recombination between the varieties might be simply a consequence of interspecific reproduction barriers and not a sign of asexuality. The genome sequences of the four reclassified Aureobasidium species reveal additional details about their possible mating strategies.
The sexual reproduction of ascomycetous fungi is orchestrated by the presence of different arrangements of mating-type genes that encode the key transcription factors at one or more of the MAT loci: the MAT1-1-1 gene encodes a protein with an alpha1 domain, and the MAT1-2-1 gene encodes a protein with a high mobility group (HMG) box [137].
One mating type locus can be clearly recognised in each of the four investigated Aureobasidium species. The mating-type locus in all four of these species has a conserved organisation, and contains one of each of the MAT1-1-1 and MAT1-2-1 homologues in opposite orientations (Figure 6). This structure of the homothallic MAT locus, where two MAT idiomorphs are linked but not fused on the same locus, is also present in some other Dothideomycetes (e.g. Cochliobolus species, Stemphylium species) [137, 138]. Another set of genes in a syntenic region of all four of these Aureobasidium species (Additional file 12) might have the role of the second mating type locus, MAT2.

Configuration of the homothallic Aureobasidium species MAT loci and adjacent genes. Names of the four new Aureobasidium species are indicated above the gene models. Black boxes, exons; arrows, direction of transcription of the gene.
There is an open reading frame of an unknown function between the MAT1-1-1 and MAT1-2-1 genes in three of the studied genomes, with the genome of A. subglaciale being the exception. In A. subglaciale, this open reading frame is still present as a pseudogene, but due to a substantial degeneration, its structure can be discerned only approximately. In Sordariomycetes and Eurotiomycetes, the MAT locus frequently lies between the homologues of the S. cerevisiae APN2 and SLA2 genes [54, 137, 139]. In the here investigated Aureobasidium species, this is the case for the APN2 homologues (Figure 6); while the homologues of SLA2 are located on the same scaffold, they are separated from the MAT genes by a substantial distance (70 to 500 kbp).
The single-copy homologues of other genes involved in mating were found outside of the mating loci, including G-protein-coupled receptor for alpha-factor (Ste2 in S. cerevisiae) and a-factor (Ste3), and the transcription factor that activates genes involved in mating or pseudohyphal/ invasive growth pathways (Ste12). Out of nine meiosis-specific genes listed by Malik et al. [140], five were identified (homologues of S. cerevisiae SPO11, MSH4, MSH5, MER3, REC8). Interestingly, the homologue of MER3, a DNA helicase that is involved in crossing over [141], is present in three copies in A. melanogenum, and in two copies in A. pullulans, A. subglaciale and A. namibiae.
The roles of absent genes might be provided by alternative proteins. For example, the species A. pullulans contains two homologues of Rad51, which is a protein involved in the repair of double-strand DNA breaks. This might replace Dmc1, which performs the same function, but only in meiosis. Furthermore, the absence of the DMC1 homologue in sexual fungi is not unusual [142].
According to their genetic design, all four of the Aureobasidium species can be considered as homothallic (self-compatible, or self-fertile), which implies that a single strain is capable of sexual reproduction even in an axenic culture. However, the existence of mating genes is not conclusive evidence for sexual recombination, as these genes might also have other roles, as has been reported in other species of fungi [143]. As a teleomorph of the previously recognised A. pullulans has not been described yet, the ability of these four Aureobasidium species to undergo sexual reproduction needs to be confirmed experimentally.
Conclusions
Aureobasidium pullulans, as it was previously defined, was considered a species of exceptional adaptability. This was reflected in its great phenotypic plasticity, polyextremotolerance, and the ability to survive across a wide variety of habitats. A part of this diversity can be explained by the relatively wide definition of this species. However, with the accumulation of new knowledge, it had become clear that smaller, well-defined groups of strains can be recognised within A. pullulans. These were initially described as varieties [19], but this comparison of their genomic data shows that the differences between these varieties are large enough to justify their redefinition as four separate Aureobasidium species: A. pullulans, A. melanogenum, A. subglaciale and A. namibiae.
Even single strains of A. pullulans can show wide adaptability, and this is reflected in the great diversity of several groups of genes. This diversity can reach, or even surpass, the gene diversity of more specialised related species, even though almost all of them have larger genomes. The investigated genomes of these four Aureobasidium species contain an abundance of different families of extracellular enzymes, which are important for the degradation of the plant cell-wall material. Their diversity is comparable to the phylogenetically related plant pathogens. The differences in the abundance of the extracellular enzymes and also in the higher numbers of sugar transporters reflect the ecological preferences of the four studied Aureobasidium species, especially for the plant-associated lifestyle of A. pullulans. Furthermore, for example, all four of the Aureobasidium genomes presented here have genes for the alternative K+ importers Hak and Acu, which are not found even in the extremely halotolerant H. werneckii or in the halophilic W. ichthyophaga. Also, when considering the mating strategy, these four Aureobasidium species appear to be more flexible than the specialised H. werneckii and W. ichthyophaga: the four Aureobasidium genomes all contain a homothallic mating type locus, whereas H. werneckii is heterothallic, and W. ichthyophaga lacks the mating genes altogether.
The genome sequences of the described four Aureobasidium species are expected to facilitate the exploitation of the substantial biotechnological potential of these fungi. For most of the interesting genes, at least four different genome copies are now available for study, with potentially different traits of the proteins that they encode. Additionally, we propose the elimination of the decade-old annotation error in the putative pullulan synthase gene. Furthermore, the genes for the degradation of a variety of compounds from plant polysaccharides to plastics and aromatic compounds should lead to more efficient industrial use of these Aureobasidium species, as well as help in the selection or construction of the best strains for any given purpose.
The polyextremotolerant character of the investigated Aureobasidium species indicates that they have efficient stress-combating mechanisms, some of which have been described here. These mechanisms can potentially be used in the future to improve the stress tolerance of economically important microorganisms or plants. Last but not least, as the opportunistic human pathogens belong only to A. melanogenum, the phylogenetic redefinition of the previously recognised A. pullulans will facilitate the identification of the more problematic strains and help in limiting their use in applications in which they might represent potential health risks for workers or consumers.
Methods
Strains, growth conditions and microscopy
Aureobasidium pullulans var. pullulans (EXF-150, CBS 100280) was isolated from the hypersaline waters of the Sečovlje solar saltern (Slovenia). Aureobasidium pullulans var. melanogenum (EXF-3378, CBS 110374) was isolated from a public fountain in Bangkok (Thailand). Aureobasidium pullulans var. subglaciale (type strain; EXF-2481, CBS 123387) was isolated from the subglacial ice of the Kongsvegen glacier on Spitsbergen (Svalbard, Norway). Aureobasidium pullulans var. namibiae (type strain; EXF-3398, CBS 147.97) was isolated from dolomitic marble in the Namib Desert (Namibia). All of these strains are preserved in the Ex Culture Collection of the Department of Biology, Biotechnical Faculty, University of Ljubljana (Infrastructural Centre Mycosmo, MRIC UL) and at the Centraalbureau voor Schimmelcultures (CBS, Utrecht, The Netherlands).
These cultures were grown at 28°C in a rotary shaker (180 rpm) in the defined yeast nitrogen base medium (YNB) of 0.17% (w/v) yeast nitrogen base, 0.08% (w/v) complete supplement mixture (both Qbiogene), 0.5% (w/v) ammonium sulphate, and 2% (w/v) glucose, in deionised water. The pH was adjusted to 7.0 prior to autoclaving. For the isolation of RNA for transcriptome sequencing of A. pullulans var. pullulans and A. pullulans var. subglaciale, which was used for gene annotation, a variety of conditions was used: (1) YNB; (2) YNB with additional 10% (w/v) NaCl; (3) YNB with 2% (w/v) raffinose or (4) 31.3% (w/v) glycerol instead of glucose; (5) YNB with 49% (w/v) sorbitol; (6) yeast peptone glucose medium (1% [w/v] yeast extract, 2% [w/v] peptone, and 2% [w/v] glucose, in deionised water); (7) minimal medium (2% [w/v] glucose, macroelements [w/v]: 0.6% NaNO3, 0.15% KH2PO4, 0.05% MgSO4 × 7H2O, 0.05% KCl, microelements [w/v]: 0.01% EDTA, 0.0044% ZnSO4, 0.001% MnCl2 × 4H2O, 0.00032% CoCl2 × 6H2O, 0.00032% CuSO4 × 5H2O, 0.00022% (NH4)6Mo7O24 × 4H2O, 0.00147% CaCl2 × 2H2O, 0.001% FeSO4 × 7H2O, in deionised water); (8) minimal medium with 2% (w/v) cellulose instead of glucose; (9) YNB and an incubation temperature of 12°C; (10) YNB and incubation until mid-exponential phase followed by heat shock at 42°C for 45 min; (11) YNB and incubation until stationary phase; (12) YNB and incubation until mid-exponential phase, followed by addition of 0.32 mM H2O2 for 5 min; and (13) YNB and incubation until mid-exponential phase, followed by centrifugation and further resuspension either in YNB at pH 2 and a 4-h incubation (one generation), or (14) in Tris base at pH 10 for 30 min. Unless otherwise noted, the cells were harvested in the mid-exponential growth phase, using a 10-min centrifugation at 5000× g, and immediately frozen in liquid nitrogen.
For photography of the macromorphology, the four A. pullulans varieties were grown on malt extract agar (MEA) according to Blakeslee, containing 2% malt extract, 0.1% peptone, 2% glucose and 2% agar (all Merck), for 30 days at 24°C.
For microscopic images, slide cultures were prepared on MEA blocks in moist chambers, and they were incubated for 7 days at 25°C in the dark. The cover slips were carefully removed and mounted in 60% lactic acid for microscopic observation under the Olympus BX51 microscope, using an Olympus DP12 camera, and the DP software.
DNA and RNA isolation
Cells frozen in liquid nitrogen were homogenised using a pestle and a mortar. Their DNA was isolated using the phenol/ chloroform/ isoamyl alcohol method, modified for DNA isolation from filamentous fungi, as described previously [144]. The quality and quantity of the DNA was evaluated spectrophotometrically with NanoDrop 2000 (Thermo Fisher Scientific, USA), and on standard 1% agarose gel electrophoresis, with the molecular weight also checked.
The RNA was isolated using TRI Reagent (Sigma-Aldrich, Germany), according to the manufacturer instructions. Possible DNA contamination was removed with DNAse I (Thermo Fisher Scientific – Fermentas, Lithuania), and the integrity and purity of the RNA was evaluated spectrophotometrically with NanoDrop 2000 (Thermo Fisher Scientific, USA) and by capillary electrophoresis (Agilent 2100 Bioanalyser; Agilent Technologies, USA).
Genome sequencing and assembly
The draft genome of A. pullulans var. pullulans was sequenced with Illumina HiSeq using two libraries, fragment, 270-bp-insert size (2× 150 bp reads) and CLIP, 4-kbp-insert size (2× 100 bp reads). Each fastq file was QC filtered for artifacts/ process contamination, and subsequently assembled together with AllPathsLG version R38445 [145], to produce a 172× coverage 29.6 Mbp assembly in 186 scaffolds (L50 = 1.2 Mbp) and 209 contigs (L50 = 779.8 kbp). The 37121 bp mitochondrion genome was assembled separately in a single contig with AllPathsLG.
The genomes of A. pullulans var. namibiae, A. pullulans var. melanogenum, and A. pullulans var. subglaciale were sequenced using a single fragment Illumina library with 270-bp-insert size (2× 150 bp reads). After filtering for artifacts/ process contamination, the sequence data were assembled with Velvet 1.2.03 [146]. The resulting assembly was used to create a long mate-pair library with an insert of 3000 +/-300 bp, which was then assembled together with the original Illumina library with AllPathsLG release version R42328 [145]. The assembly statistics are summarised in Table 1.
Transcriptome sequencing and assembly
mRNA was purified from total RNA using Absolutely mRNA™ purification kits (Stratagene) and Dynabeads® mRNA purification kits (Invitrogen), and chemically fragmented to 200 bp to 250 bp (Ambion). The mRNA was reverse transcribed with SuperScript II, using random hexamers. Second-strand cDNA was synthesised using a dNTP/dUTP mix (Thermo Scientific), E. coli DNA ligase, E. coli DNA polymerase I, and E coli RnaseH (Invitrogen). The fragmented cDNA was treated with end-pair, A-tailing, and adapter ligation, using TruSeq Sample Preparation kits (Illumina) and KAPA-Illumina library creation kits (KAPA biosystems). Second-strand cDNA was removed by AmpErase UNG (Applied Biosystems), to generate strandedness. qPCR was used to determine the concentrations of the unamplified libraries. The libraries were sequenced on Illumina Hiseq.
In all, 120 million paired-end 100-bp Illumina HiSeq 2000 reads of stranded RNA-seq data were used as input for the de-novo assembly of A. pullulans var. subglaciale EXF-2481. One-hundred and sixty-four million paired-end 150-bp Illumina HiSeq 2000 reads of stranded RNA-seq data were used as input for the de-novo assembly of A. pullulans var. pullulans EXF-150. The reads were assembled into consensus sequences using Rnnotator 2.4.14 [147], which takes short read sequences as input, and outputs assembled transcript contigs. The reads were trimmed and filtered for quality, low-complexity, adapter and duplicates, and assembled with Velvet 1.2.03 [146]. The minimum contig length was set at 100 bp. The read depth minimum was set to 3 reads. Redundant contigs were removed using Vmatch 2.1.3, and contigs with significant overlap were further assembled using Minimus2, with a minimum overlap of 40 bp. Contig post-processing included splitting misassembled contigs, and contig extension and polishing using the strand information of the reads. Single base errors were corrected by aligning the reads back to each contig with BWA, to generate a consensus nucleotide sequence. Post-processed contigs were clustered into loci and putative transcript precursors were identified. The accuracy, completeness and contiguity of the assembled contigs were checked using Blat alignment [148] of the contigs to the available genomic reference. Here, 95.87% of the EXF-2481 contigs mapped to the genome, and 90.24% of the EXF-150 contigs mapped to the genome (threshold ≥95% of the contigs).
Genome annotation
All four of these genome assemblies of A. pullulans were annotated using the Pipeline JGI annotation [149], which combines several gene prediction and annotation methods, and integrates the annotated genome into the web-based fungal resource MycoCosm [150], for comparative genomics.
Before gene prediction, assembly scaffolds were masked using RepeatMasker [151] and RepBase library [152], with the most frequent (>150 times) repeats recognised by RepeatScout [153]. The following combination of gene predictors was run on the masked assembly: ab-initio Fgenesh [154] and GeneMark [155], trained for specific genomes; homology-based Fgenesh + [154] and Genewise [156], seeded by BLASTx alignments [157] against the NCBI-NR protein database; and transcriptome-based CombEST (Zhou et al., personal communication). In addition to protein-coding genes, tRNAs were predicted using tRNAscan-SE [158]. All of the predicted proteins were functionally annotated using SignalP [159] for signal sequences, TMHMM [160] for transmembrane domains, InterProScan [161] for the integrated collection of functional and structured protein domains, and protein alignments to NCBI-NR, SwissProt (http://www.expasy.org/sprot/), KEGG [162] for metabolic pathways, and KOG [163] for eukaryotic clusters of orthologues. Interpro and SwissProt hits were also used to map the gene-ontology terms [164]. For each genomic locus, the best representative gene model was selected based on a combination of protein similarity and EST support.
Shared, unique and duplicated genes
The numbers of shared and unique proteins of all four of the A. pullulans varieties were determined using all-against-all blastp (included in blast 2.2.25+) of their whole proteomes, with the e-value cut-off at 10-10[157]. Additionally, multigene families were predicted with the MCL, to cluster the proteins based on sequence similarity, using blastp alignment scores between proteins as a similarity metric [165]. The numbers of unique and shared clusters were retrieved from MycoCosm [166].
Mating loci, secreted proteins, membrane transporters
Mating loci were identified by using the JGI online blastp programme (e-value cut-off, 10-5). The S. cerevisiae Mat1-1-1 and Mat1-2-1 proteins were used as sequence queries. Additionally, other proteins characteristic of mating type loci were identified for each of the A. pullulans varieties, as described in [91]. A blastp search was performed for pheromone precursors (α and α-factors), MAPK scaffold (Ste5), transcription factors (Ste12) and pheromone factor receptors (Ste2, Ste3) [167], and also for the representative set of homologues of meiotic genes from S. cerevisiae (i.e., a ‘meiosis detection toolkit’) [140].
The prediction of secreted proteins was performed in several steps. First SignalP [168] was used to search for signal peptides in all of the predicted proteins of each of the four A. pullulans varieties, with the default cut-off D-value of 0.43. Proteins that were predicted by TargetP [169] to localise in other cellular compartments, as well as those containing predicted transmembrane regions by TMHMM [170], were removed from the selection. This produced a list of proteins that was expanded with the help of the multigene families predicted with the Markov clustering algorithm (MCL), as described below. Clusters that contained at least half of the proteins predicted as secreted (above) were automatically included in the list of secreted proteins (2506 proteins). This covered the cases where homologues of one or two of the A. pullulans varieties were not included in the original list due to annotation errors, or for other reasons. Clusters that contained less than half of the predicted secreted proteins (2274 proteins) were further analysed with online predictors CELLO [171] and WegoLoc [172], the latter with fungal BaCelLo and Höglund databases, with an e-value threshold of 1 × e-10 and a multiplex threshold of 1. The proteins were considered as secreted if they were predicted as such by at least three of four methods (our method described above, and three online predictors). The functions of predicted secreted proteins were assigned manually with the help of the available data about each of these proteins: placement into multigene families, gene ontology terms, presence of PFAM domains, functions of similar proteins from the NCBI-NR protein database, and results of CAZymes Analysis Toolkit [173] and MEROPS [72].
The analysis of the major facilitator transporters included all of the A. pullulans predicted protein sequences containing the PFAM domain (PF00083), which is characteristic of sugar transport proteins. The proteins were classified into sugar-specific families based on the phylogenetic analyses (performed as described below), which included the related proteins from other species. These were identified with blastp against the NCBI-NR protein database [157], or were retrieved from the Transporter Classification Database (http://www.tcdb.org/superfamily.php, 1. 8. 2013), and the MFS family sugar transporters of S. cerevisiae S288C were retrieved from the TransportDB database (http://www.membranetransport.org/, 1. 8. 2013).
Genes for alkali-metal cation transporters from Mycosphaerella graminicola, S. cerevisiae and C. neoformans were recovered from public databases, as described in [54], while the genes from H. werneckii were published in [54]. Homologues from other Dothideomycetes with sequenced genomes (the same as those used for phylogenetic analyses; see below) were recovered from the Genome Portal of the JGI, and were considered homologous to the analysed proteins from A. pullulans if they formed the same MCL cluster [166]. A second set of trees (for comparison purposes; not shown) was generated by applying a maximum parsimony method as implemented in the Mega software, version 5.05 [174]. The specificity of P-type pumps was inferred by phylogenetic analysis of all of the P-type cation transporters found from A. pullulans and their homologues with known function from other fungi, as described in [54].
Melanin polyketide synthase genes were identified by searching for a homologue of ALM1 from Alternaria alternata [GenBank:AB665444]. Homologues from Magnaporthe grisea [GenBank:L22309, GenBank:AY846877] were used as queries in the search for the trihydroxynaphthalene reductase-like gene and tetrahydroxynaphthalene reductase-like genes. Protein sequences of orthodox aquaporins Aqy1p and Aqy2p, and of aquaglyceroporins Fps1p and Yfl054p, from S. cerevisiae (sequences from the Saccharomyces genome database; http://www.yeastgenome.org/), were used as queries for the aquaporin genes. Other genes discussed were found with the help of blastp and MCL clustering data on the Genome Portal of the JGI [166], and analysed with various search strategies against the NCBI-NR protein database and the Saccharomyces Genome database.
Phylogenetic analyses
For the analysis of phylogenetic relationships of the four A. pullulans varieties and related species, a super alignment of the selected fungal proteomes was constructed with the Hal pipeline [175], allowing for no missing data. In addition to all four of the A. pullulans varieties, several other proteomes available on the Genome Portal of the Department of Energy Joint Genome Institute [166] were included: Baudoinia compniacensis[51], C. fulvum[176], C. heterostrophus[51], Hysterium pulicare[51], M. graminicola[177], N. crassa[82], Sclerotinia sclerotiorum[178], and Tuber melanosporum[179]. A. nidulans[180] was used as an outgroup. Hal produced a conservative alignment of 812692 amino acids. Poorly aligned positions and positions with gaps were removed with Gblocks 0.91b [181]. Stringent parameters were used: the maximum number of contiguous non-conserved positions was limited to 5 amino acids, and the minimum length of a block to 15 amino acids. This produced a 373159-bp-long alignment, which was used for the estimation of phylogeny.
The best protein evolution model was estimated with ProtTest 3.2.1 [182]. The species tree was generated with the PhyML 3.1 software [183]. Approximate Bayes (aBayes) branch supports were calculated. The analysis was run using the LG model of evolution. The ProtTest estimate of the α-parameter of the γ-distribution of six substitution rate categories (0.791), and the determined proportion of invariable sites (0.305) were used. The tree was then calibrated with the r8s software [184], by assigning the root of the tree to an arbitrary value of 1.
Gene phylogenies (alkali-metal cation transporters, major facilitator superfamily transporters, bacteriorhodopsins, components of HOG, aquaporins, scytalone dehydratases) were estimated by aligning the protein sequences with the MAFFT software in the ‘--auto’ mode [185]. The tree was generated with the PhyML 3.1 software [183], as described above, by using the model of protein evolution, the α-parameter, and the proportion of invariable sites, as estimated by ProtTest 3.2.1 [182].
Genome alignment and genome distances
The whole genome alignments between all of the possible pairs of these four genomes were calculated with the promer algorithm, as implemented in Mummer 3.23, and plotted with the mummerplot utility [186]. All of the parameters were the same as described by Hane et al. [187], except that instead of discarding scaffolds <500 kbp, only scaffolds <200 kbp were discarded.
The distances between these four A. pullulans varieties and between four Saccharomyces species (for comparison purposes) were estimated by using two methods. First, the K r distances were calculated with the genomediff software included in the GenomeTools library [188]. This alignment-free calculation produces Jukes-Cantor corrected divergences between the pairs of genomes as output. As a second approach, the proteomes were aligned with the Hal pipeline [175]. The conservative alignment was used for the estimation of the number of amino-acid substitutions per site, in Mega software version 5.05 [174]. The Poisson correction model was used, and all of the positions containing gaps or missing data were removed prior to analysis, which resulted in a 698915-amino-acid-long alignment.
CAFE analyses
Analysis of protein family expansions and contractions was performed with the CAFE 3.0 software [71]. Pfam domains of selected fungal proteomes were identified with a stand-alone Pfam scanner and a database downloaded on 30.1.2013 [189]. Additionally, multigene families predicted with the MCL, as described above, were used as a second variant of grouping the predicted proteins. This was used to produce a Table with the numbers of proteins belonging to specific protein families in each of the analysed species. The Table was used as the input of CAFE, together with the chronogram constructed from the whole proteomes, as described above.
This produced a list of groups of proteins with significant expansions/contractions. This was manually checked, and the groups with potential roles in stress tolerance or biotechnological use were selected for further analyses.
antiSMASH analysis
The antiSMASH 2.0 software [190] was used for the identification of the secondary metabolite biosynthesis clusters in the genome sequences. antiSMASH is the first comprehensive pipeline that can identify biosynthetic loci that cover the whole range of known secondary metabolite compound classes (polyketides, non-ribosomal peptides, terpenes, aminoglycosides, aminocoumarins, indolocarbazoles, lantibiotics, bacteriocins, nucleosides, β-lactams, butyrolactones, siderophores, melanins, and others) [66]. Nucleotide sequences of all four of the A. pullulans varieties were uploaded to the antiSMASH online site (http://antismash.secondarymetabolites.org/) in FASTA format, and the parameters were set to default, with the additional setting for the DNA of eukaryotic origin.
Data access
The genome assemblies and annotations can be interactively accessed through the JGI fungal genome portal MycoCosm [150] at http://genome.jgi-psf.org/Aurpu_var_pul1/Aurpu_var_pul1.home.html, http://genome.jgi-psf.org/Aurpu_var_sub1/Aurpu_var_sub1.home.html, http://genome.jgi-psf.org/Aurpu_var_nam1/Aurpu_var_nam1.home.html, and http://genome.jgi-psf.org/Aurpu_var_mel1/Aurpu_var_mel1.home.html, and have also been deposited with DDBJ/EMBL/GenBank under accession [GenBank:AYEO00000000, GenBank:AYYB00000000, GenBank:AYEM00000000, GenBank:AYEN00000000], for A. pullulans, A. subglaciale, A. namibiae, A. melanogenum, respectively.
Abbreviations
- antiSMASH:
-
Antibiotics and secondary metabolites analysis shell
- CAFE:
-
Computational analysis of gene family evolution
- DHN:
-
Dihydroxynaphthalene
- GH:
-
Glycoside hydrolases
- GT:
-
Glucosyltransferase
- HOG:
-
High osmolarity glycerol
- MFS:
-
Major facilitator superfamily
- MIP:
-
Major intrinsic protein
- MAPK:
-
Mitogen-activated protein kinase
- NCBI-NR:
-
National center for biotechnology information non-redundant
- PL:
-
Polysaccharide lyases.
References
Gostinčar C, Grube M, Gunde-Cimerman N: Evolution of fungal pathogens in domestic environments?. Fungal Biol. 2011, 115: 1008-1018.
Leathers TD: Biotechnological production and applications of pullulan. Appl Microbiol Biotechnol. 2003, 62: 468-473.
Cheng KC, Demirci A, Catchmark JM: Pullulan: biosynthesis, production, and applications. Appl Microbiol Biotechnol. 2011, 92: 29-44.
Tada R, Tanioka A, Ishibashi K, Adachi Y, Tsubaki K, Ohno N: Involvement of branched units at position 6 in the reactivity of a unique variety of beta-D-glucan from Aureobasidium pullulans to antibodies in human sera. Biosci Biotechnol Biochem. 2009, 73: 908-911.
Muramatsu D, Iwai A, Aoki S, Uchiyama H, Kawata K, Nakayama Y, Nikawa Y, Kusano K, Okabe M, Miyazaki T: β-glucan derived from Aureobasidium pullulans is effective for the prevention of influenza in mice. Plos One. 2012, 7:
Molnarova J, Vadkertiova R, Stratilova E: Extracellular enzymatic activities and physiological profiles of yeasts colonizing fruit trees. J Basic Microbiol. 2013, 53:
Buzzini P, Martini A: Extracellular enzymatic activity profiles in yeast and yeast-like strains isolated from tropical environments. J Appl Microbiol. 2002, 93: 1020-1025.
Chi Z, Wang F, Yue L, Liu G, Zhang T: Bioproducts from Aureobasidium pullulans, a biotechnologically important yeast. Appl Microbiol Biotechnol. 2009, 82: 793-804.
Rich JO, Leathers TD, Anderson AM, Bischoff KM, Manitchotpisit P: Laccases from Aureobasidium pullulans. Enzyme Microb Technol. 2013, 53: 33-37.
Takesako K, Ikai K, Haruna F, Endo M, Shimanaka K, Sono E, Nakamura T, Kato I, Yamaguchi H: Aureobasidins, new antifungal antibiotics taxonomy, fermentation, isolation, and properties. J Antibiot (Tokyo). 1991, 44: 919-924.
Sharma RR, Singh D, Singh R: Biological control of postharvest diseases of fruits and vegetables by microbial antagonists: A review. Biol Control. 2009, 50: 205-221.
Price NPJ, Manitchotpisit P, Vermillion KE, Bowman MJ, Leathers TD: Structural characterization of novel extracellular liamocins (mannitol oils) produced by Aureobasidium pullulans strain NRRL 50380. Carbohydr Res. 2013, 370: 24-32.
Andrews JH, Spear RN, Nordheim EV: Population biology of Aureobasidium pullulans on apple leaf surfaces. Can J Microbiol. 2002, 48: 500-513.
Grube M, Schmid F, Berg G: Black fungi and associated bacterial communities in the phyllosphere of grapevine. Fungal Biol. 2011, 115: 978-986.
Olstorpe M, Schnurer J, Passoth V: Microbial changes during storage of moist crimped cereal barley grain under Swedish farm conditions. Anim Feed Sci Technol. 2010, 156: 37-46.
Maciel NOP, Pilo FB, Freitas LFD, Gomes FCO, Johann S, Nardi RMD, Lachance MA, Rosa CA: The diversity and antifungal susceptibility of the yeasts isolated from coconut water and reconstituted fruit juices in Brazil. Int J Food Microbiol. 2013, 160: 201-205.
Gunde-Cimerman N, Zalar P, de Hoog S, Plemenitaš A: Hypersaline waters in salterns - natural ecological niches for halophilic black yeasts. FEMS Microbiol Ecol. 2000, 32: 235-240.
Oren A, Gunde-Cimerman N: Fungal life in the Dead Sea. Biology of Marine Fungi. Edited by: Raghukumar C. 2012
Zalar P, Gostinčar C, de Hoog GS, Uršič V, Sudhadham M, Gunde-Cimerman N: Redefinition of Aureobasidium pullulans and its varieties. Stud Mycol. 2008, 61: 21-38.
Branda E, Turchetti B, Diolaiuti G, Pecci M, Smiraglia C, Buzzini P: Yeast and yeast-like diversity in the southernmost glacier of Europe (Calderone Glacier, Apennines, Italy). FEMS Microbiol Ecol. 2010, 72: 354-369.
Vaz ABM, Rosa LH, Vieira MLA, de Garcia V, Brandao LR, Teixeira LCRS, Moline M, Libkind D, van Broock M, Rosa CA: The diversity, extracellular enzymatic activities and photoprotective compounds of yeasts isolated in Antarctica. Braz J Microbiol. 2011, 42: 937-947.
D'Elia T, Veerapaneni R, Theraisnathan V, Rogers SO: Isolation of fungi from Lake Vostok accretion ice. Mycologia. 2009, 101: 751-763.
Vadkertiova R, Slavikova E: Killer activity of yeasts isolated from the water environment. Can J Microbiol. 1995, 41: 759-766.
Pitt JI, Hocking AD: Fungi and food spoilage. 1999, Gaithersburg, Maryland: Aspen Publishers, Inc., 2
Nisiotou AA, Chorianopoulos N, Nychas GJE, Panagou EZ: Yeast heterogeneity during spontaneous fermentation of black Conservolea olives in different brine solutions. J Appl Microbiol. 2010, 108: 396-405.
Lotrakul P, Deenarn P, Prasongsuk S, Punnapayak H: Isolation of Aureobasidium pullulans from bathroom surfaces and their antifungal activity against some Aspergilli. Afr J Microbiol Res. 2009, 3: 253-257.
Kaarakainen P, Rintala H, Vepsalainen A, Hyvarinen A, Nevalainen A, Meklin T: Microbial content of house dust samples determined with qPCR. Sci Total Environ. 2009, 407: 4673-4680.
Zalar P, Novak M, De Hoog GS, Gunde-Cimerman N: Dishwashers - A man-made ecological niche accommodating human opportunistic fungal pathogens. Fungal Biol. 2011, 115: 997-1007.
Arvanitidou M, Kanellou K, Constantinides TC, Katsouyannopoulos V: The occurrence of fungi in hospital and community potable waters. Lett Apl Microbiol. 1999, 29: 81-84.
Zhang ES, Tanaka T, Tajima M, Tsuboi R, Nishikawa A, Sugita T: Characterization of the skin fungal microbiota in patients with atopic dermatitis and in healthy subjects. Microbiol Immunol. 2011, 55: 625-632.
Rauch ME, Graef HW, Rozenzhak SM, Jones SE, Bleckmann CA, Kruger RL, Naik RR, Stone MO: Characterization of microbial contamination in United States Air Force aviation fuel tanks. J Ind Microbiol Biotechnol. 2006, 33: 29-36.
Cappitelli F, Sorlini C: Microorganisms attack synthetic polymers in items representing our cultural heritage. Appl Environ Microbiol. 2008, 74: 564-569.
Shah AA, Hasan F, Hameed A, Ahmed S: Biological degradation of plastics: a comprehensive review. Biotechnol Adv. 2008, 26: 246-265.
Hawkes M, Rennie R, Sand C, Vaudry W: Aureobasidium pullulans infection: fungemia in an infant and a review of human cases. Diagn Microbiol Infect Dis. 2005, 51: 209-213.
Chan GF, Puad MSA, Chin CF, Rashid NAA: Emergence of Aureobasidium pullulans as human fungal pathogen and molecular assay for future medical diagnosis. Folia Microbiol (Praha). 2011, 56: 459-467.
Chan GF, Bamadhaj HM, Gan HM, Rashid NAA: Genome sequence of Aureobasidium pullulans AY4, an emerging opportunistic fungal pathogen with diverse biotechnological potential. Eukaryot Cell. 2012, 11: 1419-1420.
Gostinčar C, Grube M, de Hoog GS, Zalar P, Gunde-Cimerman N: Extremotolerance in fungi: evolution on the edge. FEMS Microbiol Ecol. 2010, 71: 2-11.
Ranta HM: Effect of simulated acid rain on quantity of epiphytic microfungi on Scots pine (Pinus sylvestris L.) needles. Environ Pollut. 1990, 67: 349-359.
Shiomi N, Yasuda T, Inoue Y, Kusumoto N, Iwasaki S, Katsuda T, Katoh S: Characteristics of neutralization of acids by newly isolated fungal cells. J Biosci Bioeng. 2004, 97: 54-58.
Onofri S: Antarctic Microfungi. Enigmatic microorganisms and life in extreme environments. Edited by: Seckbach J. 1999, Dordrecht; London: Kluwer Academic, 323-336. Cellular origin and life in extreme habitats; v.1,
Kogej T, Ramos J, Plemenitas A, Gunde-Cimerman N: The halophilic fungus Hortaea werneckii and the halotolerant fungus Aureobasidium pullulans maintain low intracellular cation concentrations in hypersaline environments. Appl Environ Microbiol. 2005, 71: 6600-6605.
Managbanag JR, Torzilli AP: An analysis of trehalose, glycerol, and mannitol accumulation during heat and salt stress in a salt marsh isolate of Aureobasidium pullulans. Mycologia. 2002, 94: 384-391.
Kogej T, Gostinčar C, Volkmann M, Gorbushina AA, Gunde-Cimerman N: Mycosporines in extremophilic fungi — novel complementary osmolytes?. Environ Chem. 2006, 3: 105-110.
Turk M, Mejanelle L, Šentjurc M, Grimalt JO, Gunde-Cimerman N, Plemenitaš A: Salt-induced changes in lipid composition and membrane fluidity of halophilic yeast-like melanized fungi. Extremophiles. 2004, 8: 53-61.
Gostinčar C, Turk M, Trbuha T, Vaupotič T, Plemenitaš A, Gunde-Cimerman N: Expression of fatty-acid-modifying enzymes in the halotolerant black yeast Aureobasidium pullulans (de Bary) G. Arnaud under salt stress. Stud Mycol. 2008, 61: 51-59.
Bermejo JM, Dominguez JB, Goni FM, Uruburu F: Influence of pH on the transition from yeast-like cells to chlamydospores in Aureobasidium pullulans. Antonie Van Leeuwenhoek. 1981, 47: 385-392.
Crous PW, Summerell BA, Swart L, Denman S, Taylor JE, Bezuidenhout CM, Palm ME, Marincowitz S, Groenewald JZ: Fungal pathogens of Proteaceae. Persoonia. 2011, 27: 20-45.
Peterson SW, Manitchotpisit P, Leathers TD: Aureobasidium thailandense sp. nov. isolated from leaves and wooden surfaces. Int J Syst Evol Microbiol. 2013, 63: 790-795.
Manitchotpisit P, Leathers TD, Peterson SW, Kurtzman CP, Li XL, Eveleigh DE, Lotrakul P, Prasongsuk S, Dunlap CA, Vermillion KE, Punnapayak H: Multilocus phylogenetic analyses, pullulan production and xylanase activity of tropical isolates of Aureobasidium pullulans. Mycol Res. 2009, 113: 1107-1120.
Slepecky RA, Starmer WT: Phenotypic plasticity in fungi: a review with observations on Aureobasidium pullulans. Mycologia. 2009, 101: 823-832.
Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, Barry KW, Condon BJ, Copeland AC, Dhillon B, Glaser F, Hesse CN, Kosti I, Labutti K, Lindquist EA, Lucas S, Salamov AA, Bradshaw RE, Ciuffetti L, Hamelin RC, Kema GH, Lawrence C, Scott JA, Spatafora JW, Turgeon BG, de Wit PJ, Zhong S, Goodwin SB, Grigoriev IV: Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog. 2012, 8: e1003037-
Gerstein AC, Chun HJE, Grant A, Otto SP: Genomic convergence toward diploidy in Saccharomyces cerevisiae. PLoS Genet. 2006, 2: 1396-1401.
Dhar R, Sagesser R, Weikert C, Yuan J, Wagner A: Adaptation of Saccharomyces cerevisiae to saline stress through laboratory evolution. J Evol Biol. 2011, 24: 1135-1153.
Lenassi M, Gostinčar C, Jackman S, Turk M, Sadowski I, Nislow C, Jones S, Birol I, Gunde-Cimerman N, Plemenitaš A: Whole genome duplication and enrichment of metal cation transporters revealed by de novo genome sequencing of extremely halotolerant black yeast Hortaea werneckii. PLoS ONE. 2013, 8: e71328-
Duan XH, Chi ZM, Wang L, Wang XH: Influence of different sugars on pullulan production and activities of alpha-phosphoglucose mutase, UDPG-pyrophosphorylase and glucosyltransferase involved in pullulan synthesis in Aureobasidium pullulans Y68. Carbohydr Polym. 2008, 73: 587-593.
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B: The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42: D490-D495.
Kang BK, Yang HJ, Choi NS, Ahn KH, Park CS, Yoon BD, Kim MS: Production of pure beta-glucan by Aureobasidium pullulans after pullulan synthetase gene disruption. Biotechnol Lett. 2010, 32: 137-142.
Zhang DP, Spadaro D, Valente S, Garibaldi A, Gullino ML: Cloning, characterization, expression and antifungal activity of an alkaline serine protease of Aureobasidium pullulans PL5 involved in the biological control of postharvest pathogens. Int J Food Microbiol. 2012, 153: 453-464.
Chi Z, Wang XX, Ma ZC, Buzdar MA, Chi ZM: The unique role of siderophore in marine-derived Aureobasidium pullulans HN6.2. Biometals. 2012, 25: 219-230.
Takesako K, Kuroda H, Inoue T, Haruna F, Yoshikawa Y, Kato I, Uchida K, Hiratani T, Yamaguchi H: Biological properties of aureobasidin A, a cyclic depsipeptide antifungal antibiotic. J Antibiot (Tokyo). 1993, 46: 1414-1420.
Slightom JL, Metzger BP, Luu HT, Elhammer AP: Cloning and molecular characterization of the gene encoding the aureobasidin A biosynthesis complex in Aureobasidium pullulans BP-1938. Gene. 2009, 431: 67-79.
Wang WL, Chi ZM, Chi Z, Li J, Wang XH: Siderophore production by the marine-derived Aureobasidium pullulans and its antimicrobial activity. Bioresour Technol. 2009, 100: 2639-2641.
Johnson L: Iron and siderophores in fungal-host interactions. Mycol Res. 2008, 112: 170-183.
Brakhage AA: Regulation of fungal secondary metabolism. Nat Rev Microbiol. 2013, 11: 21-32.
Scharf DH, Brakhage AA: Engineering fungal secondary metabolism: A roadmap to novel compounds. J Biotechnol. 2013, 163: 179-183.
Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R: antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39: 339-346.
Zhong XY, Tian YQ, Niu GQ, Tan HR: Assembly and features of secondary metabolite biosynthetic gene clusters in Streptomyces ansochromogenes. Sci China Life Sci. 2013, 56: 609-618.
Knerr PJ, van der Donk WA: Discovery, biosynthesis, and engineering of lantipeptides. Annu Rev Biochem. 2012, 81: 479-505.
Choi J, Park J, Kim D, Jung K, Kang S, Lee YH: Fungal Secretome Database: Integrated platform for annotation of fungal secretomes. BMC Genomics. 2010, 11:
Zhao ZT, Liu HQ, Wang CF, Xu JR: Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics. 2013, 14:
Han MV, Thomas GWC, Lugo-Martinez J, Hahn MW: Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol. 2013, 30: 1987-1997.
Rawlings ND, Barrett AJ, Bateman A: MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012, 40: D343-D350.
Belmares R, Contreras-Esquivel JC, Rodriguez-Herrera R, Coronel AR, Aguilar CN: Microbial production of tannase: an enzyme with potential use in food industry. Lebensm Wiss Technol. 2004, 37: 857-864.
Isola D, Selbmann L, de Hoog GS, Fenice M, Onofri S, Prenafeta-Boldu FX, Zucconi L: Isolation and screening of black fungi as degraders of volatile aromatic hydrocarbons. Mycopathologia. 2013, 175: 369-379.
Prenafeta-Boldu FX, Guivernau M, Gallastegui G, Vinas M, de Hoog GS, Elias A: Fungal/bacterial interactions during the biodegradation of TEX hydrocarbons (toluene, ethylbenzene and p-xylene) in gas biofilters operated under xerophilic conditions. FEMS Microbiol Ecol. 2012, 80: 722-734.
Webb JS, Nixon M, Eastwood IM, Greenhalgh M, Robson GD, Handley PS: Fungal colonization and biodeterioration of plasticized polyvinyl chloride. Appl Environ Microbiol. 2000, 66: 3194-3200.
Howard GT: Biodegradation of polyurethane: a review. Int Biodeterior Biodegradation. 2002, 49: 245-252.
Shivakumar S: Poly-beta-hydroxybutyrate (PHB) Depolymerase from Fusarium solani Thom. J Chem. 2013
Reddy VS, Shlykov MA, Castillo R, Sun EI, Saier MH: The major facilitator superfamily (MFS) revisited. FEBS J. 2012, 279: 2022-2035.
Saier MH, Tran CV, Barabote RD: TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006, 34: D181-D186.
Saier MH: Families of transmembrane sugar transport proteins. Mol Microbiol. 2000, 35: 699-710.
Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, FitzHugh W, Ma LJ, Smirnov S, Purcell S, Rehman B, Elkins T, Engels R, Wang S, Nielsen CB, Butler J, Endrizzi M, Qui D, Ianakiev P, Bell-Pedersen D, Nelson MA, Werner-Washburne M, Selitrennikoff CP, Kinsey JA, Braun EL, Zelter A, Schulte U, Kothe GO, Jedd G, Mewes W, et al: The genome sequence of the filamentous fungus Neurospora crassa. Nature. 2003, 422: 859-868.
Leandro MJ, Fonseca C, Goncalves P: Hexose and pentose transport in ascomycetous yeasts: an overview. FEMS Yeast Res. 2009, 9: 511-525.
Hohmann S: Control of high osmolarity signalling in the yeast Saccharomyces cerevisiae. FEBS Lett. 2009, 583: 4025-4029.
Bahn YS: Master and commander in fungal pathogens: the two-component system and the HOG signaling pathway. Eukaryot Cell. 2008, 7: 2017-2036.
Krantz M, Becit E, Hohmann S: Comparative genomics of the HOG-signalling system in fungi. Curr Genet. 2006, 49: 137-151.
Musacchio A, Gibson T, Rice P, Thompson J, Saraste M: The PH Domain - a common piece in the structural patchwork of signaling proteins. Trends Biochem Sci. 1993, 18: 343-348.
Gostinčar C, Muggia L, Grube M: Polyextremotolerant black fungi: oligotrophism, adaptive potential, and a link to lichen symbioses. Front Microbiol. 2012, 3: 390-
Robinson CH: Cold adaptation in Arctic and Antarctic fungi. New Phytol. 2001, 151: 341-353.
Ansell R, Granath K, Hohmann S, Thevelein JM, Adler L: The two isoenzymes for yeast NAD(+)-dependent glycerol-3-phosphate dehydrogenase encoded by GPD1 and GPD2 have distinct roles in osmoadaptation and redox regulation. EMBO J. 1997, 16: 2179-2187.
Zajc J, Liu Y, Dai W, Yang Z, Hu J, Gostinčar C, Gunde-Cimerman N: Genome and transcriptome sequencing of the halophilic fungus Wallemia ichthyophaga: haloadaptations present and absent. BMC Genomics. 2013, 14: 617-
Lenassi M, Zajc J, Gostinčar C, Gorjan A, Gunde-Cimerman N, Plemenitaš A: Adaptation of the glycerol-3-phosphate dehydrogenase Gpd1 to high salinities in the extremely halotolerant Hortaea werneckii and halophilic Wallemia ichthyophaga. Fungal Biol. 2011, 115: 959-970.
Jung S, Marelli M, Rachubinski RA, Goodlett DR, Aitchison JD: Dynamic changes in the subcellular distribution of Gpd1p in response to cell stress. J Biol Chem. 2010, 285: 6739-6749.
Francois J, Parrou JL: Reserve carbohydrates metabolism in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev. 2001, 25: 125-145.
Ruijter GJG, Bax M, Patel H, Flitter SJ, van de Vondervoort PJI, de Vries RP, van Kuyk PA, Visser J: Mannitol is required for stress tolerance in Aspergillus niger conidiospores. Eukaryot Cell. 2003, 2: 690-698.
Silva SS, Felipe MGA, Mancilha IM: Factors that affect the biosynthesis of xylitol by xylose-fermenting yeasts - A review. Appl Biochem Biotechnol. 1998, 70–72: 331-339.
Granstrom TB, Izumori K, Leisola M: A rare sugar xylitol. Part I: the biochemistry and biosynthesis of xylitol. Appl Microbiol Biotechnol. 2007, 74: 277-281.
Sugai JK, Veiga LA: Purification and properties of the xylitol dehydrogenase from Pullularia pullulans. An Acad Bras Cienc. 1981, 53: 183-193.
Siehr DJ: Melanin biosynthesis in Aureobasidium pullulans. J Coating Tech. 1981, 53: 23-25.
Bell AA, Wheeler MH: Biosynthesis and functions of fungal melanins. Annu Rev Phytopathol. 1986, 24: 411-451.
Kogej T, Stein M, Volkmann M, Gorbushina AA, Galinski EA, Gunde-Cimerman N: Osmotic adaptation of the halophilic fungus Hortaea werneckii: role of osmolytes and melanization. Microbiol. 2007, 153: 4261-4273.
Butler MJ, Day AW: Fungal melanins: a review. Can J Microbiol. 1998, 44: 1115-1136.
Taborda CP, da Silva MB, Nosanchuk JD, Travassos LR: Melanin as a virulence factor of Paracoccidioides brasiliensis and other dimorphic pathogenic fungi: a minireview. Mycopathologia. 2008, 165: 331-339.
Yurlova NA, de Hoog GS, Fedorova LG: The influence of ortho- and para-diphenoloxidase substrates on pigment formation in black yeast-like fungi. Stud Mycol. 2008, 39-49.
Kogej T, Wheeler MH, Lanisnik Rizner T, Gunde-Cimerman N: Evidence for 1,8-dihydroxynaphthalene melanin in three halophilic black yeasts grown under saline and non-saline conditions. FEMS Microbiol Lett. 2004, 232: 203-209.
Langfelder K, Streibel M, Jahn B, Haase G, Brakhage AA: Biosynthesis of fungal melanins and their importance for human pathogenic fungi. Fungal Genet Biol. 2003, 38: 143-158.
Takano Y, Kubo Y, Kawamura C, Tsuge T, Furusawa I: The Alternaria alternata melanin biosynthesis gene restores appressorial melanization and penetration of cellulose membranes in the melanin-deficient albino mutant of Colletotrichum lagenarium. Fungal Genet Biol. 1997, 21: 131-140.
Takano Y, Kubo Y, Shimizu K, Mise K, Okuno T, Furusawa I: Structural analysis of Pks1, a polyketide synthase gene involved in melanin biosynthesis in Colletotrichum lagenarium. Mol Gen Genet. 1995, 249: 162-167.
Zhang A, Lu P, Dahl-Roshak AM, Paress PS, Kennedy S, Tkacz JS, An Z: Efficient disruption of a polyketide synthase gene (pks1) required for melanin synthesis through Agrobacterium-mediated transformation of Glarea lozoyeasis. Mol Genet Genomics. 2003, 268: 645-655.
Tsai HF, Wheeler MH, Chang YC, Kwon-Chung KJ: A developmentally regulated gene cluster involved in conidial pigment biosynthesis in Aspergillus fumigatus. J Bacteriol. 1999, 181: 6469-6477.
Borgnia MJ, Agre P: Reconstitution and functional comparison of purified GlpF and AqpZ, the glycerol and water channels from Escherichia coli. Proc Natl Acad Sci USA. 2001, 98: 2888-2893.
Alleva K, Chara O, Amodeo G: Aquaporins: Another piece in the osmotic puzzle. FEBS Lett. 2012, 586: 2991-2999.
Borgnia M, Nielsen S, Engel A, Agre P: Cellular and molecular biology of the aquaporin water channels. Annu Rev Biochem. 1999, 68: 425-458.
Hohmann S, Bill RM, Kayingo G, Prior BA: Microbial MIP channels. Trends Microbiol. 2000, 8: 33-38.
Pettersson N, Filipsson C, Becit E, Brive L, Hohmann S: Aquaporins in yeasts and filamentous fungi. Biol Cell. 2005, 97: 487-500.
Xu H, Cooke JEK, Zwiazek JJ: Phylogenetic analysis of fungal aquaporins provides insight into their possible role in water transport of mycorrhizal associations. Botany-Botanique. 2013, 91: 495-504.
Arino J, Ramos J, Sychrova H: Alkali metal cation transport and homeostasis in yeasts. Microbiol Mol Biol Rev. 2010, 74: 95-120.
Shabala S, Cuin TA: Potassium transport and plant salt tolerance. Physiol Plant. 2008, 133: 651-669.
Ko CH, Gaber RF: Trk1 and Trk2 encode structurally related K+ transporters in Saccharomyces cerevisiae. Mol Cell Biol. 1991, 11: 4266-4273.
Ketchum KA, Joiner WJ, Sellers AJ, Kaczmarek LK, Goldstein SA: A new family of outwardly rectifying potassium channel proteins with two pore domains in tandem. Nature. 1995, 376: 690-695.
Haro R, Garciadeblas B, Rodriguez-Navarro A: A novel P-type ATPase from yeast involved in sodium-transport. FEBS Lett. 1991, 291: 189-191.
Garciadeblas B, Rubio F, Quintero FJ, Banuelos MA, Haro R, Rodriguez-Navarro A: Differential expression of two genes encoding isoforms of the ATPase involved in sodium efflux in Saccharomyces cerevisiae. Mol Gen Genet. 1993, 236: 363-368.
Wieland J, Nitsche AM, Strayle J, Steiner H, Rudolph HK: The PMR2 gene-cluster encodes functionally distinct isoforms of a putative Na+ pump in the yeast plasma-membrane. EMBO J. 1995, 14: 3870-3882.
Ramos J, Arino J, Sychrova H: Alkali-metal-cation influx and efflux systems in nonconventional yeast species. FEMS Microbiol Lett. 2011, 317: 1-8.
Serrano R, Kiellandbrandt MC, Fink GR: Yeast plasma-membrane ATPase is essential for growth and has homology with (Na++K+), K+- and Ca2+-ATPases. Nature. 1986, 319: 689-693.
Ambesi A, Miranda M, Petrov VV, Slayman CW: Biogenesis and function of the yeast plasma-membrane H+-ATPase. J Exp Biol. 2000, 203: 155-160.
Waschuk SA, Bezerra AG, Shi L, Brown LS: Leptosphaeria rhodopsin: Bacteriorhodopsin-like proton pump from a eukaryote. Proc Natl Acad Sci USA. 2005, 102: 6879-6883.
Gostinčar C, Lenassi M, Gunde-Cimerman N, Plemenitaš A: Fungal adaptation to extremely high salt concentrations. Adv Appl Microbiol. 2011, 77: 71-96.
Wagner NL, Greco JA, Ranaghan MJ, Birge RR: Directed evolution of bacteriorhodopsin for applications in bioelectronics. J R Soc Interface. 2013, 10:
Zhang F, Vierock J, Yizhar O, Fenno LE, Tsunoda S, Kianianmomeni A, Prigge M, Berndt A, Cushman J, Polle J, Magnuson J, Hegemann P, Deisseroth K: The microbial opsin family of optogenetic tools. Cell. 2011, 147: 1446-1457.
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Bolchacova E, Voigt K, Crous PW, Miller AN, Wingfield MJ, Aime MC, An KD, Bai FY, Barreto RW, Begerow D, Bergeron MJ, Blackwell M, Boekhout T, Bogale M, Boonyuen N, Burgaz AR, Buyck B, Cai L, Cai Q, Cardinali G, Chaverri P, Coppins BJ, Crespo A, et al: Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA. 2012, 109: 6241-6246.
Manitchotpisit P, Skory CD, Peterson SW, Price NPJ, Vermillion KE, Leathers TD: Poly(beta-L-malic acid) production by diverse phylogenetic clades of Aureobasidium pullulans. J Ind Microbiol Biotechnol. 2012, 39: 125-132.
Spatafora JW, Sung GH, Johnson D, Hesse C, O'Rourke B, Serdani M, Spotts R, Lutzoni F, Hofstetter V, Miadlikowska J, Reeb V, Gueidan C, Fraker E, Lumbsch T, Lucking R, Schmitt I, Hosaka K, Aptroot A, Roux C, Miller AN, Geiser DM, Hafellner J, Hestmark G, Arnold AE, Budel B, Rauhut A, Hewitt D, Untereiner WA, Cole MS, Scheidegger C, et al: A five-gene phylogeny of Pezizomycotina. Mycologia. 2006, 98: 1018-1028.
Liu YJ, Hall BD: Body plan evolution of ascomycetes, as inferred from an RNA polymerase II, phylogeny. Proc Natl Acad Sci USA. 2004, 101: 4507-4512.
Schmitt I, Crespo A, Divakar PK, Fankhauser JD, Herman-Sackett E, Kalb K, Nelsen MP, Nelson NA, Rivas-Plata E, Shimp AD, Widhelm T, Lumbsch HT: New primers for promising single-copy genes in fungal phylogenetics and systematics. Persoonia. 2009, 23: 35-40.
de Hoog GS, Guarro J, Gene J, Figueras MJ: Atlas of clinical fungi. 2000, Utrecht: ASM Press, 2
Debuchy R, Turgeon BG: Mating-type structure, evolution, and function in Euascomycetes. The Mycota: A Comprehensive Treatise on Fungi as Experimental Systems for Basic and Applied Research: Growth, Differentiation and Sexuality, Volume 1. Edited by: Kües U, Fischer R. 2006, Berlin, Heidelberg: Springer, 293-323.
Ni M, Feretzaki M, Sun S, Wang XY, Heitman J: Sex in Fungi. Annu Rev Genet. 2011, 45: 405-430.
Butler G, Kenny C, Fagan A, Kurischko C, Gaillardin C, Wolfe KH: Evolution of the MAT locus and its Ho endonuclease in yeast species. Proc Natl Acad Sci USA. 2004, 101: 1632-1637.
Malik SB, Pightling AW, Stefaniak LM, Schurko AM, Logsdon JM: An expanded inventory of conserved meiotic genes provides evidence for sex in Trichomonas vaginalis. Plos One. 2008, 3:
Nakagawa T, Kolodner RD: Saccharomyces cerevisiae Mer3 is a DNA helicase involved in meiotic crossing over. Mol Cell Biol. 2002, 22: 3281-3291.
Gioti A, Nystedt B, Li WJ, Xu J, Andersson A, Averette AF, Munch K, Wang XY, Kappauf C, Kingsbury JM, Kraak B, Walker LA, Johansson HJ, Holm T, Lehtio J, Stajich JE, Mieczkowski P, Kahmann R, Kennell JC, Cardenas ME, Lundeberg J, Saunders CW, Boekhout T, Dawson TL, Munro CA, de Groot PWJ, Butler G, Heitman J, Scheynius A: Genomic insights into the atopic eczema-associated skin commensal yeast Malassezia sympodialis. Mbio. 2013, 4:
Srikantha T, Daniels KJ, Pujol C, Sahni N, Yi S, Soll DR: Nonsex genes in the mating type locus of Candida albicans play roles in a/alpha biofilm formation, including impermeability and fluconazole resistance. PLoS Pathog. 2012, 8:
Rozman D, Komel R: Isolation of genomic DNA from filamentous fungi with high glucan level. BioTechniques. 1994, 16: 382-383.
Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, Berlin AM, Aird D, Costello M, Daza R, Williams L, Nicol R, Gnirke A, Nusbaum C, Lander ES, Jaffe DB: High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA. 2011, 108: 1513-1518.
Zerbino DR, Birney E: Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18: 821-829.
Martin J, Bruno VM, Fang ZD, Meng XD, Blow M, Zhang T, Sherlock G, Snyder M, Wang Z: Rnnotator: an automated de novo transcriptome assembly pipeline from stranded RNA-Seq reads. BMC Genomics. 2010, 11:
Kent WJ: BLAT - The BLAST-like alignment tool. Genome Res. 2002, 12: 656-664.
Grigoriev I, Martines D, Salamov A: Fungal genomic annotation. Applied mycology and biotechnology, Volume 6. Edited by: Aurora D, Berka R, Singh G. 2006, Amsterdam London: Elsevier, 123-142.
Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, Shabalov I: MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014, 42: D699-D704.
RepeatMasker Open-3.0. http://www.repeatmasker.org,
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J: Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005, 110: 462-467.
Price AL, Jones NC, Pevzner PA: De novo identification of repeat families in large genomes. Bioinformatics. 2005, 21: I351-I358.
Salamov AA, Solovyev VV: Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000, 10: 516-522.
Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M: Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18: 1979-1990.
Birney E, Clamp M, Durbin R: GeneWise and Genomewise. Genome Res. 2004, 14: 988-995.
Altschul SF, Madden TL, Shaffer AA, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402.
Lowe TM, Eddy SR: tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25: 955-964.
Nielsen H, Engelbrecht J, Brunak S, von Heijne G: Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997, 10: 1-6.
Melen K, Krogh A, von Heijne G: Reliability measures for membrane protein topology prediction algorithms. J Mol Biol. 2003, 327: 735-744.
Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R: InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33: W116-W120.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y: KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36: D480-D484.
Koonin EV, Fedorova ND, Jackson JD, Jacobs AR, Krylov DM, Makarova KS, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Rogozin IB, Smirnov S, Sorokin AV, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA: A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5:
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G, Consortium GO: Gene Ontology: tool for the unification of biology. Nat Genet. 2000, 25: 25-29.
Enright AJ, Van Dongen S, Ouzounis CA: An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30: 1575-1584.
Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D, Kuo A, Minovitsky S, Nikitin R, Ohm RA, Otillar R, Poliakov A, Ratnere I, Riley R, Smirnova T, Rokhsar D, Dubchak I: The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 2012, 40: 26-32.
Merlini L, Dudin O, Martin SG: Mate and fuse: how yeast cells do it. Open Biology. 2013, 3:
Petersen TN, Brunak S, von Heijne G, Nielsen H: SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011, 8: 785-786.
Emanuelsson O, Nielsen H, Brunak S, von Heijne G: Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000, 300: 1005-1016.
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL: Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol. 2001, 305: 567-580.
Yu CS, Lin CJ, Hwang JK: Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004, 13: 1402-1406.
Chi SM, Nam D: WegoLoc: accurate prediction of protein subcellular localization using weighted Gene Ontology terms. Bioinformatics. 2012, 28: 1028-1030.
Park BH, Karpinets TV, Syed MH, MR L u, Uberbacher EC: CAZymes Analysis Toolkit (CAT): Web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology. 2010, 20: 1574-1584.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739.
Robbertse B, Yoder RJ, Boyd A, Reeves J, Spatafora JW: Hal: an automated pipeline for phylogenetic analyses of genomic data. PLoS Currents. 2011, 3: RRN1213-
de Wit PJGM, van der Burgt A, Okmen B, Stergiopoulos I, Abd-Elsalam KA, Aerts AL, Bahkali AH, Beenen HG, Chettri P, Cox MP, Datema E, de Vries RP, Dhillon B, Ganley AR, Griffiths SA, Guo YA, Hamelin RC, Henrissat B, Kabir MS, Jashni MK, Kema G, Klaubauf S, Lapidus A, Levasseur A, Lindquist E, Mehrabi R, Ohm RA, Owen TJ, Salamov A, Schwelm A: The genomes of the fungal plant pathogens Cladosporium fulvum and Dothistroma septosporum reveal adaptation to different hosts and lifestyles but also signatures of common ancestry. PLoS Genet. 2012, 8:
Goodwin SB, Ben M'Barek S, Dhillon B, Wittenberg AHJ, Crane CF, Hane JK, Foster AJ, Van der Lee TAJ, Grimwood J, Aerts A, Antoniw J, Bailey A, Bluhm B, Bowler J, Bristow J, van der Burgt A, Canto-Canche B, Churchill ACL, Conde-Ferraez L, Cools HJ, Coutinho PM, Csukai M, Dehal P, De Wit P, Donzelli B, van de Geest HC, Van Ham RCHJ, Hammond-Kosack KE, Henrissat B, Kilian A, et al: Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 2011, 7:
Amselem J, Cuomo CA, van Kan JAL, Viaud M, Benito EP, Couloux A, Coutinho PM, de Vries RP, Dyer PS, Fillinger S, Fournier E, Gout L, Hahn M, Kohn L, Lapalu N, Plummer KM, Pradier JM, Quevillon E, Sharon A, Simon A, ten Have A, Tudzynski B, Tudzynski P, Wincker P, Andrew M, Anthouard V, Beever RE, Beffa R, Benoit I, Bouzid O, et al: Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7:
Martin F, Kohler A, Murat C, Balestrini R, Coutinho PM, Jaillon O, Montanini B, Morin E, Noel B, Percudani R, Porcel B, Rubini A, Amicucci A, Amselem J, Anthouard V, Arcioni S, Artiguenave F, Aury JM, Ballario P, Bolchi A, Brenna A, Brun A, Buee M, Cantarel B, Chevalier G, Couloux A, Da Silva C, Denoeud F, Duplessis S, Ghignone S, et al: Périgord black truffle genome uncovers evolutionary origins and mechanisms of symbiosis. Nature. 2010, 464: 1033-1038.
Arnaud MB, Chibucos MC, Costanzo MC, Crabtree J, Inglis DO, Lotia A, Orvis J, Shah P, Skrzypek MS, Binkley G, Miyasato SR, Wortman JR, Sherlock G: The Aspergillus Genome Database, a curated comparative genomics resource for gene, protein and sequence information for the Aspergillus research community. Nucleic Acids Res. 2010, 38: 420-427.
Talavera G, Castresana J: Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007, 56: 564-577.
Darriba D, Taboada GL, Doallo R, Posada D: ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011, 27: 1164-1165.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O: New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol. 2010, 59: 307-321.
Sanderson MJ: r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003, 19: 301-302.
Katoh K, Toh H: Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008, 9: 286-298.
Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL: Versatile and open software for comparing large genomes. Genome Biol. 2004, 5:
Hane JK, Rouxel T, Howlett BJ, Kema GHJ, Goodwin SB, Oliver RP: A novel mode of chromosomal evolution peculiar to filamentous Ascomycete fungi. Genome Biol. 2011, 12:
Gremme G, Steinbiss S, Kurtz S: GenomeTools: A comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans Comput Biol Bioinform. 2013, 10: 645-656.
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer ELL, Eddy SR, Bateman A, Finn RD: The Pfam protein families database. Nucleic Acids Res. 2012, 40: 290-301.
Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T: antiSMASH 2.0-a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013, 41: 204-212.
Acknowledgements
The work conducted by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The authors acknowledge financial support from the state budget through the Slovenian Research Agency (Infrastructural Centre Mycosmo, MRIC UL, Postdoctoral Project Z4-5531 to CG, and Young Researcher Grant to JZ). The study was also partly financed via operation “Centre of excellence for integrated approaches in chemistry and biology of proteins” number OP13.1.1.2.02.0005, financed by European Regional Development Fund (85% share of financing) and by the Slovenian Ministry of Higher Education, Science and Technology (15% share of financing). The authors would like to thank Chris Berrie for language editing assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CG performed the phylogenetic and CAFE analyses, analysed the genes encoding the alkali-metal cation transporters, and predicted secreted proteins; performed genome alignments; participated in the coordination of the project; and coordinated and participated in the writing of the manuscript. RAO performed the genome annotations and clustering analyses. TK isolated the DNA, analysed the genes for melanin biosynthesis, aquaporins, and secondary biosynthesis gene clusters; and wrote the corresponding parts of the manuscript. SS isolated the RNA, analysed the genes for pullulan synthesis, siderophores, and biodegradation of plastic and aromatic compounds; and wrote the corresponding parts of the manuscript. MT isolated the RNA, analysed the genes for sugar transporters and the high osmolarity pathway; and wrote the corresponding parts of the manuscript. JZ analysed the mating-type and meiosis-related genes, predicted secreted proteins and genes involved in the management of compatible solutes; and wrote the corresponding parts of the manuscript. PZ analysed the taxonomic markers; and wrote the corresponding parts of the manuscript and the taxonomic descriptions. MG analysed the bacteriorhodopsin-like genes; and wrote the corresponding parts of the manuscript. HS and JH assembled the genomes. AS, CYN and JC were involved in sequencing of the genomes and transcriptomes. AL assembled the transcriptomes. KB and IVG coordinated the genome project. NGC conceived the study and participated in its coordination. All authors have read and approved the final manuscript.
Electronic supplementary material
12864_2014_7061_MOESM1_ESM.pdf
Additional file 1: Alignment of putative pullulan synthetases. Homologues from the sequenced genomes of the four A. pullulans varieties and a previously published gene ([GenBank:AF470619] [57]). ApP, A. pullulans var. pullulans; ApS, A. pullulans var. subglaciale; ApN, A. pullulans var. namibiae; ApM, A. pullulans var. melanogenum. JGI protein IDs are included. Red, predicted exones; green, introns; blue (arrow), one-nucleotide frameshift insertion. (PDF 13 KB)
12864_2014_7061_MOESM2_ESM.xlsx
Additional file 2: Secondary metabolite biosynthetic clusters of the four Aureobasidium pullulans varieties.(XLSX 23 KB)
12864_2014_7061_MOESM3_ESM.xlsx
Additional file 3: Numbers of carbohydrate-active enzymes in the predicted secretomes of the four Aureobasidium pullulans varieties.(XLSX 12 KB)
12864_2014_7061_MOESM4_ESM.pdf
Additional file 4: Different CAZy glucoside hydrolase families that are involved in degradation of plant cell walls in different fungal genomes. Blue bars, degradation of hemicellulose (GH10, GH11, GH27, GH29, GH35, GH36, GH39, GH43, GH51, GH53, GH54, GH62, GH67, GH93, GH115); red bars, degradation of cellulose (GH6, GH7, GH12, GH45, GH74, GH94, AA9 (previously GH61)); dark yellow vertical line (left), adapted from [51]; green vertical line (left), adapted from [70]. (PDF 37 KB)
12864_2014_7061_MOESM5_ESM.pdf
Additional file 5: Protein tree of scytalone dehydratases. GenBank accession numbers of individual proteins are listed in the tree. Different gene clusters are marked with different colours. Scytalone dehydratases of the A. pullulans varieties are marked in red. (PDF 38 KB)
12864_2014_7061_MOESM6_ESM.pdf
Additional file 6: Protein tree of the aquaporins. The GenBank accession numbers of the individual proteins are listed in the tree. Different gene clusters are marked with different colours, which correspond to previously recognised phylogenetic groups [116]. Aquaporins of A. pullulans varieties are marked in red. (PDF 55 KB)
12864_2014_7061_MOESM7_ESM.pdf
Additional file 7s: Protein trees of various membrane transporters of Na+and K+. Protein trees marked with the names of the S. cerevisiae homologues (except for Acu and Hak K+ transporters, which are not found in S. cerevisiae). The trees (except for Acu and Hak) were rooted with homologous proteins from C. neoformans (Trk: [GenBank:XP_570017] and [GenBank:XP_569339]; Tok: [GenBank:XP_568987] and [GenBank:XP_568988]; Nha: [GenBank:XP_569560]; Ena: [GenBank:XP_572412], [GenBank:XP_568029] and [GenBank:XP_570160]; Pho: [GenBank:XP_568082]; Nhx: [GenBank:XP_570596]; Kha: [GenBank:XP_571501]; Vnx: [GenBank:XP_569752]; Mrs: [GenBank:XP_569566]; Pma: [GenBank:XP_568571]) and the root location is marked with an arrow. In addition to genes from A. pullulans, homologues from the following fungi were used: H. werneckii (Hw), M. graminicola (Mg) and S. cerevisiae (Sc). For Acu, homologues from Ustilago maydis (Um) and Millerozyma farinosa (Mf; Pichia farinosa) were used; for Hak, homologues from N. crassa (Nc) and C. albicans (Ca) were used. The GenBank accession numbers of the individual proteins are listed in the trees. Putative gene duplications leading to the present diversity of these genes in A. pullulans are marked with double arrows. ApP, A. pullulans var. pullulans; ApS, A. pullulans var. subglaciale; ApN, A. pullulans var. namibiae; ApM, A. pullulans var. melanogenum.(PDF 35 KB)
12864_2014_7061_MOESM8_ESM.xlsx
Additional file 8: Alkali-metal cation transporters of the four Aureobasidium pullulans varieties and related fungi, divided into subgroups as determined by the phylogenetic analysis (Figure 4).(XLSX 11 KB)
12864_2014_7061_MOESM9_ESM.pdf
Additional file 9: Phylogenetic analysis of bacteriorhodopsins from the four Aureobasidium pullulans varieties and related fungi. Numbers before species names are protein ID numbers of the Joint Genome Institute Genome Portal. The structures of the genes are represented by horizontal bars (exons) and spaces between them (introns). Lengths of the bars are proportional to the lengths of the feature represented. (PDF 22 KB)
12864_2014_7061_MOESM10_ESM.xlsx
Additional file 10: Distances between the four sequenced genomes of Aureobasidium species and selected Saccharomyces species.(XLSX 12 KB)
12864_2014_7061_MOESM11_ESM.pdf
Additional file 11: Dot-plot comparison of Aureobasidium species scaffolds longer than 200 kbp. Six-frame translations of scaffolds aligned with Mummer 3.23. Homologous regions are plotted as dots. Scaffolds of each species are displayed ordered by decreasing size along the X and Y axes. Diagonal lines of dots in individual boxes represent syntenic regions. (PDF 7 MB)
12864_2014_7061_MOESM12_ESM.pdf
Additional file 12: Putative second mating type locus, MAT2-2. Putative MAT2-2-1 genes with clearly recognisable HMG box domains (PF00505). Downstream of the putative MAT2-2-1 there is a gene with the Mob1/phocein family (Mob1 kin. reg.; PF03637), and taurine catabolism dioxygenase (TauD; PF02668), and a gene with the major facilitator superfamily (MFS_1; PF07690) domain. Upstream of the putative MAT2-2-1 there are two genes, the first encoding a hypothetical protein most similar to GDSL lipase (with no Pfam domain), and the second encoding SNF-2 helicase (PF00176). (PDF 16 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Gostinčar, C., Ohm, R.A., Kogej, T. et al. Genome sequencing of four Aureobasidium pullulans varieties: biotechnological potential, stress tolerance, and description of new species. BMC Genomics 15, 549 (2014). https://doi.org/10.1186/1471-2164-15-549
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-549