
Abstract
Background
Common bean (Phaseolus vulgaris L.) is the most important legume cropped worldwide for food production and its agronomic performance can be greatly improved if the benefits from symbiotic nitrogen fixation are maximized. The legume is known for its high promiscuity in nodulating with several Rhizobium species, but those belonging to the Rhizobium tropici “group” are the most successful and efficient in fixing nitrogen in tropical acid soils. Rhizobium leucaenae belongs to this group, which is abundant in the Brazilian “Cerrados” soils and frequently submitted to several environmental stresses. Here we present the first high-quality genome drafts of R. leucaenae, including the type strain CFN 299T and the very efficient strain CPAO 29.8. Our main objective was to identify features that explain the successful capacity of R. leucaenae in nodulating common bean under stressful environmental conditions.
Results
The genomes of R. leucaenae strains CFN 299T and CPAO 29.8 were estimated at 6.7–6.8 Mbp; 7015 and 6899 coding sequences (CDS) were predicted, respectively, 6264 of which are common to both strains. The genomes of both strains present a large number of CDS that may confer tolerance of high temperatures, acid soils, salinity and water deficiency. Types I, II, IV-pili, IV and V secretion systems were present in both strains and might help soil and host colonization as well as the symbiotic performance under stressful conditions. The symbiotic plasmid of CPAO 29.8 is highly similar to already described tropici pSyms, including five copies of nodD and three of nodA genes. R. leucaenae CFN 299T is capable of synthesizing Nod factors in the absence of flavonoids when submitted to osmotic stress, indicating that under abiotic stress the regulation of nod genes might be different.
Conclusion
A detailed study of the genes putatively related to stress tolerance in R. leucaenae highlighted an intricate pattern comprising a variety of mechanisms that are probably orchestrated to tolerate the stressful conditions to which the strains are submitted on a daily basis. The capacity to synthesize Nod factors under abiotic stress might follow the same regulatory pathways as in CIAT 899T and may help both to improve bacterial survival and to expand host range to guarantee the perpetuation of the symbiosis.
Similar content being viewed by others


Background
Biological nitrogen fixation (BNF) is a key process for global N inputs, and greatly contributes to avoidance of soil-nutrient impoverishment, to recovery of fertility of degraded areas, and also to reduce the emission of greenhouse gases related to application of chemical fertilizers. The greatest contribution of BNF occurs by means of symbiotic diazotrophic bacteria—collectively known as rhizobia—associated with several legumes, partially or fully supplying plant’s N needs [1, 2].
Considering the nutritional needs of growing populations, particularly in developing countries, undoubtedly common bean (Phaseolus vulgaris L.) represents the most important legume cropped for food purposes. A strategic goal of research with the legume is to maximize BNF through the selection and/or breeding of both plant and rhizobial genotypes [2–4]. Common bean is well known for its high promiscuity in associating with a variety of rhizobial species [5, 6], and in acid tropical soils of South America strains related to the Rhizobium tropici “group” represent the most efficient microsymbionts [6–9].
R. tropici was first described in 1991 [5], and its main features include its ability to nodulate both common bean and leucaena (Leucaena spp.), its high tolerance of stressful environmental tropical conditions, and higher genetic stability of the symbiotic plasmid in comparison to other common bean rhizobia (e.g. [4, 5, 9–11]). Although R. tropici is thought to have originated in South America [5], it is also found in Europe, Australia, Africa, and North and Central America [9]. The species has also been isolated from other host legumes, including Gliricidia spp., Acaciella angustissima, Mimosa caesalpiniifolia, Bolusanthus spp., Aspartium spp., and Lotus tenuis [9]. In addition to this promiscuity, also intriguing is the capacity of R. tropici to synthesize a variety of nodulation (Nod) factors [12, 13], even in the absence of plant molecular signals [12–15].
Despite long-standing reports of high phenetic and genetic diversity of strains within the R. tropici species [5], it was only two decades later that a group of strains named as R. tropici type A was split from the type-B group and reclassified as Rhizobium leucaenae [6]. The new species shares typical properties with R. tropici, such as high genetic stability of the symbiotic plasmid and tolerance of stressful environmental conditions [6].
R. tropici, R. leucaenae and two other new species previously classified as R. tropici—Rhizobium freirei [7] and Rhizobium paranaense [8]—encompass strains very effective in fixing nitrogen with common bean, representing an important source of inoculants for sustainable agriculture in the tropics (e.g. [4, 9]). However, the only available genomes of the group are of R. tropici CIAT 899T and R. freirei PRF 81T [11]. Here we present two genomes of R. leucaenae, including the type strain CFN 299T and strain CPAO 29.8, very effective in fixing nitrogen with common bean. Our main objective was to search for genes that could help to explain the successful symbiotic performance of R. leucaenae under environmentally stressful conditions.
Results and discussion
General characteristics and comparison of R. leucaenae genomes
Sequencing of CFN 299T and CPAO 29.8 resulted in high-quality draft genomes with average coverages of 251-fold and 155-fold, respectively. In CFN 299T, the genome was assembled in 95 contigs with an N50 size of 296,137 bp, whereas in CPAO 29.8 there are 179 contigs with an N50 size of 219,636 bp.
Both strains of R. leucaenae present genomes of similar size and composition. CFN 299T has a 6,694,130-bp and CPAO 29.8 a 6,850,073-bp genome. General features and statistics of the genomes are presented in Table 1. The chromosomes of both strains contain three ribosomal operons. CFN 299T has four plasmids of approximately 1.9 Mb, 500 kb, 220 kb and 190 kb, and apparently the same for CPAO 29.8. Both strains have a megaplasmid and the second largest replicon is the symbiotic plasmid (pSym).
Totals of 7015 and 6899 coding DNA sequences (CDS) were predicted for CFN 299T and CPAO 29.8, respectively (Table 1). Putative functions could be assigned to around 66 and 72 % of the CDS from both strains, respectively.
After combining the annotation obtained by RAST and a detailed manual curation process, the information about each contig, gene product and localization in the genomes of CFN 299T and CPAO 29.8 was included in Additional file 1: Table S1. In addition, we made comparisons with other genomes of related Rhizobium species, including R. tropici CIAT 899T, R. freirei PRF 81T, R. rhizogenes K84, R. etli CFN 42T, R. phaseoli CIAT 652, R. grahanii CCGE 502T, R. leguminosarum 3841, R. mesoamericanum STM 3625T, and other genomes.
Functional classification in COG of the CDS was very similar in both strains, but the two highest categories were of general function and unknown functions (Table 2), highlighting our still poor knowledge about bacterial genomes. Similar percentages of genes with unknown function were revealed by the RAST functional classification system.
The chromosomes coded for most genes assigned to functionally important classes, predominantly of central metabolism, cellular processes, DNA metabolism, and several CDS related to transport. The great majority of the genes related to stress tolerance that will be commented upon are also in the chromosome, although there were few, but important genes in the pSym. The megaplasmid encoded several hypothetical genes, conjugal transfer tra and trb operons, and several other classes of genes, including many transporters. pSym encoded all nodulation and symbiosis-related genes, in addition to many hypothetical CDS and transporters (Additional file 1: Table S1).
Genome comparisons indicated high similarities between CFN 299T and CPAO 29.8 strains, with 6264 CDS in common (Fig. 1). CFN 299T presented 751 unique CDS, 70 % of which with hypothetical functions, while CPAO 29.8 presented 635 unique CDS, 57 % with hypothetical functions. Genes unique to CFN 299T included conjugation genes and sugar transporters and catabolic genes, and several of these genes were located in the smallest plasmid of this strain. On the other hand, CPAO 29.8 specific loci included a prophage, a cluster of flagellum-related genes, conjugation genes, and carbohydrate transporters. The phylogeny of this interesting group of strains classified in the “R. tropici group” is shown in Fig. 2, and considering the comparison of CFN 299T with R. tropici CIAT 899T, R. freirei PRF 81T and R. rhizogenes K84, we found that CFN 299T carries a high number of exclusive CDS—2333 (Fig. 3).

Venn diagram showing the number of orthologous gene clusters shared by Rhizobium leucaenae strains CFN 299T and CPAO 29.8. Based on RAST predicted genes and manual curation

Neighbor joining phylogenetic tree based on a concatenated alignment of recA, glnII and gyrB sequences of Rhizobium leucaenae strains from this study and other type/reference strains. Bootstrap support values 70 % or greater are shown at tree nodes

Venn diagram showing the number of orthologous gene clusters shared by Rhizobium leucaenae strains CFN 299T and Rhizobium tropici CIAT 899T, Rhizobium freirei PRF 81T and Rhizobium rhizogenes K84. Based on RAST predicted genes and manual curation
Searching for genes related to the high tolerance of environmental stress in R. leucaenae
One major limiting factor in agriculture results from edaphoclimatic stressful conditions, in the tropics represented by high temperatures, acidic soils, salinity and water deficiency, all affecting both the host plant and the bacterium; the symbiosis of common bean-rhizobium seems particularly sensitive to environmental stresses [16, 17]. Both CFN 299T and CPAO 29.8 are capable of tolerating environmental stresses, including high temperatures (37 °C) and acidity (pH 4.0) [6, 9], and the species apparently encompasses the great majority of strains isolated from nodules on common bean and leucaena in the Cerrados region [6, 10, 18], an edaphic type of savannah covering 207 million hectares of Brazilian land (25 %) that are frequently exposed to harsh environmental stressful conditions. Therefore, investigating genes related to the stress-tolerance features of R. leucaenae may not only help to understand the successful saprophytic and symbiotic strategies used by the species, but also contribute to the identification of genes with biotechnological potential. In Additional file 2: Table S2, we included the genes related to stress tolerance in both strains, and below we discuss the main categories.
pH stress
Besides the need to survive in acid soils, rhizobial strains may face pH stresses in the process of establishment of the symbioses, and also when the bacterium is inside the symbiosome; in both situations, the challenge is to maintain the intracellular pH around 7.2 to 7.5 [19]. To ensure intracellular pH homeostasis when submitted to acid conditions, bacteria may take advantage of different mechanisms. One major strategy consists of proton transport mediated by proton pumps, proton-coupled ATPases and cation-proton antiporters. CFN 299T and CPAO 29.8 genomes present a set of genes—phaA, phaB, phaC, phaD, phaE, phaF and phaG—related to different Na+/H+ antiporter subunits. This antiporter system has been described in cell defenses in alkaline conditions, in which intracellular H+ is replaced by Na+ [19, 20]. The accumulation of positively charged ions, such as Na+ and K+, added to the efflux of H+ also seems important when cells are exposed to acid stress [21].
Glutathione synthase (gshB) is involved in acid tolerance of R. tropici CIAT 899T, since an auxotrophic mutant showed reduced growth at pH 5.0, with the suggestion that it could be related to a lower concentration of intracellular K+ compared to the wild-type strain [22]. The same authors suggested that the low intracellular K+ content may be related to the high activity of the KefB/KefC glutathione-regulator K+ efflux transporter verified in the absence of glutathione [22]. Muglia et al. [23] also emphasized the importance of glutathione to the acid tolerance of R. tropici CIAT 899T, once the transcription of gshB is activated at low pH. Both glutathione synthase and K+-efflux transporter genes ghsB and kefB are present in the genomes of CFN 299T and CPAO 29.8.
As in R. tropici CIAT 899T and R. freirei PRF 81T genomes [11], R. leucaenae stains CPAO 29.8 and CFN 299T possess two copies of the gene cfa, encoding a cyclopropane-fatty-acyl-phospholipid synthase protein. In Escherichia coli, this gene converts unsaturated fatty acids to saturated counterparts, modifying the inner-membrane phospholipids, and leading to an acid-tolerant phenotype [24]. One possible mechanism would be that the changes in the inner membrane reduce the permeability to H+ [25]. Genes coding for ClC-type chloride channels have also been reported as playing a role in acid-pH tolerance by controlling the intracellular H+ concentration. In a low-pH environment, chloride channels accomplish proton extrusion, maintaining the cell homeostasis [26]. Three genes of the widespread ClC-family are present in the chromosomes of CPAO 29.8 and CFN 299T. These paralogous genes may present different functions in addition to their protective roles in acid-pH stress, since the knockout of sycA, a gene from the ClC-family, results in a symbiotically defective phenotype [27].
The cellular membrane is a crucial component for avoidance of pH stress, and tolerance may be related to structural modifications, either by changes in the lipid composition, or by allocating several proteins involved in transport of compounds to maintain the intracellular pH homeostasis. It is equally true that some genes involved in membrane composition and with a role in pH-stress defense can also contribute to the success of the symbiosis. Thus, when CIAT 899T is submitted to low pH conditions, an increase of ornithine lipids (OL) and their hydroxylation are observed in the outer membrane [28]. The gene coding for ornithine lipid biosynthesis, olsC, is present in the chromosomes of CFN 299T and CPAO 29.8 and, according to Rojas-Jiménez et al. [27], is responsible for the OL biosynthesis and for the addition of the hydroxyl group, suggesting a protective role to acid stress. Indeed, a CIAT 899T olsC mutant showed significant growth reduction at pH 4.5, as well as generated symbiotic defects, resulting in poor development of the nodules and a two-fold reduction in nitrogen fixation [28].
In CIAT 899T, the genes lipA and atvA were up-regulated when the bacterium was exposed to an experimental acid condition [29]. Among the CFN 299T and CPAO 29.8 genes, we found lipA, which is required for lysyl-phosphatidylglycerol biosynthesis, showing a relation with antimicrobial resistance and competitiveness [30], while atvA participates in the membrane lipids metabolism [29], both studied in CIAT 899T. Complex pleiotropic phenotypes were observed in CIAT 899T atvA auxotroph, including acid sensitivity [30]. However, the acid-sensitive mutants were not affected in competitiveness or nitrogen fixation capacity, but in contrast, the lipA CIAT 899T mutant exhibited a seven-fold reduction in competitiveness of nodulation in comparison to the wild type [29].
High-temperature stress
Although the optimum growth temperature for most rhizobia ranges from 25 to 30 °C, outlier temperatures are often experienced at the rhizosphere, affecting both growth and saprophytic competence. Besides affecting survival in the free-living state, heat stress negatively affects molecular-signal exchange, root infection, nodulation and several steps of the nitrogen fixation process [9, 16, 17, 31], and one major feature of R. leucaenae and R. tropici is their superior symbiotic performance under high temperature conditions in comparison to species that do not belong to the “R. tropici group” (Additional file 3: Table S3).
The ability of R. leucaenae to grow at high temperature (37 °C) [6] might be related to several genes that were identified in CFN 299T and CPAO 29.8 genomes. In the closely related species R. freirei PRF 81T, Gomes et al. [32] identified 54 proteins in response to heat stress, 38 of which were now recognized in R. leucaenae. This set of genes comprises the molecular bases of temperature-stress responses, being responsible for rapid physiological changes, and many of them are under the transcription control of the RpoH alternative sigma factor [33].
Heat-shock proteins (HSP) play key roles in repair of heat-stress damages. These proteins are distributed in different groups, including the major chaperone-system proteins, the small heat-shock proteins and the chaperone-like proteins, all of which are represented by clusters of genes in both of the CFN 299T and CPAO 29.8 genomes. The major chaperones, DnaK, DnaJ and GrpE, compose the DnaK system, which is the most versatile chaperone system, responsible for de novo protein folding, protein transport and for the increase on RpoH stability during heat stress, indirectly assisting the gene expression aimed at cell defense against high temperatures. In complement, the GroEL system, comprising the GroES protein, can routinely rescue more than 80 % of the proteins damaged by heat [33, 34].
The small heat-shock proteins (sHSP), as their denomination indicates, present low molecular mass and are involved in reversing protein aggregations generated under high temperatures, keeping them in a folding-competent state [35]. A cluster of small HSPs genes is harbored in the chromosomes of the R. leucaenae genomes, especially those correlated with the HSP20 family, among them, ibpA, hspG and hspH, hspH; the last of which is up-regulated in Bradyrhizobium japonicum under heat-stress conditions [36]. In E. coli, for example, only two of these sHSP genes are presen, but different from most bacteria, the occurrence of several genes encoding these small proteins seem to be typical of rhizobial species [11, 35]. In another study, the comparison of heat-tolerant and -sensitive Rhizobium strains revealed the overexpression of different sHSP by the tolerant strain in response to increased temperature [37]. The same is expected for CFN 299T and CPAO 29.8 strains, once they are capable of growing at temperatures above the optimum conditions for most rhizobial species. Curiously, two genes of HSP20 are located at the CFN 299T and CPAO 29.8 symbiotic plasmids, as well as in CIAT 899T and PRF 81T, suggesting a probable role in cell defense during the symbiosis [11].
Apart from the classical chaperones, other proteins can display a heat-stress-protective role in addition to their main function. Within this group of chaperone-like proteins are the translation factors EF-Tu, EF-G and IF2, which help in protein folding and prevent unfolding proteins from forming aggregates during high-temperature conditions [38, 39]. The genes that encode these chaperone-like proteins are up-regulated in PRF 81T at 37 °C [32]. Gene-deletion studies have improved understanding of how the cell endures the heat stress by revealing new genes that are indirectly associated with high-temperature tolerance, but confer tolerant phenotypes [31]. For example, R. etli relA and R. tropici guaB mutants had their ability to tolerate high temperatures decreased [40]. Both relA and guaB, in addition to the genes coding for translation factors, EF-Tu, EF-G and IF2, which were up-regulated in PRF 81T, are present in the CFN 299T and CPAO 29.8 genomes; therefore, they might be related to the heat tolerance of this strain.
Osmotic stress
Osmotic stress can affect bacteria by loss of intracellular water or by excessive water influx, depending on the environmental conditions, and soil desiccation and salinity are the main factors related to osmotic stress. Salinity, for example, affects almost 40 % of land globally, potentially negatively impacting soil bacteria [2, 41]. In rhizobia, desiccation and salinity also affect the nitrogen-fixation process [41]. To overcome osmotic stress, the cells have protective mechanisms and, in the genomes of R. leucaenae strains CPAO 29.8 and CFN 299T, we found more than 30 genes related to osmotolerance.
The initial response of Rhizobium strains to cope with osmotic stress relies on the uptake and accumulation of potassium (K+) [42]. A high-affinity K+ (Kup) system was previously reported in R. tropici and R. freirei [11, 43] and the gene is present in the chromosomes of CFN 299T and CPAO 29.8. After uptake, the K+ ion acts as a secondary messenger driving other responses to osmotic stress, and in CFN 299T and in CPAO 29.8, we found genes encoding the KdpABC K+ transporting ATPase, also present in strains CIAT 899T and PRF 81T [11].
Another common strategy for coping with hyperosmotic environments is the synthesis and accumulation of compatible solutes, including trehalose, glycine, betaine, proline and ectoine [44]. Such compounds are called compatible because they are not harmful to macromolecules and their diversity is important for bacterial adaptation to environmental changes, which involve the amount of water and, in the rhizosphere, the salts and other exudates from plants [45]. Trehalose, for example, was described as an osmoprotectant in Sinorhizobium meliloti [46], besides being involved in protection against other abiotic stresses [44]. It is also implicated in the nodulation process of S. meliloti [43], B. japonicum [47], R. etli [48] and R. leguminosarum bv. trifolii [49]. R. leucaenae presents the genes otsA and otsB that encode enzymes involved in trehalose biosynthesis from UDP-glucose and glucose 6-phosphate [49]. The additional pathway that involves trehalose synthase (TreS), an enzyme present in CIAT 899T that catalyzes trehalose synthesis from maltose [50], is also present in the chromosomes of CFN 299T and CPAO 29.8.
Among the osmoprotectants, glycine betaine can be taken up or synthesized from choline [51]. The presence of this compatible solute in the medium improves the growth rate of R. tropici, S. meliloti, S. fredii and R. galegae when submitted to 300 mM of NaCl [51]. In such conditions, the enzyme activity for glycine betaine degradation decreases, while the enzymes’ activities converting choline to glycine betaine— choline dehydrogenase (BetA) and betaine aldehyde dehydrogenase (BetB)—increase [51]. The external choline could be taken by the ChoXWV ABC-type transporters [52], also encoded in R. leucaenae strains CPAO 29.8 and CFN 299T. Genes betA and betB are also present in the genomes of both strains of R. leucaenae, as well as betC, which encodes choline sulfatase and may use choline sulfate as a precursor to glycine betaine synthesis [11]. The uptake of proline betaine performed by the transporters genes—prbA, prbB, prbC and prbD—can also overcome the osmotic stress.
Contrasting with hyper-osmotic stress, less is known of how bacteria cope with hypo-osmotic shock. Dylan et al. [53] reported that a low-osmolarity medium leads to a striking increase in the level of β-(1 → 2)-glucans in S. meliloti periplasm space. This finding is directly related with the ndv locus, which includes two genes, ndvA and ndvB [54], both harbored in the chromosome of strains CFN 299T and CPAO 29.8. NdvB and NdvA are responsible for the synthesis of cyclic beta glucans (CbG) and their translocation to the periplasmic space, roles that are essential for nodulation [54]. Mutants in ndvA and ndvB genes implied in the absence of CbG, generate disturbances associated with hypo-osmotic adaptation, motility, root attachment and infection [53]. Increases in the membrane turgor pressure, caused by the movement of water into the cell cytoplasm in a low-osmotic environment, induce the activation of mechanosensitive (MS) channels [54, 55]. There are two major classes of bacterial MS channels, MscS and MscL, which, under hypo-osmotic conditions, perform the release of internal solutes to equilibrate intra and extracellular osmolarity [55]. Whereas the MscL channel activity is usually the product of a single gene, many bacteria possess multiple MscS homologues, including R. leucaenae CFN 299T and CPAO 29.8, as well as R. tropici CIAT 899T, which presents five genes encoding MscS. Multiple genes for MS channels may offer the cell higher tolerance to hypo-osmotic shocks.
Changes in the osmolarity activate fluxes of solutes and water along the concentration gradient. In addition to solutes, water flows through the membranes of living cells by two distinct mechanisms, i.e. by simple diffusion and by water-selective channel proteins, namely aquaporins [56]. Widespread in a variety of organisms, the occurrence of aquaporins in bacteria suggests that they could be involved in osmo-adaptation responses, which was confirmed in E. coli under hypo-osmotic conditions [57] and in Brucella abortus exposed to a hypertonic environment [56]. As observed in R. tropici CIAT 899T [11], DNA sequences for two aquaporins were identified in the genomes of the CFN 299T and CPAO 29.8, one in the chromosome and the other in the symbiotic plasmid.
Oxidative stress
Aerobic bacteria, including rhizobia, have to deal with reactive oxygen species (ROS) such as the superoxide anion (O2 −) and hydrogen peroxide (H2O2), which are produced mainly by cellular respiration [58]. To avoid the detrimental effects generated by oxidative stress agents, R. leucaenae strains CPAO 29.8 and CFN 299T possess a cluster of genes the products of which are related to the detoxification of ROS. Additionally, rhizobia are exposed to oxidant agents in the rhizosphere when in the free-living stage, during the infection process [59] and inside the nodules [60]. This reflects the importance of tolerating and overcoming oxidative stress as one essential feature for achieving an effective symbiosis.
Different mechanisms of protection against the ROS have been described, including catalases, superoxide dismutases (SODs), peroxidases and other enzymes such as peroxiredoxin [59–61]. The oxidative-stress responses to superoxide compounds are induced by the SoxR transcriptional activator [11], leading to the expression of genes for superoxide dismutase. Together with soxR, two genes for SOD, sodC and sodM, are harbored in the genomes of CFN 299T and CPAO 29.8, as well as in CIAT 899T and R. freirei PRF 81T [11]. Regarded as the most important factor in protection and maintenance of oxidative homeostasis in bacteria, SOD catalyzes the dismutation of O2 − to H2O2 and O2 [59]. The responses to H2O2, in turn, are driven by the OxyR transcription factor [62], a key regulator encoded by the CPAO 29.8 and CFN 299T genomes. Responses to oxidative stress include also the expression of catalases, which break down H2O2 to oxygen and water [61, 63]. Two genes encoding catalases were found in the genomes of R. leucaenae, katG for a bifunctional catalase-peroxidase (HPI) [64] and another encoding a monofuntional catalase. The expression of catalases is crucial during plant infection [65], because hydrogen peroxide is one of the plant defenses experienced by symbiotic microorganisms [66]. In this adverse environment, rhizobia have to tolerate the oxidative stress, but should also maintain a correct balance of H2O2 for the establishment of a successful symbiosis [61]. This was demonstrated by over-expressing the katB gene of S. meliloti, inducing a lower number of nodules on M. sativa, probably reflecting the intense reduction of H2O2 in comparison to the wild-type strain [61].
Organic hydroperoxides (OHRs) are highly harmful to cells and, and together with H2O2, represent an important component of plant defense during bacterial infection [67]. Organic hydroperoxide resistance (ohr) proteins are related to hydroperoxide detoxification, and as in PRF 81T [11], CFN 299T and CPAO 29.8 present three ohr copies, in addition to their putative organic peroxide-inducible transcription repressor (ohrR) gene. Resistance to organic hydroperoxides in S. meliloti requires ohr and ohrR gene expression; however, their gene products are not essential for overcoming the oxidative condition faced during the nodulation, since ohr and ohrR auxotroph mutants form effective nodules [67]. It has been suggested that the alkyl hydroperoxidase reductase protein detoxifies the plant’s OHRs during the symbiosis establishment [67]. Two genes encoding putative alkyl hydroperoxidase reductase proteins, members of the peroxiredoxin family, were detected in CFN 299T and CPAO 29.8. Such peroxiredoxines represent the main enzymes for the detoxification of organic peroxides, reducing them to alcohols [68]. Another peroxiredoxin gene (prxS) was found in the symbiotic plasmid of the two R. leucaenae strains. Its induction has already been reported in rhizobial bacteroids, detected both by transcriptomics and proteomics [63, 69], suggesting the involvement of this gene product with ROS detoxification during the nitrogen-fixation process [60].
As explained, bacteria possess several mechanisms to avoid damage caused by oxidative stresses. However, if ROS are not efficiently detoxified, it is necessary that the damaged molecules return to their original redox states. Methionine sulfoxide reductase activity (Msr) may repair damaged proteins during oxidative stress [70]. msrA and msrB genes, present in two copies on the CFN 299T and CPAO 29.8 genomes, encode Msr proteins that provide the reversion of methionine from its oxidized to the reduced state [71]; they were also detected in R. tropici and R. freirei genomes [11].
Secretion systems
Protein secretion by the type-I secretion system (T1SS), a Sec-independent system to export proteins (usually proteases) from Gram-negative bacteria, occurs through an oligomeric protein channel composed of an inner membrane ATP-binding cassette (ABC) protein, a periplasmic membrane fusion protein (MFP, the HylD protein), and a pore-forming outer-membrane protein (OMP) [72]. Ten copies of the hylD genes were found in the CFN 299T and CPAO 29.8 genomes. However, a single copy of the TolC OMP was found in these genomes, close to a cluster that included HlyD and one ATPase. In the case of CFN 299T, this region was close to a gene that encodes an RTX protein (repeats in toxin), that is secreted by the T1SS [73]. This protein is not present in the genome of CPAO 29.8. The CFN 299T RTX protein contains nine T1SS_rpt_143 domains and the typical carboxy-terminal, glycine- and aspartate-rich repeats.
Proteins secreted by the T2SS depend on the Sec or Tat systems for initial transport into the periplasm. Once there, they pass through the outer membrane via a multimeric (12–14 subunits) complex of pore-forming secreting proteins. Both genomes present the TadBCDE proteins with 100 % identity to each other, and 91, 94, 89 and 85 % identical, respectively, to these proteins in CIAT 899T. The tad genes (from tigh adherence) encode functions necessary for the biogenesis of the Flp subfamily of type IVb [74].
Type-IV pili are virulence factors in various bacteria and mediate, among other functions, the colonization of surfaces in different genera of Gram-negative bacteria, including Haemophilus, Pasteurella, Pseudomonas and Yersinia [75]. The tad-like genes found in the genome of Micrococcus luteus are also required for genetic transformation in this actinobacterial species [76]. Both strains of R. leucaenae harbor an identical cpaABCDEF cluster, which encodes the collagen-binding pilus component. The genes that encode these proteins are also present in the genome of CIAT 899T, with identities of 69, 82, 90, 73, 86 and 91 %, respectively.
Type-IV secretion systems (T4SS) are able to transfer proteins or nucleoprotein complexes across membranes [77]. We found T4SSs in the CFN 299T and CPAO 29.8 genomes. In both strains we found virD4 and virB1/virB11 genes that are not present in the CIAT 899Tgenome, but virD4 is present in the PRF 81T genome. virD4 and virB3-B11 of R. leucaenae are located in the same cluster, whereas virB1virB2 and virb7-like are located in separate regions. The vir system of Agrobacterium tumefaciens is responsible for the transference of tumorigenic DNA (T-DNA) into plant cells [78], while in Mesorhizobium loti R7A, a VirB/VirD4 system acts in the translocation of effector proteins into host cells, affecting the symbiosis in a host-dependant manner [79]. In R. leucaenae, we did not find homologues to the two-component regulatory system VirA/VirG that controls expression of virB/virD4 systems in A. tumefaciens and M. loti. However, we found in both strains three regions containing the F-type tra/trb T4SS genes, required for conjugal transfer of plasmids. The first of these regions, located in pA, contained the cluster traGDCAFBH, adjacent to the cluster traMRtrbHIGFLJEDCBtraI. However, in CPAO 29.8, the genes traRtrbHIGFLJE and traI were not present, suggesting that the conjugation system may not be functional. This region is similar to the conjugation machineries present in R. etli CFN 42T and Mim1, R. tropici CIAT 899T and S. fredii GR64. The second region contains traGDCAFBH close to trbIHGFLKJEDCB, and only trbD is not present in CPAO 29.8. The tra genes of this second region are similar to those present in S. meliloti 41, S. fredii NGR234, and A. tumefaciens plasmid pRiA4b; the trb region is similar to that found in R. etli CFN 42T, S. fredii NGR234 and S. meliloti 41. The third region is located in the pSym and contains the genes traGDCAFBHMRtrbIHGFLJEDCBtraI, and only the conjugational transfer transcriptional repressor TraR is not present in CPAO 29.8, which may represent a constitutive repression of the conjugation system in this strain. This pSym region is highly homologous to the one in the pSym of R. tropici CIAT 899T and also showed high homology with regions present in R. etli CFN 42T and S. fredii GR4. The presence of traI suggests that the transcription of these genes is regulated by quorum-sensing mechanisms involving N-acyl homoserine lactones. In the pSym of CIAT 899T, an IS256 inserted upstream of traR may have disrupted its promoter, leading to a constitutive repression of the conjugation system of this plasmid. However, this transposase is not present in the CFN 299T pSym and the region is not constitutively repressed. All three F-type T4SSs identified in the CFN 299T and CPAO 29.8 genomes were adjacent to repABC genes. However, in the first T4SS region of CPAO 29.8, we have not found repC, although we still have a draft genome.
The type-V secretion system (T5SS) allows secretion of large proteins that act as virulence factors [80]. In the genomes of CFN 299T and CPAO 29.8, a channel-forming transporter/cytolysins activator of the TpsB family was found, but not the TpsA protein that encodes a filamentous hemagglutinin-like T5SS-secreted protein. TpsBs are 100 % identical to each other in R. leucaenae and 79 % to R. tropici CIAT 899T, and form a beta-barrel domain for T5SS-secretion proteins. However, only one auto-transporter, a pertactin-like passenger domain (virulence factors), C-terminal, subgroup 2, is present in the genome of CFN 299T but not in CPAO 29.8.
In conclusion, both genomes of R. leucaenae are rich in CDS for genes of secretion systems. Type I, II and V can help in the exportation/secretion of proteins, and might be implied in giving competitive ability to the bacteria, especially under stressful conditions. Type-IV pili can help in colonization and, therefore, lead to a successful competitive ability in relation to other soil microorganisms, facilitating root infection and nodulation. Type-IV genes are particularly interesting for their role as effector proteins in host cells. It is interesting that such effectors were found to be expressed at high temperatures in R. freirei PRF 81T [32], enabling tolerance of stresses, and better establishment of the symbiosis under such conditions deserves fuller investigation.
Symbiotic features
The symbiotic plasmid
It is worth remembering that legume nodulation requires a cascade of molecular signals exchanged between the host plant and the rhizobium. This molecular dialogue begins with the exudation of molecules—mostly flavonoids—from the legume, which are recognized by the bacterium. When induced by the plant-host molecules, rhizobia synthesize lipochitooligosaccharides (LCOs), also known as Nod factors, responsible for launching the nodulation process, and the whole process is orchestrated by a set of nodulation (nod) genes [81]. In Rhizobium, nod genes are present in the symbiotic plasmid, which, in R. leucaenae CFN 299T and CPAO 29.8, showed high similarity (>99 %) with the pSym of R. tropici CIAT 899T. This finding gives strength to the suggestion of a conserved symbiotic plasmid defining the symbiovar tropici of species belonging to the R. tropici “group” [11]. Figure 4 displays the conservation between the pSym of R. tropici CIAT 899T, R. leucaenae CFN 299T and R. leucaenae CPAO 29.8.

Conservation between the symbiotic plasmids of R. tropici CIAT 899T, R. leucaenae CFN 299T, R. leucaenae CPAO 29.8 and R. gallicum R602T, reinforcing the suggestion of a common symbiotic plasmid defining the symbiovar tropici. Circles from innermost to outermost depict BLASTN matches between CIAT 899 and CFN 299 (blue), CPAO 29.8 (green) or R602 (red)
Comparisons of similarities of each nod gene of R. leucaenae CFN 299T with those of strains CPAO 29.8 and R. tropici CIAT 899T are shown in Additional file 4: Table S4. Few differences were detected, and included full similarity but slightly lower coverage, of nodA1 and nodA3 genes, and apparently CFN 299T carries both a truncated and a full copy of the nodS gene. The synthesis of the Nod factor-backbone chitin oligosaccharide structure is driven by nodC, and nodS is one of the genes responsible for the decoration of the basic structure, the methylation, an important property to define host range [82]. However, possible evolutionary events and biological implications of an additional truncated nodS are still to be determined.
Interestingly, when nod genes of R. leucaenae CPAO 29.8 were compared with CFN 299T and R. tropici CIAT 899T, a difference was detected in nodD3, with full identity but lower coverage than the other homologues (Additional file 5: Table S5). Analysing the genomic region, we verified that this occurred due to the presence of a mobile element, that in CIAT 899T corresponds to one putative IS21 family transposase OrfA y OrfB” (>90 % identity), that has been interposed inside the gene. Recently, nodD1 of CIAT 899T was recognized as the most important regulatory nodD gene for nodulation of common bean and leucaena [13], and nodD3 was shown to be an activator of nodD1 [12]. However, in CPAO 29.8, apparently the transposon is not affecting the symbiotic performance, as the strain is highly effective in nodulating and fixing nitrogen with common bean (Additional file 3: Table S3) and also with leucaena (data not shown).
Synthesis of Nod factors under abiotic stress
The most important agronomic feature of the two R. leucaenae strains from our study is their high capacity of nodulating and fixing nitrogen with common bean even under stressful environmental conditions. These properties might be at least partially related to the capacity of producing Nod factors under adverse conditions, as observed in R. tropici CIAT 899T under saline stress [12–15]. We have recently raised the hypothesis that the release of a large set of Nod factors by R. tropici CIAT 899T under saline stress and in the absence of plant signals might represent a strategy: i) to nodulate a broad range of hosts under stressful conditions, as an evolutionary strategy to perpetuate the symbiosis; ii) to confer stress tolerance to the bacterium, in a mechanism not yet elucidated [12, 13].
To get a better understanding of the biosynthesis of Nod factors under abiotic stress, we made comparisons with other rhizobial strains symbionts of common bean. TLC chromatographic profiles of Nod factors in the presence and absence of flavonoids and abiotic stresses were obtained for R. leucaenae CFN 299T, R. tropici CIAT 899T, R. freirei PRF 81T, R. leguminosarum bv phaseoli strain TAL1121, R. etli strains CFN 42T, Sc15, ISp19 and ISP36, R. giardinii H152T and R. gallicum R602T. None of the studied strains synthesized Nod factors in the absence of the nod-gene inducer apigenin when grown at high temperature (37 °C), but it is noteworthy that, in comparison to growth at 28 °C, heat also decreased drastically Nod-factor synthesis even when induced by flavonoids (data not shown). Indeed, nodulation of common bean is highly affected by heat stress [16, 17], and our results indicate that inhibition may start from the release of Nod factors.
Under acid stress (pH 5.0), although substantially reduced, R. leucaenae CFN 299T, R. tropici CIAT 899T, R. leguminosarum bv phaseoli TAL1121, R. etli CFN 42T, and R. gallicum strains R602T were able to synthesize Nod factors in the absence of flavonoids, but not R. giardinii H152T, but none was able to synthesize Nod factors under alkaline stress (Additional file 6: Figure S1). However, under osmotic stress (saline) only CFN 299T, CIAT 899T and PFR 81T, all belonging to the “R. tropici group” and R602T synthesized Nod factors in the absence of flavonoids. Figure 5 displays a comparison of Nod-factor profiles in CFN 299T and R602T induced by different concentrations of NaCl.

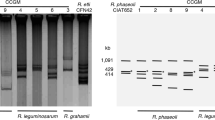
TLC analysis obtained with 14C labeled N-acetylglucosamine of Nod factors produced by R. gallicum bv. gallicum R602T and R. leucaenae CFN 299T grown under saline stress. Bacteria were induced (+) or not (−) with a flavonoid nod-gene inducer (apigenin, 3.7 μM), under different levels of saline stress (concentration of NaCl). Lines 1 and 2 represent the control without saline stress
In view of the results of synthesis of Nod factors under saline stress, we concentrated the comparison of nodulation genes on the rhizobial species carrying the pSym-tropici, including the two strains from our study, and also R. gallicum bv. gallicum R602T, because this was the only species not belonging to the “R. tropici” group able to synthesize Nod factors under saline conditions and in the absence of flavonoids. The homology of the pSym-tropici with R602T was of about 92 %. In relation to the main regulatory nodD genes, in the pSym of R. tropici CIAT 899T, R. leucaenae CFN 299T and CPAO 29.8 there were five nodD copies showing 100 % of homology. R. gallicum R602T carries four copies of nodD genes, considerably different from the pSym-tropici (Fig. 6a). From recent studies performed with mutants of the five nodD genes of R. tropici, it has been determined that although all nodD copies play a role in the synthesis of Nod factors under abiotic stress, nodD2 could be a major protagonist [12, 13]. However, none of the nodD genes of R. gallicum R602T showed high similarity with nodD2 of R. tropici. Another gene that might participate in the nod-gene induction by abiotic stress is nodA, but, again, in R602T the similarity of the two copies and a third nodA-like copy with nodA genes of R. tropici CIAT 899T, CFN 299T, and CPAO29.3 (identical to each other) was lower (Fig. 6b). Altogether, these comparisons seem to indicate that, in R. gallicum R602T, the regulatory induction of Nod-factor synthesis by abiotic stress might be different from that in R. tropici.

a Phylogenetic tree of representatives nodD genes of R. leucaenae CFN 299T and CPAO 29.8, R. tropici CIAT 899T and R. gallicum R602T. The branches length represents the evolutionary lineages changing over time. The length of the brach represents the amount of changes and it is proportional to the number of nucleotide substitutions per site. The bar at the bottom of the figure provides a scale for the evolution. b Phylogenetic tree of the three copies of nodA genes of R. leucaenae CFN 299T and CPAO 29.8, R. tropici CIAT 899T and the two nodA copies and a third nodA-like of R. gallicum R602T. The branches length represents the evolutionary lineages changing over time. The length of the brach represents the amount of changes and it is proportional to the number of nucleotide substitutions per site. The bar at the bottom of the figure provides a scale for the evolution
Conclusions
A detailed study of the genes putatively related to stress tolerance in R. leucaenae highlighted an intricated pattern comprising a variety of mechanisms that are probably orchestrated to tolerate the stressful conditions to which the strains are submitted on a daily basis. The capacity to synthesize Nod factors under abiotic stress might follow the same regulatory pathways as in CIAT 899T and may help both to improve bacterial survival and to expand host range to guarantee the perpetuation of the symbiosis.
Methods
Bacterial strains and growth conditions
R. leucaenae strains CFN 299T and CPAO 29.8 were obtained and are deposited at the “Diazotrophic and Plant Growth Promoting Bacteria Culture Collection of Embrapa Soja” (WFCC Collection # 1213, WDCM Collection # 1054), at Londrina, Paraná, Brazil. Several previous studies have reported properties of CFN 299T, culminating in its choice as the type strain of the R. leucaenae species [6]. Phenetic and genetic properties of CPAO 29.8 were also compiled before [6, 10]. Bacterial growth conditions and DNA extraction for genome sequencing were performed as described before [10].
Sequencing, assembly and gap closure
R. leucaenae CFN 299T whole-genome sequence reads were generated by MiSeq Illumina and 454 Roche (3 kb paired end library) sequencing at LNCC, Petropolis, Brazil. Hybrid de novo assemblies were generated using a combination of 454 and Illumina reads with the programs SPAdes [83] and Newbler (454 Life Sciences). Additionally, assemblies with only Illumina reads or only 454 reads were generated with the same programs. Raw Illumina reads were quality-trimmed before assembly using Trimmomatic [84]. Assemblies were merged using CISA [85], and then any possible misassemble was identified by read mapping with Bowtie [86] or Newbler. A set of primer pairs previously designed to close gaps in the symbiotic plasmid of R. tropici CIAT 899T [11] was used to amplify gap regions in the symbiotic plasmid of R. leucaenae CFN 299T. PCR products were Sanger-sequenced and manually combined with the assembly using SeqMan Pro (DNASTAR Inc.).
Whole-genome sequences of R. leucaenae strain CPAO 29.8 were generated by MiSeq Illumina and 454 Roche (3 Kb paired end library) sequencing at LNCC, Petrópolis, Brazil. De novo assembled were generated using Newbler.
A previously described strategy [11] to assign contigs to specific replicons was used. Chromosomal contigs were identified by mapping each sequence to closed chromosomes of other Rhizobium strains as these molecules are highly conserved in this genus. Likewise, contigs of the symbiotic plasmids were identified by sequence comparison with the previously reported and highly conserved tropici pSyms [11]. The smallest plasmid of CFN 299 was recovered as a single contig harboring teu genes previously mapped to that replicon.
Annotation and genome comparisons
Gene prediction and annotation were performed using the Rapid Annotation using Subsystem Technology (RAST) server [87] using the following options: RAST gene caller, FIGfam release 70 for annotation, automatically error fixing, gap backfilling and no frameshift fixing. Genome sequences were aligned with BLASTN [88] with an E-value cutoff of 1e-5. Alignments were visualized with the Artemis comparison tool [89]. Orthologues were identified with PanOCT [90] using the results of all-versus-all alignments performed with BLASTP ’140].
Symbiotic performance under regular and high temperature
A greenhouse experiment was performed with common bean cultivar Pérola (colored seeds) inoculated with eight main rhizobial strains microsymbionts of this legume: R. leucaenae CFN 299T and CPAO 29.8, R. tropici CIAT 899T, R. freirei PRF 81T, R. paranaense PRF 35T, R. etli CFN 42T, R. leguminosarum bv. phaseoli TAL1121 and R. gallicum R602T. The experiment was performed in Leonard jars as described before [17] and inoculant preparation and seed inoculation were also performed as described before [17]. Non-inoculated controls without and with (70 mg N plant−1 week−1). Plants were grown at 28/23 or 35/23 °C (day/night). At early flowering stage, 30 days after seedling emergence, plants were harvested for the determination of nodulation (nodule number). At the same harvest shoot dry weight and concentration of N (N-Kjeldhal) in shoots were determined as described before [17], to estimate the value of total N accumulated in shoots.
TLC analysis of nod factors
TLC analysis of Nod factors was performed as described before [15], in which Nod factors were labeled in vivo with 1 μCi of 14C-glucosamine hydrochloride (specific activity 52 mCi mmol−1) (Amersham Pharmacia Biotech, Buckinghamshire, England) at a final concentration of 1 μM in the medium and, after growth till exponential growth phase, extraction of the supernatant with water-saturated n-butanol and submitted to the TLC analysis. Synthesis of Nod factors was always verified in the presence or absence of flavonoids (apigenin, 3.7 μM). Abiotic stresses verified were temperature (28 and 37 °C), pH (5.0, 7.0, 9.0), and saline (up to 300 mM of NaCl). Growth conditions to verify the synthesis of Nod factors were as described before [13].
Abbreviations
ABC, ATP-binding cassette; BNF, biological nitrogen fixation; CbG, cyclic beta glucans; CDS, coding sequence; COG, cluster of orthologous groups; HSP, heat-shock protein; LCOs, lipochitooligosaccharides; MFP, membrane fusion protein; MS, mechanosensitive; OHR, organic hydroperoxide; OMP, outer-membrane protein; RAST, rapid annotation using subsystem technology; ROS, reactive oxygen species; RTX, repeats in toxin; Sec, (general) secretion pathway; sHSP, small heat-shock protein; SOD, superoxide dismutase; T1SS, type-I secretion system; T4SS, Type-IV secretion system; T5SS, Type-V secretion system; Tat, twin-arginine translocation pathway; T-DNA, tumorigenic DNA; UDP, uridine diphosphate
References
Ormeño-Orrillo E, Hungria M, Martínez-Romero E. Dinitrogen-fixing prokaryotes. In: Roseberg E, De Long EF, Lory S, Stackebrandt E, Thopmson F, editors. The Prokaryotes: Prokaryotic Physiology and Biochemistry. Berlin Heidelberg: Springer; 2013. p. 427–51.
Dwivedi SL, Sahrawat KL, Upadhyaya HD, Mengoni A, Galardini M, Bazzicalupo M, Biondi EG, Hungria M, Kaschuk G, Blair MW, Ortiz R. Advances in host plant and Rhizobium genomics to enhance symbiotic nitrogen fixation in grain legumes. Adv Agron. 2015;129:1–116.
Hungria M, Neves MCP. Cultivar and Rhizobium strain effect on nitrogen fixation and transport in Phaseolus vulgaris L. Plant Soil. 1987;103:111–21.
Hungria M, Andrade DS, Chueire LMO, Probanza A, Gutiérrez-Mañero FJ, Megías M. Isolation and characterization of new efficient and competitive bean (Phaseolus vulgaris L.) rhizobia from Brazil. Soil Biol Biochem. 2000;32:1515–28.
Martínez-Romero E, Segovia L, Mercante FM, Franco AA, Graham PH, Pardo MA. Rhizobium tropici, a novel species nodulating Phaseolus vulgaris L. beans and Leucaena sp. trees. Int J Syst Bacteriol. 1991;41:417–26.
Ribeiro RA, Rogel MA, López-López A, Ormeño-Orrillo E, Barcellos FG, Martínez J, Thompson FL, Martínez-Romero E, Hungria M. Reclassification of Rhizobium tropici type A strains as Rhizobium leucaenae sp. nov. Int J Syst Evol Microbiol. 2012;62:1179–84.
Dall’Agnol RF, Ribeiro RA, Ormeño-Orrillo E, Rogel MA, Delamuta JRM, Andrade DS, Martínez-Romero E, Hungria M. Rhizobium freirei, a symbiont of Phaseolus vulgaris very effective in fixing nitrogen. Int J Syst Evol Microbiol. 2013;63:4167–73.
Dall’Agnol RF, Ribeiro RA, Delamuta JRM, Ormeno-Orrillo E, Rogel MA, Andrade DS, Martínez-Romero E, Hungria M. Rhizobium paranaense sp. nov. an effective N2-fixing symbiont of common bean (Phaseolus vulgaris L.) with broad geographical distribution in Brazil. Int J Syst Evol Microbiol. 2014;64:3222–29.
Gomes DF, Ormeño-Orrillo E, Hungria M. Biodiversity, symbiotic efficiency and genomics of Rhizobium tropici and related species. In: de Bruijin F, editor. Biological nitrogen fixation. Hoboken: Wiley; 2015. p. 747–57. doi:10.1002/9781119053095.ch74.
Pinto FGS, Hungria M, Mercante FM. Polyphasic characterization of Brazilian Rhizobium tropici strains effective in fixing N2 with common bean (Phaseolus vulgaris L.). Soil Biol Biochem. 2007;39:1851–64.
Ormeño-Orrillo E, Menna P, Gonzaga Almeida L, Ollero FJ, Nicolás MF, Pains Rodrigues E, Shigueyoshi Nakatani A, Silva Batista JS, Oliveira Chueire LM, et al. Genomic basis of broad host range and environmental adaptability of Rhizobium tropici CIAT 899 and Rhizobium sp. PRF 81 which are used in inoculants for common bean (Phaseolus vulgaris L.). BMC Genomics. 2012;13:735.
del Cerro P, Rolla-Santos AA, Gomes DF, Marks BB, Espuny MR, Rodriguez-Carvajal MA, Soria-Diaz E, Nakatani AS, Hungria M, Ollero FJ, Megías M. Opening the “black box” of nodD3, nodD4 and nodD5 genes of Rhizobium tropici strain CIAT 899. BMC Genomics. 2015;16:864.
del Cerro P, Rolla-Santos AA, Gomes DF, Marks BB, Pérez-Montano F, Rodriguez-Carvajal MA, Nakatani AS, Gil-Serrano A, Megías M, Ollero FJ, Hungria M. Regulatory nodD1 and nodD2 genes of Rhizobium tropici strain CIAT 899 and their roles in the early stages of molecular signaling and host-legume nodulation. BMC Genomics. 2015;16:251.
Estévez J, Soria-Díaz ME, de Córdoba FF, Morón B, Manyani H, Gil A, Thomas-Oates J, van Brussel AAN, Dardanelli MS, Sousa C, Megías M. Different and new Nod factors produced by Rhizobium tropici CIAT899 following Na + stress. FEMS Microbiol Lett. 2009;293:220–31.
Guasch-Vidal B, Dardanelli MS, Soria-Díaz ME, de Córdoba FF, Balog CI, Manyani H, Gil-Serrano A, Thomas-Oates J, Hensbergen PJ, Deelder AM, Megías M, van Brussel AA. High NaCl concentrations induce the nod genes of Rhizobium tropici CIAT899 in the absence of flavonoid inducers. Mol Plant Microbe Interact. 2013;26:451–60.
Hungria M, Franco AA. Effects of high temperatures on nodulation and N2 fixation by Phaseolus vulgaris L. Plant Soil. 1993;149:95–102.
Hungria M, Kaschuk G. Regulation of N2 fixation and NO3 −/NH4 + assimilation in nodulated and N-fertilized Phaseolus vulgaris L. exposed to high-temperature stress. Environ Exp Bot. 2014;98:32–9.
Mercante FM, Cunha CO, Straliotto R, Ribeiro-Junior W, Vanderleyden J, Franco AA. Leucaena leucocephala as a trap-host for Rhizobium tropici strain from the Brazilian cerrado region. Rev Microbiol. 1998;29:49–58.
Krulwich TA, Sachs G, Padan E. Molecular aspects of bacterial pH sensing and homeostasis. Nat Rev Microbiol. 2011;9:330–43.
Hunte C, Screpanti E, Venturi M, Rimon A, Padan E, Michel H. Structure of a Na+/H+ antiporter and insights into mechanism of action and regulation by pH. Nature. 2005;435:1197–202.
Foster JW. Escherichia coli acid resistance: tales of an amateur acidophile. Nat Rev Microbiol. 2004;2:898–907.
Riccillo PM, Muglia CI, Bruijn FJDE, Roe AJ, Booth IR, Aguilar OM. Glutathione is involved in environmental stress responses in Rhizobium tropici, including acid tolerance. J Bacteriol. 2000;182:1748–53.
Muglia CI, Grasso DH, Aguilar OM. Rhizobium tropici response to acidity involves activation of glutathione synthesis. Microbiology. 2007;153:1286–96.
Chang YY, Cronan Jr JE. Membrane cyclopropane fatty acid content is a major factor in acid resistance of Escherichia coli. Mol Microbiol. 1999;33:249–59.
Shabala L, Ross T. Cyclopropane fatty acids improve Escherichia coli survival in acidified minimal media by reducing membrane permeability to H+ and enhanced ability to extrude H+. Res Microbiol. 2008;159:458–61.
Iyer R, Iverson TM, Accardi A, Miller C. A biological role for prokaryotic ClC chloride channels. Nature. 2002;419:715–7.
Rojas-Jiménez K, Sohlenkamp C, Geiger O, Martínez-Romero E, Werner D, Vinuesa P. A ClC chloride channel homolog and ornithine-containing membrane lipids of Rhizobium tropici CIAT899 are involved in symbiotic efficiency and acid tolerance. Mol Plant Microbe Interact. 2005;18:1175–85.
Vences-Guzmán MA, Guan Z, Ormeño-Orrillo E, González-Silva N, López-Lara IM, Martínez-Romero E, Geiger O, Sohlenkamp C. Hydroxylated ornithine lipids increase stress tolerance in Rhizobium tropici CIAT899. Mol Microbiol. 2011;79:1496–514.
Vinuesa P, Neumann-Silkow F, Pacios-Bras C, Spaink HP, Martínez-Romero E, Werner D. Genetic analysis of a pH-regulated operon from Rhizobium tropici CIAT899 involved in acid tolerance and nodulation competitiveness. Mol Plant Microbe Interact. 2003;16:159–68.
Sohlenkamp C, Galindo-Lagunas KA, Guan Z, Vinuesa P, Robinson S, Thomas-Oates J, Raetz CR, Geiger O. The lipid lysyl-phosphatidylglycerol is present in membranes of Rhizobium tropici CIAT899 and confers increased resistance to polymyxin B under acidic growth conditions. Mol Plant Microbe Interact. 2007;20:1421–30.
Alexandre A, Oliveira S. Response to temperature stress in rhizobia. Crit Rev Microbiol. 2013;39:219–28.
Gomes DF, Batista JSDS, Schiavon AL, Andrade DS, Hungria M. Proteomic profiling of Rhizobium tropici PRF 81: identification of conserved and specific responses to heat stress. BMC Microbiol. 2012;12:84.
Georgopoulos C, Liberek K, Zylicz M, Ang D. The Biology of heat shock proteins and molecular chaperones. Monograph 26. Cold Spring: Cold Spring Harbor Laboratory; 1994. p. 209–49.
Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–81.
Ehrnsperger M, Gräber S, Gaestel M, Buchner J. Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J. 1997;16:221–9.
Münchbach M, Dainese P, Staudenmann W, Narberhaus F, James P. Proteome analysis of heat shock protein expression in Bradyrhizobium japonicum. Eur J Biochem. 1999;264:39–48.
Michiels J, Verreth C, Vanderleyden J. Effects of temperature stress on bean nodulating Rhizobium strains. Appl Environ Microbiol. 1994;60:1206–12.
Caldas TD, Yaagoubi A, Richarme G. Chaperone properties of bacterial elongation factor EF-Tu. J Biol Chem. 1998;273:11478–82.
Caldas T, Laalami S, Richarme G. Chaperone properties of bacterial elongation factor EF-G and initiation factor IF2. J Biol Chem. 2000;275:855–60.
Braeken K, Fauvart M, Vercruysse M, Beullens S, Lambrichts I, Michiels J. Pleiotropic effects of a rel mutation on stress survival of Rhizobium etli CNPAF512. BMC Microbiol. 2008;8:219.
Zahran HH. Rhizobium-Legume symbiosis and nitrogen fixation under severe conditions and in an arid climate. Microbiol Mol Biol Rev. 1999;63:968–89.
Yap SF, Lim ST. Response of Rhizobium sp UMKL-20 to sodium-chloride stress. Arch Microbiol. 1983;135:224–8.
Nogales J, Campos R, BenAbdelkhalek H, Olivares J, Lluch C, Sanjuán J. Rhizobium tropici genes involved in free-living salt tolerance are required for the establishment of efficient nitrogen-fixing symbiosis with Phaseolus vulgaris. Mol Plant Microbe Interact. 2002;15:225–32.
Domínguez-Ferreras A, Soto MJ, Pérez-Arnedo R, Olivares J, Sanjuán J. Importance of trehalose biosynthesis for Sinorhizobium meliloti osmotolerance and nodulation of alfalfa roots. J Bacteriol. 2009;191:7490–9.
Fernández-Aunión C, Ben Hamouda T, Iglesias-Guerra F, Argandoña M, Reina-Bueno M, Nieto JJ, Aouani ME, Vargas C. Biosynthesis of compatible solutes in rhizobial strains isolated from Phaseolus vulgaris nodules in Tunisian fields. BMC Microbiol. 2010;10:192.
Smith LT, Pocard JA, Bernard-Smith LT, Smith GM. An osmoregulated dipeptide in stressed Rhizobium meliloti. J Bacteriol. 1989;171:4714–7.
Sugawara M, Cytryn EJ, Sadowsky MJ. Functional role of Bradyrhizobium japonicum trehalose biosynthesis and metabolism genes during physiological stress and nodulation. Appl Environ Microbiol. 2010;76:1071–81.
Suárez R, Wong A, Ramírez M, Barraza A, Orozco MC, Cevallos MA, Lara M, Hernández G, Iturriaga G. Improvement of drought tolerance and grain yield in common bean by overexpressing trehalose-6-phosphate synthase in rhizobia. Mol Plant Microbe Interact. 2008;21:958–66.
McIntyre HJ, Davies H, Hore TA, Miller SH, Dufour JP, Ronson CW. Trehalose biosynthesis in Rhizobium leguminosarum bv. trifolii and its role in desiccation tolerance. Appl Environ Microbiol. 2007;73:3984–92.
Domínguez-Ferreras A, Muñoz S, Olivares J, Soto MJ, Sanjuán J. Role of potassium uptake systems in Sinorhizobium meliloti osmoadaptation and symbiotic performance. J Bacteriol. 2009;191:2133–43.
Boncompagni E, Osteras M, Poggi MC, le Rudulier D. Occurrence of choline and glycine betaine uptake and metabolism in the family Rhizobiaceae and their roles in osmoprotection. Appl Environ Microbiol. 1999;65:2072–7.
Aktas M, Jost KA, Fritz C, Narberhaus F. Choline uptake in Agrobacterium tumefaciens by the high-affinity ChoXWV transporter. J Bacteriol. 2011;193:5119–29.
Dylan T, Helinski DR, Ditta GS. Hypoosmotic adaptation in Rhizobium meliloti requires β(1 → 2)-Glucan. J Bacteriol. 1990;172:1400–8.
Breedveld MW, Miller KJ. Cyclic beta-glucans of members of the family Rhizobiaceae. Microbiol Rev. 1994;58:145–61.
Martinac B, Kloda A. Evolutionary origins of mechanosensitive ion channels. Prog Biophys Mol Biol. 2003;82:11–24.
Hernández-Castro R, Rodríguez MC, Seoane A, García Lobo JM. The aquaporin gene aqpX of Brucella abortus is induced in hyperosmotic conditions. Microbiology. 2003;149:3185–92.
Calamita G, Kempf B, Bonhivers M, Bishai WR, Bremer E, Agre P. Regulation of the Escherichia coli water channel gene aqpZ. Proc Natl Acad Sci U S A. 1998;95:3627–31.
Santos R, Herouart D, Puppo A, Touati D. Critical protective role of bacterial superoxide dismutase in Rhizobium–legume symbiosis. Mol Microbiol. 2000;38:750–9.
Santos R, Herouart D, Sigaud S, Touati D, Puppo A. Oxidative burst in alfalfa-Sinorhizobium meliloti symbiotic interaction. Mol Plant Microbe Interact. 2001;14:86–9.
Dombrecht B, Heusdens C, Beullens S, Verreth C, Mulkers E, Proost P, Vanderleyden J, Michiels J. Defense of Rhizobium etli bacteroids against oxidative stress involves a complexly regulated atypical 2-Cys peroxiredoxin. Mol Microbiol. 2005;55:120–1221.
Jamet A, Sigaud S, van de Sype G, Puppo A, Herouart D. Expression of the bacterial catalase genes during Sinorhizobium meliloti-Medicago sativa symbiosis and their crucial role during the infection process. Mol Plant Microbe Interact. 2003;16:217–25.
Bauer CE, Elsen S, Bird TH. Mechanisms for redox control of gene expression. Annu Rev Microbiol. 1999;53:495–523.
Vargas MC, Encarnación S, Dávalos A, Reyes-Pérez A, Mora Y, García-de los Santos A, Brom S, Mora J. Only one catalase, katG, is detectable in Rhizobium etli, and is encoded along with the regulator OxyR on a plasmid replicon. Microbiology. 2003;149:1165–76.
Triggs-Raine BL, Doble BW, Mulvey MR, Sorby PA, Loewen PC. Nucleotide sequence of katG, encoding catalase HPI of Escherichia coli. J Bacteriol. 1988;170:4415–9.
Orikasa Y, Nodasaka Y, Ohyama T, Okuyama H, Ichise N, Yumoto I, Morita N, Wei M, Ohwada T. Enhancement of the nitrogen fixation efficiency of genetically-engineered Rhizobium with high catalase activity. J Biosci Bioeng. 2010;110:397–402.
Rubio MC, James EK, Clemente MR, Bucciarelli B, Fedorova M, Vance CP, Becana M. Localization of superoxide dismutases and hydrogen peroxide in legume root nodules. Mol Plant Microbe Interact. 2004;17:1294–305.
Fontenelle C, Blanco C, Arrieta M, Dufour V, Trautwetter A. Resistance to organic hydroperoxides requires ohr and ohrR genes in Sinorhizobium meliloti. BMC Microbiol. 2011;11:100.
Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–52.
Perret X, Freiberg C, Rosenthal A, Broughton WJ, Fellay R. High-resolution transcriptional analysis of the symbiotic plasmid of Rhizobium sp. NGR234. Mol Microbiol. 1999;32:415–25.
Ezraty B, Aussel L, Barras F. Methionine sulfoxide reductases in prokaryotes. Biochim Biophys Acta. 2005;1703:221–9.
Grimaud R, Ezraty B, Mitchell JK, Lafitte D, Briand C, Derrick PJ, Barras F. Repair of oxidized proteins. Identification of a new methionine sulfoxide reductase. J Biol Chem. 2001;276:48915–20.
Thanabalu T, Koronakis E, Hughes C, Koronakis V. Substrate-induced assembly of a contiguous channel for protein export from E. coli: reversible bridging of an innermembrane translocase to an outer membrane exit pore. EMBO J. 1998;17:6487–96.
Linhartova I, Bumba L, Masin J, Basler M, Osicka R, Kamanova J, Prochazkova K, Adkins I, Hejnova-Holubova J, Sadilkova L, et al. RTX proteins: a highly diverse family secreted by a common mechanism. FEMS Microbiol Rev. 2010;34:1076–112.
Schilling J, Wagner K, Seekircher S, Greune L, Humberg V, Schmidt MA, Heusipp G. Transcriptional activation of the tad type IVb pilus operon by PypB in Yersinia enterocolitica. J Bacteriol. 2010;192:3809–21.
Tomich M, Planet PJ, Figurski DH. The tad locus: postcards from the widespread colonization island. Nat Rev Microbiol. 2007;5:363–75.
Angelov A, Bergen P, Nadler F, Hornburg F, Lichev A, Übelacker M, Pachl F, Kuster B, Liebl W. Novel Flp pilus biogenesis-dependent natural transformation. Front Microbiol. 2015;6:84.
Juhas M, Crook DW, Hood DW. Type IV secretion systems: tools of bacterial horizontal gene transfer and virulence. Cell Microbiol. 2008;10:2377–86.
Christie PJ, Atmakuri K, Krishnamoorthy V, Jakubowski S, Cascales E. Biogenesis, architecture, and function of bacterial type IV secretion systems. Annu Rev Microbiol. 2005;59:451–85.
Hubber A, Vergunst AC, Sullivan JT, Hooykaas PJ, Ronson CW. Symbiotic phenotypes and translocated effector proteins of the Mesorhizobium loti strain R7A VirB/D4 type IV secretion system. Mol Microbiol. 2004;54:561–74.
Hayes CS, Aoki SK, Low DA. Bacterial contact-dependent delivery systems. Annu Rev Genet. 2010;44:71–90.
Denarié J, Debellé F, Promé JC. Rhizobium lipo-chitinoligosaccharide nodulation factors: signaling molecules mediating recognition and morphogenesis. Annu Rev Biochem. 1996;65:503–35.
Waelkens F, Voets T, Vlassak K, Vanderleyden J, van Rhijn P. The nodS gene of Rhizobium tropici strain CIAT899 is necessary for nodulation on Phaseolus vulgaris and on Leucaena leucocephala. Mol Plant Microbe Interact. 1995;8:147–54.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.
Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Lin SH, Liao YC. CISA: Contig Integrator for Sequence Assembly of bacterial genomes. PLoS ONE. 2013;8:e60843.
Langmead B, Trapnell C, Pop M, Salzberg S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
Aziz RK, Bartels D, Best A, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75.
Camacho C, Coulouris G, Avagyan V, Papadopoulos N, Bealer JK, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421.
Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3.
Fouts DE, Brinkac L, Beck E, Inman J, Sutton G. PanOCT: automated clustering of orthologs using conserved gene neighborhood for pan-genomic analysis of bacterial strains and closely related species. Nucleic Acids Res. 2012;40:e172.
Acknowledgements
The Brazilian group belongs to the MCTI/CNPq/CAPES/FAPS (INCT-MPCPAgro). The authors acknowledge Dr. Jesiane S. S. Batista and Dr. Esperanza Martínez-Romero for help with some analyses, and to Dr. Allan R. J. Eaglesham for English review. DFG acknowledges a postdoc fellowship from CAPES-Embrapa. MH and ATRV are also researcher fellows from CNPq. PC is recipient of the FPU fellowship of the Ministerio de Economía y Competitividad (FPU14_00160).
Funding
The study was partially supported by CNPq (National Council for Scientific and Technological Development, Science without Borders (400205/2012-5), Universal (470515/2012-0), Embrapa (02.13.08.001.00.00) and Ministerio de Economía y Competitividad from de Spanish Government (AGL2012-38831).
Availability of data and material
The genome sequences of R. leucaenae CFN 299T and CPAO 29.8 have been deposited in the GenBank database under accession numbers LNCJ00000000 and LMZF00000000, respectively.
Authors’ contributions
EOO, FJO, MM and MH conceived and designed the experiments. The experiments were performed by EOO, ATRV, CC, LGPA, FJO, MM, MH. Data were analyzed by EOO, DFG, PC, ATRV, CC, LGPA, FMM, FJO, MM, MH. ATRV, FMM, FJO, MM and MH contributed reagents/materials/analysis tools. The paper was written by DFG, PC, FJO, MM, MH. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Genome annotation of Rhizobium leucaenae strain CFN 299T and correspondence with CDS of strain CPAO 29.8 and other related rhizobial genomes. Based on RAST and manual curation. (XLSX 3313 kb)
Additional file 2: Table S2.
Genome annotation of CDS related to stress tolerance in the genomes of Rhizobium leucaenae strains CFN 299T and CPAO 29.8. Based on RAST and manual curation. (XLSX 17 kb)
Additional file 3: Table S3.
Symbiotic performance of Rhizobium leucaenae CFN 299T and CPAO 29.8 under high temperature in comparison to other microsymbionts of common bean. Plants grown under greenhouse controlled condition, at 28/23 °C and 37/23 °C (day/night) and harvested at early flowering (30 days after seedling emergency). (DOCX 16 kb)
Additional file 4: Table S4.
Homology obtained in the comparison of nodulation genes of R. leucaenae CFN 299T in comparison to strain CPAO 29.8 and R. tropici CIAT 899T. (DOCX 12 kb)
Additional file 5: Table S5.
Homology obtained in the comparison of nodulation genes of R. leucaenae CPAO 29.8 in comparison to strain CFN 299T and R. tropici CIAT 899T. (DOCX 12 kb)
Additional file 6: Figure S1.
TLC analysis obtained with 14C labeled N-acetylglucosamine of Nod factors produced by rhizobial strains microsymbionts of Phaseolus vulgaris grown under acid (pH 4.0) or alkaline (pH 9.0) stress. Bacteria were induced (+) or not (−) with a flavonoid nod-gene inducer (apigenin, 3.7 μM). Profiles obtained for R. tropici CIAT 899T were identical to those of R. leucaenae CFN 299T. (PPTX 1271 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ormeño-Orrillo, E., Gomes, D.F., del Cerro, P. et al. Genome of Rhizobium leucaenae strains CFN 299T and CPAO 29.8: searching for genes related to a successful symbiotic performance under stressful conditions. BMC Genomics 17, 534 (2016). https://doi.org/10.1186/s12864-016-2859-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-016-2859-z