
Abstract
Background
Anguillicola crassus is an economically and ecologically important parasitic nematode of eels. The native range of A. crassus is in East Asia, where it infects Anguilla japonica, the Japanese eel. A. crassus was introduced into European eels, Anguilla anguilla, 30 years ago. The parasite is more pathogenic in its new host than in its native one, and is thought to threaten the endangered An. anguilla across its range. The molecular bases for the increased pathogenicity of the nematodes in their new hosts is not known.
Results
A reference transcriptome was assembled for A. crassus from Roche 454 pyrosequencing data. Raw reads (756,363 total) from nematodes from An. japonica and An. anguilla hosts were filtered for likely host contaminants and ribosomal RNAs. The remaining 353,055 reads were assembled into 11,372 contigs of a high confidence assembly (spanning 6.6 Mb) and an additional 21,153 singletons and contigs of a lower confidence assembly (spanning an additional 6.2 Mb). Roughly 55% of the high confidence assembly contigs were annotated with domain- or protein sequence similarity derived functional information. Sequences conserved only in nematodes, or unique to A. crassus were more likely to have secretory signal peptides. Thousands of high quality single nucleotide polymorphisms were identified, and coding polymorphism was correlated with differential expression between individual nematodes. Transcripts identified as being under positive selection were enriched in peptidases. Enzymes involved in energy metabolism were enriched in the set of genes differentially expressed between European and Asian A. crassus.
Conclusions
The reference transcriptome of A. crassus is of high quality, and will serve as a basis for future work on the invasion biology of this important parasite. The polymorphisms identified will provide a key tool set for analysis of population structure and identification of genes likely to be involved in increased pathogenicity in European eel hosts. The identification of peptidases under positive selection is a first step in this programme.
Similar content being viewed by others
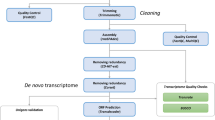

Background
The nematode Anguillicola crassus Kuwahara, Niimi et Itagaki, 1974 is a native parasite of the Japanese eel Anguilla japonica[1]. Adults localise to the swim bladder where they feed on blood [2]. Larvae are transmitted via crustacean intermediate hosts [3]. Originally endemic to East Asian populations of An. japonica, A. crassus has attracted interest due to recent anthropogenic expansion of its geographic and host ranges to Europe and the European eel, Anguilla anguilla. A. crassus was recorded for the first time in Europe in North-West Germany in 1982 [4], where it was most likely introduced through the live-eel trade [5, 6]. A. crassus has subsequently spread rapidly through populations of its newly acquired host [7], and has been found in all An. anguilla populations except those in Iceland [8]. A. crassus can thus be regarded as a model for the introduction and spread of invasive parasites [9].
In An. anguilla, both prevalence and mean intensity of infection by A. crassus are higher than in An. japonica[10, 11]. In An. anguilla infections, the adult nematodes are larger, have an earlier onset of reproduction, a greater egg output [12] and induce increased pathology, including thickening and inflammation of the swim bladder wall [13]. It has been suggested that the life history modifications and changed virulence observed in A. crassus in the new host are due to an inadequate immune response in An. anguilla[14]. An. japonica is capable of killing histotropic larvae of the parasite after vaccination [15] or under high infection pressure [16], but this does not happen in A. anguilla.
The genus Anguillicola is placed in the nematode suborder Spirurina (clade III sensu[17]) [18, 19]. The Spirurina are exclusively parasitic and include important human pathogens (the causative agents of filariasis and ascariasis) as well as prominent veterinary parasites. Molecular phylogenetic analyses place Anguillicola in a clade of spirurine nematodes (Spirurina B of [20]) that have a freshwater or marine intermediate host, but infect a wide range of carnivorous definitive hosts. Spirurina B is sister to the main Spirurina C, including the agents of filariasis and ascariasis, and thus A. crassus may be used as an outgroup taxon to understand the evolution of parasitic phenotypes in these species.
The differences in the biology of A. crassus in An. japonica (coevolved) and An. anguilla (recently captured) eel hosts is likely to result from differential interactions between host genetics and parasite genetics. While genetic differences between the host species are expected, it is not known what part, if any, genetic differentiation between the invading European and endemic Asian parasites plays. European A. crassus are less genetically variable than parasites taken from Asian hosts [21], reflecting the derived nature of the invading populations and the likely population bottlenecks this entailed. As part of a programme to understand the invasiveness of A. crassus in An. anguilla, we are investigating differences in gene expression and genetic distinction between invading European and endemic Asian A. crassus exposed to the two host species.
Recent advances in sequencing technology (often termed next generation sequencing) provide the opportunity for rapid and cost-effective generation of genome-scale data. The Roche 454 platform [22] is particularly suited to transcriptomics of previously unstudied species [23]. Here we describe the generation of a reference transcriptome for A. crassus based on Roche 454 data, and explore patterns of gene expression and diversity within the nematode.
Methods
Nematode samples, RNA extraction, cDNA synthesis and Sequencing
A. crassus from An. japonica were sampled from Kao-Ping river and an adjacent aquaculture in Taiwan as described in [16]. Nematodes from An. anguilla were sampled from Sniardwy Lake, Poland and from the Linkenheimer Altrhein, Germany. After determination of the sex of adult nematodes, they were stored in RNA-later (Quiagen, Hilden, Germany) until extraction of RNA. RNA was extracted from individual adult male and female nematodes and from a population of second stage larvae (L2) (Table 1). For host contamination screening a liver sample from an uninfected An. japonica was also processed. RNA was reverse transcribed and amplified into cDNA using the MINT-cDNA synthesis kit (Evrogen, Moscow, Russia). Emulsion PCR and library preparation were performed for each cDNA library according to the manufacturer’s protocols (Roche/454 Life Sciences), and sequenced on a Roche 454 Genome Sequencer FLX.
Raw sequencing reads are archived under study-accession number SRP010313 in the NCBI Sequence Read Archive (SRA; http://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP010313) [24]. All samples were sequenced using the FLX Titanium chemistry, except for the Taiwanese female sample T1, which was sequenced using FLX standard chemistry, to generate between 99,000 and 209,000 raw reads per sample. For the L2 library, which had a larger number of non-A. crassus, non-Anguilla reads, we confirmed that these data were not laboratory contaminants by screening Roche 454 data produced on the same run in independent sequencing lanes.
Trimming, quality control and assembly
Raw sequences were extracted in FASTA format (with the corresponding qualities files) using sffinfo (Roche/454) and screened for MINT adapter sequences using cross-match [25] (with parameters -minscore 20 -minmatch 10). Seqclean [26] was used to identify and remove poly-A-tails, low quality, low complexity and short (<100 base) sequences. All reads were compared to a set of screening databases using BLAST [27] (expect value cutoff E <1e-5, low complexity filtering turned off: -F F). The databases used were (a) a host sequence database comprising an assembly of the An. japonica Roche 454 data, a unpublished assembly of An. anguilla Sanger dideoxy sequenced expressed sequence tags (made available to us by Gordon Cramb, University of St Andrews) and transcripts from EeelBase [28], a publicly available transcriptome database for the European eel; (b) a database of ribosomal RNA (rRNA) sequences from eel species derived from our Roche 454 data and EMBL-Bank; and (c) a database of rRNA sequences identified in our A. crassus data by comparing the reads to known nematode rRNAs from EMBL-Bank. This last database notably also contained cobiont rRNA sequences. Reads with matches to one of these databases over more than 80% of their length and with greater than 95% identity were removed from the dataset. Screening and trimming information was written back into sff-format using sfffile (Roche 454). The filtered and trimmed data were assembled using the combined assembly approach [23]: Two assemblies were generated, one using Newbler v2.6 [22] (with parameters -cdna -urt), the other using Mira v3.2.1 [29] (with parameters–job=denovo,est,accurate,454). The resulting two assemblies were combined into one using Cap3 [30] at default settings and contigs were labeled by whether they derived from both assemblies (high confidence assembly; highCA), or one assembly only (lowCA; for a detailed analysis of the assembly categories see the supporting Methods file). The superset of highCA contigs, lowCA contigs and the remaining unassembled reads defines the set of tentatively unique genes (TUGs).
Post-assembly classification and taxonomic assignment of contigs
We rescreened the assembly for host and other contamination by comparing it (using BLAST) to the three databases defined above, and also to NEMBASE4, a nematode transcriptome database derived from whole genome sequencing and EST assemblies [31, 32]. For each contig, the highest-scoring match was recorded, if it spanned more than 50% of the contig. We also compared the contigs to the NCBI non-redundant nucleotide (NCBI-nt) and protein (NCBI-nr) databases, recording the taxonomy of all best matches with expect values better than 1e-05. Sequences with a best hit to non-Metazoans or to Chordata within Metazoa were excluded from further analysis.
Protein prediction and annotation
Protein translations were predicted from the contigs using prot4EST (version 3.0b) [33]. Proteins were predicted either by joining single high scoring segment pairs (HSPs) from a BLAST search of uniref100 [34], or by ESTscan [35], using as training data the Brugia malayi complete proteome [36] back-translated using a codon usage table derived from the BLAST HSPs, or, if the first two methods failed, simply the longest ORF in the contig. For contigs where the protein prediction required insertion or deletion of bases in the original sequence, we also imputed an edited sequence for each affected contig. Annotations with Gene Ontology (GO), Enzyme Commission (EC) and Kyoto Encyclopedia of Genes and Genomes (KEGG) terms were inferred for these proteins using annot8r (version 1.1.1) [37], using the annotated sequences available in uniref100 [34]. Up to 10 annotations based on a BLAST similarity bitscore cut-off of 55 were obtained for each annotation set. The complete B. malayi proteome (as present in uniref100) and the complete C. elegans proteome (as present in WormBase v.220) were also annotated in the same way. SignalP V4.0 [38] was used to predict signal peptide cleavage sites and signal anchor signatures for the A. crassus transcriptome and for the proteomes of the two model nematodes. InterProScan [39] (command line utility iprscan version 4.6 with options -cli -format raw -iprlookup -seqtype p -goterms) was used to obtain domain annotations for the highCA contigs. We recorded the presence of a lethal RNAi phenotype in the C. elegans ortholog of each TUG using the biomart-interface [40] to WormBase v. 220 using the R package biomaRt [41].
Single nucleotide polymorphism analysis
We mapped the raw reads to the complete set of contigs, replacing imputed sequences for originals where relevant, using ssaha2 (with parameters -kmer 13 -skip 3 -seeds 6 -score 100 -cmatch 10 -ckmer 6 -output sam -best 1) [42]. From the ssaha2 output, pileup files were produced using samtools [43], discarding reads mapping to multiple regions. VarScan [44] (pileup2snp) was used with default parameters on pileup files to output lists of single nucleotide polymorphisms (SNPs) and their locations.
In the 10,496 SNPs thus defined, the ratio of transitions (ti; 6,908) to transversion (tv; 3,588) was 1.93. From the prot4EST predictions, 7,189 of the SNPs were predicted to be inside an ORF, with 2,322 at codon first positions, 1,832 at second positions and 3,035 at third positions. As expected, ti/tv inside ORFs (2.39) was higher than outside ORFs (1.25). The ratio of synonymous polymorphisms per synonymous site to non-synonymous polymorphisms per non-synonymous site in this unfiltered SNP set (dn/ds) was 0.45, rather high compared to other analyses. Roche 454 sequences have well-known systematic errors associated with homopolymeric nucleotide sequences [45], and the effect of exclusion of SNPs in, or close to, homopolymer regions was explored. When SNPs were discarded using different size thresholds for homopolymer runs and proximity thresholds, the ti/tv and in dn/ds ratios changed (Additional file 1: Figure S1). Based on this SNPs associated with a homopolymer run longer than 3 bases within a window of 11 bases (5 bases to the right, 5 to the left) around the SNP were discarded. There was a relationship between TUG dn/ds and TUG coverage, associated with the presence of sites with low abundance minority alleles (less than 7% of the allele calls), suggesting that some of these may be errors. Removing low abundance minority allele SNPs from the set removed this effect (Additional file 1: Figure S2). For enrichment analysis of GO terms associated with positively selected TUGs we used the R package GOstats [46].
Using Samtools [43] (mpileup -u) and Vcftools [47] (view -gcv) we genotyped individual libraries for each of the master list of SNPs. Genotype- calls were accepted at a phred-scaled genotype quality threshold of 10. In addition to the relative heterozygosity (number of homozygous sites/number of heterozygous sites) we used the R package Rhh [48] to calculate internal relatedness [49], homozygosity by locus [50] and standardised heterozygosity [51] from these data. We confirmed the significance of heterozygote-heterozygote correlation by analysing the mean and 95% confidence intervals from 1000 bootstrap replicates estimated for all measurements.
Gene expression analysis
Read-counts were obtained from the bam files generated for genotyping using the R package Rsamtoools [52]. LowCA contigs and contigs with fewer than 32 reads over all libraries were excluded from analysis. Libraries E1 and L2 had very low overall counts and thus we excluded these libraries from analysis. The statistic of Audic and Claverie [53] as implemented in ideg6 [54] was used to contrast single libraries. Differential expression between libraries from male versus female nematodes was accepted for genes that differed in expression values between all the female libraries (E2, T1 and T2; see Table 1) versus the male (M) library (p <0.01), but had no differential expression within any of the female libraries at the same threshold. Differential expression between libraries from nematodes of European An. anguilla and Taiwanese An. japonica origin was accepted for genes that differed in expression values between library E2 and both libraries T1 and T2 (p <0.01), but showed no differences between T1 and T2.
Overrepresentation analyses
The R package annotationDbi [55] was used to obtain a full list of associations (along with higher-level terms) from annot8r annotations prior to analysis of GO term overrepresentation in gene sets selected on the basis of dn/ds or expression values. The R package topGO [56] was used to traverse the annotation graph and analyse each node term for overrepresentation in the focal gene set compared to an appropriate universal gene set (all contigs with dn/ds values or all contigs analysed for gene expression) with the “classic” method and Fisher’s exact test. Terms for which an offspring term was already in the table and no additional counts supported overrepresentation were removed. Mann-Whitney u-tests were used to test the influence of factors on dn/ds values. To investigate multiple contrasts between groups (factors) Nemenyi-Damico-Wolfe-Dunn tests were used, and for overrepresentation of one group (factor) in other groups (factors) Fisher’s exact test was used.
General coding methods
The bulk of analysis (unless otherwise described) presented was carried out in R [57] using custom scripts. For visualisation we used the R packages ggplot2 [58] and VennDiagram [45].
Results
Sampling A. crassus
One female A. crassus and one male A. crassus were sampled from an An. japonica aquaculture with high infection loads in Taiwan, and an additional female was sampled from an An. japonica caught in a stream with low infection pressure adjacent to the aquaculture. A female nematode and pool of L2 were sampled from An. anguilla in the river Rhine, and one female from A. anguilla from a lake in Poland. All adult nematodes were replete with host blood. To assist in downstream filtering of host from nematode reads, we also sampled RNA from the liver of an uninfected Taiwanese An. japonica.
Assembly and post-assembly screening
A total of 756,363 raw sequencing reads were generated for A. crassus (Table 1). These were rigorously filtered (see supporting infromation) and 353,055 remaining reads (spanning 100,491,819 bases) were assembled using the combined assembler strategy [23], employing Roche 454 gsAssembler (also known as Newbler; version 2.6) and MIRA (version 3.21) [29] (Additional file 1). This coassembly will be included in future versions of nembase (nembase5) and is available at http://www.anguillicola.nematod.es (further contig data can be found in Additional file 2). It comprised 13,851 contigs supported by both assembly algorithms, 3,745 contigs supported by only one of the assembly algorithms and 22,591 singletons that not assembled by either program (Table 2). Contigs supported by both assemblers were longer than those supported by only one assembler, and were more likely to have a significant similarity to previously sequenced protein coding genes than contigs assembled by only one of the algorithms, or the remaining unassembled singletons. These constitute the highCA, while those with evidence from only one assembler and the singletons are the lowCA. These datasets were the most parsimonious (having the smallest size) for their quality (covering the largest amount of sequence in reference transcriptomes). In the highCA parsimony and low redundancy was prioritised, while in the complete assembly (highCA plus lowCA including singletons) completeness was prioritised. The 40,187 sequences (contig consensuses and singletons) in the complete assembly are referred to as tentatively unique genes (TUGs).
We screened the complete assembly for remaining host contamination, and identified 3,441 TUGs that had significant, higher similarity to eel (and/or chordate; EMBLBank Chordata proteins) than to nematode sequences [32]. Given the identification of cercozoan ribosomal RNAs in the L2 library, we also screened the complete assembly for contamination with transcripts from other taxa.
1,153 TUGs were found with highest significant similarity to Eukaryota outside of the kingdoms Metazoa, Fungi and Viridiplantae. These contigs matched genes from a wide range of protists from Apicomplexa (mainly Sarcocystidae, 28 hits and Cryptosporidiidae 10 hits), Bacillariophyta (diatoms, mainly Phaeodactylaceae, 41 hits), Phaeophyceae (brown algae, mainly Ectocarpaceae, 180 hits), Stramenopiles (Albuginaceae, 63 hits), Kinetoplasitda (Trypanosomatidae, 26 hits) and Heterolobosea (Vahlkampfidae, 38 hits). Additionally 298 TUGs had best, significant matches to genes from fungi (e.g Ajellomycetaceae, 53 hits) and 585 TUGs had best, significant matches to genes from plants. Outside the Eukaryota there were significant best matches to Bacteria (825 TUGs; mostly to members of the Proteobacteria), Archaea (8 TUGs) and viruses (9 TUGs). No TUGs had significant, best matches to Wolbachia or related Bacteria known as symbionts of nematodes and arthropods. All TUGs with highest similarity to sequences deriving from taxa outside Metazoa were excluded. The final, screened A. crassus assembly has 32,525 TUGs, spanning 12,733,095 bases (of which 11,372 are highCA-derived, and span 6,575,121 bases). All analyses reported below are based on this filtered dataset.
Annotation
For 32,418 of the A. crassus TUGs a protein was predicted using prot4EST [33] (Table 2). An apparently full-length open reading frame (ORF) was identified in 353 TUGs, while for 29,877 the 5’ ends and for 24,277 the 3’ ends were missing. In 13,383 TUGs the corrected sequence with the imputed ORF was slightly changed compared to the raw sequence by insertions or deletions necessary to obtain a continuous reading frame. Using BLAST we determined that 9,556 had significant similarity to C. elegans proteins, 9,664 TUGs matched B. malayi proteins, and 11,620 TUGs had matches in NEMPEP4 [31, 32]. Comparison to the UniProt reference identified 11,115 TUGs with significant similarities. We used annot8r [37] to assign GO terms to 6,511 TUGs, EC numbers for 2,460 TUGs and KEGG pathway annotations for 3,846 TUGs (Table 2). Additionally 5,125 highCA contigs were annotated with GO terms through InterProScan [39]. Nearly one third (6,989) of the A. crassus TUGs were annotated with at least one identifier, and 1,831 had GO, EC and KEGG annotations (Figure 1).

Annotation of the Anguilicolla crassus transcriptome. Number of annotated sequences in the transcriptome of A. crssus for all TUGs (a) and for highCA contigs (b). Annotations with Gene Ontology (GO), Enzyme Commission (EC) and Kyoto Encyclopedia of Genes and Genomes (KEGG) terms were inferred for predicted proteins using annot8r (version 1.1.1) [37]. For highCA contigs additional domain annotations obtained with InterProScan [39] are also enumerated.
We compared our A. crassus GO annotations for high-level GO-slim terms to the annotations (obtained in the same way) for the complete proteome of the spirurid filarial nematode B. malayi and the complete proteome of C. elegans (Figure 2). The occurrence of GO terms in the annotation of the partial transcriptome of A. crassus was more similar to that of the proteome of B. malayi (0.95; Spearman correlation coefficient) than to the that of the proteome of C. elegans (0.9).

Comparing high level GO-slim annotations. Comparing high level GO-slim annotations obtained through annot8r (version 1.1.1) [37] for A. crassus to those for the model nematodes C. elegans and B. malayi infered using the same pipeline. For GO categories molecular function, cellular compartment and biological process the number of terms in high level GO-slim categories is given. In the two parasitic nematodes a higher degree of congruence in annotation spectrum is observed (Spearman correlation coefficient 0.95) than in comarison to the complete proteome of C. elegans (0.90).
Despite the lack of completeness at the 5’ end suggested by peptide prediction, just over 3% of the TUGs were predicted to be secreted (920 with signal peptide cleavage sites and 65 signal peptides with a transmembrane signature). Again these predictions are more similar to predictions using the same methods for the proteome of B. malayi (742 signal peptide cleavage sites and 41 with transmembrane anchor) than for the proteome of C. elegans (4,273 signal peptide cleavage sites and 154 with transmembrane anchor).
By comparison to RNAi phenotypes for C. elegans genes [59, 60] likely to be orthologous to A. crassus TUGs, 6,029 TUGs were inferred to be essential (RNAi lethal phenotype in C. elegans).
To explore the phylogenetic conservation of A. crassus TUGs, they were classified as conserved across kingdoms, conserved in Metazoa, conserved in Nematoda, conserved in Spirurina or novel to A. crassus by comparing them to custom database subsets using BLAST (Table 3). Using a relatively strict cutoff, a quarter of the highCA contigs were conserved across kingdoms, and 10% were apparently restricted to Nematoda. Nearly half of the highCA contigs were novel to A. crassus.
Similar patterns were observed for conservation assessed at different stringency, and when assessed across all TUGs, except that a higher proportion of all TUGs were apparently unique to A. crassus.
Proteins predicted to be restricted to Nematoda and novel in A. crassus were significantly enriched in signal peptide annotation compared to conserved proteins, proteins novel in Metazoa and novel in Spirurina (Fisher’s exact test p <0.001 ; Figure 3). The proportion of lethal RNAi phenotypes was significantly higher for C. elegans presumed orthologs of TUGs conserved across kingdoms (97.23%) than for orthologs of TUGs not conserved across kingdoms (94.59%; p <0.001, Fisher’s exact test).

Enrichment of Signal-positives for categories of evolutionary conservations. Categories of evolutionary conservation recorded using the taxonomy of BLAST-matches at two different bitscore thresholds (50 or 80) are compared for the occurence of signal peptide cleavage sites and signal anchor signatures, predicted using SignalP V4.0 [38]. Contigs were categorised as conserved, novel in the kingdom Metazoa, the phylum Nematoda or Spirurina sensu[17]. TUGs without any match at a given threshold were categorised as novel in A. crassus (Ac). The highest proportions of genes predicted to have secretory signal peptides are observed in TUGs predicted to be part of gene families that arose in the last common ancestor of Nematoda or to be novel to A. crassus.
Identification and analysis of single nucleotide polymorphisms
Single nucleotide polymorphisms (SNPs) were called using VARScan [44] on the 1,100,522 bases of TUGs that had coverage of more than 8-fold available. SNPs predicted to have more than 2 alleles, or that mapped to an undetermined (N) base were excluded, as were SNP likely to be due to base calling errors close to homopolymer tracts and SNP calls resulting from apparent rare variants.
Our filtered SNP dataset includes 5,113 SNPs, with 4.65 SNPs per kb of contig sequence (Additional file 3). There were 7.95 synonymous SNPs per 1000 synonymous bases and 2.44 non-synonymous SNPs per 1000 non synonymous bases. A mean dn/ds of 0.244 was calculated for the 765 TUGs (all highCA contigs) containing at least one synonymous SNP. Positive selection can be inferred from high dn/ds ratios. Overrepresented GO ontology terms associated with TUGs with dn/ds higher than 0.5 were identified (Table 4; Additional file 4: Figure S11 a, b, c). Within the molecular function category, “peptidase activity” was the most significantly overrepresented term. Twelve of the thirteen high dn/ds TUGs annotated as peptidases each had unique orthologs in C. elegans and B. malayi. Other overrepresented categories identified subunits of the respiratory chain: “heme-copper terminal oxidase activity” and “cytochrome-c oxidase activity” in molecular function and “mitochondrion” in cellular compartment. Contigs identified as novel to Spirurina and novel in A. crassus had a significantly higher dn/ds than other contigs (Additional file 1: Figure S3).
Signal peptide containing proteins have been shown to have higher rates of evolution than cytosolic proteins in a number of nematode species. A. crassus TUGs predicted to contain signal peptide cleavage sites showed a non-significant trend towards higher dn/ds values than TUGs without signal peptide cleavage sites (p=0.22; two sided Mann-Whitney-test). Orthologs of C. elegans transcripts with lethal RNAi phenotype are expected to evolve under stronger selective constraints and the values of dn/ds showed a non-significant trend towards lower values in TUGs with orthologs with a lethal phenotype compared to a non-lethal phenotypes (p=0.815, two-sided U-test).
The genotypes of single adult nematodes were called using Samtools [43] and Vcftools [47], and 199 informative sites (where two alleles were found in at least one assured genotype at least in one of the nematodes) were identified in 152 contigs. Internal relatedness [49], homozygosity by loci [50] and standardised heterozygosity [51] all identified the Taiwanese nematode from aquaculture (sample T1) as the most heterozygous and the European nematode from Poland (sample E2) as the least heterozygous individuals (Table 5).
The genome-wide representativeness of these 199 SNP markers for the whole genome in population genetic studies was confirmed using heterozygosity-heterozygosity correlation [48]: mean internal relatedness = 0.78, lower bound of 95% confidence intervals from 1000 bootstrap replicates (cil) = 0.444; mean homozygosity by loci = 0.86, cil = 0.596; standardised heterozygosity = 0.87, cil= 0.632.
Differential gene expression
Gene expression was inferred by the unique mapping of 252,388 (71.49%) of the raw reads to the fullest assembly (including all assembled contigs as a “filter”; total contigs/all TUGs in Table 2). Non-A. crassus contigs, and all contigs with fewer than 32 reads overall were excluded. Thus 658 TUGs were analysed for differential expression using ideg6 for normalisation and the statistic of Audic and Claverie [53] for detection of differences. Of these TUGs, 54 showed expression predominantly in the male library, 56 TUGs were more highly represented in the female library (Additional file 5), 56 TUGs were primarily expressed in the libraries from Taiwan, and 22 TUGs were overrepresented in the European library (Additional file 6).
Analysis of overrepresentation of of GO terms associated with TUGs differentially expressed between male and female libraries identified ribosomal proteins, oxidoreductases and collagen processing enzyme terms (Table 6; Additional file 4: Figure S11 g, h, i). The ribosomal proteins were all overexpressed in the male library, while the oxidoreductases and collagen processing enzymes were overexpressed in female libraries. Similar analysis of overrepresentation of of GO terms associated with the TUGs differentially expressed between European nematodes and Asian nematodes identified several terms of catalytic activity related to metabolism (Table 7; Additional file 5: Figure S11 d, e, f).
TUGs annotated as acyltransferase were upregulated in the European libraries. However, the expression patterns for other TUGs with overrepresented terms connected to metabolism did not show concerted up or down-regulation. Thus for the term “steroid biosynthetic process”, 2 TUGs were downregulated and 3 contigs upregulated in European nematodes. No enrichment of signal peptide positive TUGs, of TUG conservation categories, or TUGs with C. elegans orthologs with lethal or non-lethal RNAi-phenotypes was identified. Significantly elevated dn/ds was found for TUGs differentially expressed in European versus Asian nematodes (Fisher’s exact test p=0.007; also both up- or down-regulated were significant). TUGs overexpressed in the female libraries showed elevated levels of dn/ds (Fisher’s exact test p=0.041), but contrast male overexpressed genes showed decreased levels of dn/ds (Fisher’s exact test p=0.014).
Discussion
We have generated a de novo transcriptome for A. crassus, an important invasive parasite that threatens wild stocks of the European eel An. anguilla. These data will enable a broad spectrum of molecular research on this ecologically important and evolutionarily interesting parasite.
As A. crassus lives in close association with its host, we used exhaustive filtering to remove all host-derived, and host-associated organism-derived contamination from the raw and assembled data. We generated a transcriptome dataset from the definitive host An. japonica as part of this filtering process. In addition to eel-derived transcripts, we also removed data apparently derived from protists, particularly cercozoans, that may have been co-parasites of the eels sampled. Similar taxonomic screening of transcrioptome data has been shown to be important previously [61], particularly in rejection of hypotheses of horizontal gene transfer into the focal species [62]. We were not able to use base frequency and codon usage based screening to identify contaminant data [63, 64] because contaminant sequences in our data derived from multiple genomes.
We used a combined assembly approach [23] to generate a transcriptome estimate that had low redundancy and high completeness. Projects using single assemblers often report substantially greater numbers of contigs for datasets of similar size (see e.g. [65]). The 3’ bias in the assembly likely derivesd from the use of oligod(T) in mRNA capture and cDNA synthesis and is near-ubiquitous in deep transcriptome sequencing projects (e.g. [66]). The final A. crassus TUG assembly (32,418 contig consensuses) spans 12.7 Mb, and thus likely covers most of the expected span of the transcriptome (the C. elegans transcriptome spans 30 Mb [67], and the B. malayi transcriptome 14 Mb [36]), albeit fragmented.
Comparison between free-living and parasitic nematode species can be used to identify genes that may underpin adaptations for parastism [68, 69]. Annotations were derived for a 30% of all TUGs, and over 50% of the highCA contigs using sequence similarity to known proteins. Domain annotations were derived for 45% of the highCA TUGs using InterProScan [39]. Comparison with the complete proteomes of B. malayi and C. elegans showed a remarkable degree of congruence in annotation spectrum in the two parasitic nematodes. This implies that the A. crassus transcriptome is a representative partial genome [70]. Using a taxonomically-stratified analysis of BLAST similarities, we identified more A. crassus TUGs that apparently arose in the common ancestor of Nematoda than arose in the last common ancestor of the Spirurina. As A. crassus is part of a lineage that arises basally in Spirurina, the lack of genes associated with Sprirurina may be due to phylogenetic distance obscuring relationships, particularly if the genes underpinning parasitism are, as would be expected, rapidly evolving. TUGs predicted to be part of gene families that arose in the last common ancestor of Nematoda or to be novel to A. crassus contained the highest proportion of genes predicted to have secretory signal peptides. This confirms observations made in a Nippostrongylus brasiliensis[71], where secreted and surface proteins were less conserved. Analysis of dn/ds (see below) across conservation categories favors the hypothesis of rapid evolution in proteins with more restricted phylogenetic origins.
Transcriptome data were generated from multiple individual A. crassus of Taiwanese and European origin. We identified abundant SNPs both within and between populations, but noted aberrant patterns in the ratio of transitions to transversions (ti/tv) and the ratio of non-synonymous SNPs per non-synonymous site to synonymous SNPs per synonymous site (dn/ds). Screening of SNPs in or adjacent to homopolymer regions, removing “noise” associated with common homopolymer errors [72], improved overall measurements of SNP quality, increased the ti/tv ratio to more closely resemble that of canonical datasets, and resulted in a reduced, credible dn/ds ratio distribution. The corrected ti/tv value of 1.93 (1.25 outside and 2.39 inside ORFs) is in good agreement with the overall ti/tv of Homo sapiens (2.16 [73]) or Drosophila melanogaster (2.07 [74]). The mean dn/ds ratio decreased with removal of SNPs adjacent to homopolymer regions from 0.45 to 0.24. While interpretation of dn/ds ratios within populations is not unproblematic [75], the assumption of negative (purifying) selection on most protein coding genes makes lower mean values seem more plausible.
We applied a threshold value for the minority allele of 7% for exclusion of SNPs, as approximately 10 haploid equivalents were sampled (5 individual nematodes plus negligible contributions from the L2 library and offspring within the adult female nematodes). This screening reduced the number of non-synonymous SNPs in high coverage TUGs, removed the dependence of dn/ds on coverage, and removed the need to control for sampling biased by depth (i.e. coverage; see [76, 77]).
The final dn/ds estimates seem plausible, as D. melanogaster female reproductive tract transcripts have dn/ds of 0.15 [78] and a Roche 454 transcriptomic analysis of the parasitic nematode Ancylostoma caninum reported dn/ds of 0.3 [79]. A dn/ds threshold (on coding sequence) of 0.5 has been suggested as threshold for assuming positive selection [78]. Using this we identified 144 TUGs that may be under positive selection, thirteen peptidases were under positive selection (out of 43 annotated), and the GO term peptidases was significantly overrepresented in the set of positively selected TUGs. Those thirteen peptidases are deeply conserved, as twelve had unique orthologue pairs in B. malayi and C. elgans. Peptidases have previously been proposed to have acquired prominent roles in host-parasite interactions. An A. crassus trypsin-like proteinase may be utilised by the tissue-dwelling third stage larvae to penetrate host tissue and an aspartyl proteinase may be a blood meal digestive enzyme in adults [2]. The thirteen proteinases under positive selection could be targets of adaptive immunity developed against A. crassus[15, 80], which is often only elicited against some but not all larvae [81].
A set of 199 high-credibility SNPs with high information content for population genetic studies was identified by genotyping individual nematodes. The low number of SNPs inferred reflects both the variance in allele contribution introduced in transcriptomic data and the stringency of the software used, which is targeted at higher throughput genome sequence data [82]. Nevertheless, levels of genome-wide heterozygosity found for the five adult nematodes examined are in agreement with existing microsattelite data that show reduced heterozygosity in European populations of A. crassus[21]. The Polish female nematode was the most highly inbred, while the nematode from the cultured An. japonica from Taiwan was the most highly outbred.
While our experiment was not designed to identify differential expression between conditions (due to low replication) we used methods developed for comparison of cDNA libraries [53] to infer differential gene expression according to the origin of the sequencing libraries. This approach is widely used with 454 transcriptome data (e.g. [79]). We can only tentatively infer differential expression of a gene under different conditions (sex, origin) based on identification of significantly differential expression between libraries. Genes over-expressed in the male A. crassus included major sperm proteins [83], and, surprisingly, a suite of ribosomal proteins. Collagen processing enzymes were overexpressed in the female nematodes in line with modulation of collagen synthesis in nematode embryonic development, and the ovoviviparity of this species [83]. Acetyl-CoA acetyltransferase was identified as overexpressed in European nematodes compared to the Asian one. Acetyl-CoA acetyltransferases act in fatty-acid-oxidation in peroxisomes and mitochondria [84]. Together with a change in steroid metabolism and the enrichment of mitochondrially localised enzymes these suggest changes in the energy metabolism of A. crassus from different origins. Possible explanations could include a change to more or less aerobic processes in nematodes in Europe due to their bigger size and/or increased availability of nutrients. TUGs overexpressed in the female libraries showed elevated levels of dn/ds but genes overexpressed in males had decreased levels of dn/ds. The first finding is unexpected, as genes overexpressed in female libraries will also include TUGs related to larval development (such as the collagen modifying enzymes discussed above), and these larval transcripts in turn are expected to be under purifying selection because of pleiotropic effects of genes in early development [85]. The second contrasts with findings that male specific traits and transcripts often show hallmarks of positive selection [86, 87]. In A. caninum, female-specific transcripts showed an enrichment of parasitism genes” [79] and a possible explanation would be a similar enrichment of positively selected parasitism-related genes in our dataset. For males the decreased dn/ds may be explained by the high number of ribosomal protein-encoding TUGs, which all show very low levels of dn/ds. That these TUGs were found to be differentially expressed remains puzzling. Some male-overexpressed TUGs, such as that encoding major sperm protein, showed elevated dn/ds. It is unlikely that correlation of differential expression with positive selection results from mapping artifacts, as all the ribosomal protein encoding TUGs identified overexpressed in males have very low dn/ds.
Genes differentially expressed according to the geographic origin of the nematodes showed significantly elevated levels of dn/ds. We interpret this as reflecting a correlation between sequence evolution and phenotypic modification in different host, environments or correlation between sequence evolution and evolution of gene expression. Whether expression of these genes is modified in different hosts or evolved rapidly in the contemporary divergence between European and Asian populations of A. crassus, is one focus of ongoing work building on the reference transcriptome presented here. For such an analysis it will be important to disentangle the influence of the host and the nematode population in common garden, co-inoculation experiments.
Conclusions
The A. crassus transcriptome provides a basis for a new era of molecular research on this ecologically important species. It will aid not only analysis of the invasive biology of this parasite, assisting in identifying the origins of invading populations as well as the adaptations that may be selected in the new European host, but also in the investigation of the acquisition of parasitism in the great clade of animal parasites, Spirurina. In particular, positive selection of proteinases and differences in energy metabolism between European and Asian A. crassus constitute a candidate phenotype relevant for phenotypic modification or contemporary divergent evolution as well as for the long term evolution of parasitism.
References
Kuwahara A, Niimi H, Itagaki H: Studies on a nematode parasitic in the air bladder of the eel I. Descriptions of Anguillicola crassa sp. n. (Philometridea, Anguillicolidae). Jpn J Parasitol. 1974, 23 (5): 275-279.
Polzer M, Taraschewski H: Identification and characterization of the proteolytic enzymes in the developmental stages of the eel-pathogenic nematode Anguillicola crassus. Parasitol Res. 1993, 79: 24-27. [http://www.ncbi.nlm.nih.gov/pubmed/7682326]
De Charleroy D, Grisez L, Thomas K, Belpaire C, Ollevier F: The life cycle of Anguillicola crassus. Dis Aquat Org. 1990, 8 (2): 77-84.
Neumann W: Schwimblasenparasit Anguillicolabei Aalen. Fischer und Teichwirt. 1985, 11: 322-
Koops H, Hartmann F: Anguillicola-infestations in Germany and in German eel imports. J Appl Ichthyology. 1989, 5: 41-45. [http://onlinelibrary.wiley.com/doi/10.1111/j.1439-0426.1989.tb00568.x/abstract]
Koie M: Swimbladder nematodes (Anguillicola spp.) and gill monogeneans (Pseudodactylogyrus spp.) parasitic on the European eel (Anguilla anguilla). ICES J Mar Sci. 1991, 47 (3): 391-398. [http://icesjms.oxfordjournals.org/cgi/content/abstract/47/3/391]
Kirk RS: The impact of Anguillicola crassus on European eels. Fisheries Manage Ecol. 2003, 10 (6): 385-394. 10.1111/j.1365-2400.2003.00355.x.
Kristmundsson A, Helgason S: Parasite communities of ecls Anguilla anguilla in freshwater and marine habitats in Iceland in comparison with other parasite communities of eels in Europe. Folia Parasitologica. 2007, 54 (2): 141-
Taraschewski H: Hosts and parasites as aliens. J Helminthol. 2007, 80 (02): 99-128. [http://journals.cambridge.org/action/displayAbstract?fromPage=online%26aid=713884]
Mûnderle M, Taraschewski G, Klar B, Chang CW, Shiao JC, Shen KN, He JT, Lin SH, Tzeng WN: Occurrence of Anguillicola crassus (Nematoda: Dracunculoidea) in Japanese eels Anguilla japonica from a river and an aquaculture unit in SW Taiwan. Dis Aquat Org. 2006, 71 (2): 101-108. [http://www.ncbi.nlm.nih.gov/pubmed/16956057]
Lefebvre FS, Crivelli AJ: Anguillicolosis: dynamics of the infection over two decades. Diseases Aquatic Org. 2004, 62 (3): 227-232. [http://www.ncbi.nlm.nih.gov/pubmed/15672878]
Knopf K, Mahnke M: Differences in susceptibility of the European eel (Anguilla anguilla) and the Japanese eel (Anguilla japonica) to the swim-bladder nematode Anguillicola crassus. Parasitology. 2004, 129 (Pt 4): 491-496. [http://www.ncbi.nlm.nih.gov/pubmed/15521638]
Wûrtz J, Taraschewski H: Histopathological changes in the swimbladder wall of the European eel Anguilla anguilla due to infections with Anguillicola crassus. Dis Aquatic Org. 2000, 39 (2): 121-134. [http://www.ncbi.nlm.nih.gov/pubmed/10715817]
Knopf K: The swimbladder nematode Anguillicola crassus in the European eel Anguilla anguilla and the Japanese eel Anguilla japonica: differences in susceptibility and immunity between a recently colonized host and the original host. J Helminthol. 2006, 80 (2): 129-136. [http://www.ncbi.nlm.nih.gov/pubmed/16768856]
Knopf K, Lucius R: Vaccination of eels (Anguilla japonica and Anguilla anguilla) against Anguillicola crassus with irradiated L3. Parasitology. 2008, 135 (5): 633-640. [http://www.ncbi.nlm.nih.gov/pubmed/18302804]
Heitlinger E, Laetsch D, Weclawski U, Han YS, Taraschewski H: Massive encapsulation of larval Anguillicoloides crassus in the intestinal wall of Japanese eels. Parasites and Vectors. 2009, 2: 48-[http://www.parasitesandvectors.com/content/2/1/48]
Blaxter M, De Ley P, Garey JX, Liu L, Scheldeman P, Vierstraete A, Vanfleteren J, Mackey L, Dorris M, Frisse L, Vida J, Thomas W: A molecular evolutionary framework for the phylum Nematoda. Nature. 1998, 392 (6671): 71-75. 10.1038/32160.
Nadler SA, Carreno RA, Meja-Madrid H, Ullberg JC, Pagan C, Houston R, Hugot J: Molecular phylogeny of clade III nematodes reveals multiple origins of tissue parasitism. Parasitology. 2007, 134 (10): 1421-1442. [http://journals.cambridge.org/action/displayAbstract?fromPage=online%26aid=1279744]
Wijová M, Moravec F, Horák A, Lukes J: Evolutionary relationships of Spirurina (Nematoda: Chromadorea: Rhabditida) with special emphasis on dracunculoid nematodes inferred from SSU rRNA gene sequences. Int J Parasitol. 2006, 36 (9): 1067-1075. [http://www.ncbi.nlm.nih.gov/pubmed/16753171]
Laetsch DR, Heitlinger EG, Taraschewski H, Nadler SA, Blaxter M: The phylogenetics of Anguillicolidae (Nematoda: Anguillicolidea), swimbladder parasites of eels. BMC Evol Biol. 2012, 12 (60): [http://www.biomedcentral.com/1471-2148/12/60]
Wielgoss S, Taraschewski H, Meyer A, Wirth T: Population structure of the parasitic nematode Anguillicola crassus, an invader of declining North Atlantic eel stocks. Mol Ecol. 2008, 17 (15): 3478-3495. [http://www.ncbi.nlm.nih.gov/pubmed/18727770]
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM: Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005, 437: 376-380. 10.1038/nature03959.
Kumar S, Blaxter ML: Comparing de novo assemblers for 454 transcriptome data. BMC Genomics. 2010, 11: 571-10.1186/1471-2164-11-571.
Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Federhen S, Feolo M, Fingerman IM, Geer LY, Helmberg W, Kapustin Y, Krasnov S, Landsman D, Lipman DJ, Lu Z, Madden TL, Madej T, Maglott DR, Marchler-Bauer A, Miller V, Karsch-Mizrachi I, Ostell J, Panchenko A, Phan L, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Slotta D, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Wang Y, Wilbur WJ, Yaschenko E, Ye J: Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2012, 40: 13-25. [http://www.ncbi.nlm.nih.gov/pubmed/22140104]
Green P: PHRAP informally published online documentation. 1994, [http://www.phrap.org]
Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B, Tsai J, Quackenbush J: TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003, 19: 651-652. [http://www.ncbi.nlm.nih.gov/pubmed/12651724]
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410. [http://www.ncbi.nlm.nih.gov/pubmed/2231712]
Coppe A, Pujolar JM, Maes GE, Larsen PF, Hansen MM, Bernatchez L, Zane L, Bortoluzzi S: Sequencing, de novo annotation and analysis of the first Anguilla anguilla transcriptome: EeelBase opens new perspectives for the study of the critically endangered European eel. BMC Genomics. 2010, 11: 635-[http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3012609]
Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Muller WE, Wetter T, Suhai S: Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14: 1147-1159. [http://www.ncbi.nlm.nih.gov/pmc/articles/PMC419793]
Huang X, Madan A: CAP3: A DNA sequence assembly program. Genome Res. 1999, 9: 868-877. [http://www.ncbi.nlm.nih.gov/pmc/articles/PMC310812]
Parkinson J, Whitton C, Schmid R, Thomson M, Blaxter M: NEMBASE: a resource for parasitic nematode ESTs. Nucl Acids Res. 2004, 32 (suppl_1): D427-430. [http://nar.oxfordjournals.org/cgi/content/abstract/32/suppl_1/D427]
Elsworth B, Wasmuth J, Blaxter M: NEMBASE4: The nematode transcriptome resource. Int J Parasitol. 2011, 41: 881-894. [http://www.ncbi.nlm.nih.gov/pubmed/21550347]
Wasmuth J, Blaxter M: prot4EST: Translating Expressed Sequence Tags from neglected genomes. BMC Bioinformatics. 2004, 5: 187-[http://www.biomedcentral.com/1471-2105/5/187]
Bairoch A, Bougueleret L, Altairac S, Amendolia V, Auchincloss A, Argoud-Puy G, Axelsen K, Baratin D, Blatter MC, Boeckmann B, Bolleman J, Bollondi L, Boutet E, Quintaje SB, Breuza L, Bridge A, deCastro E, Ciapina L, Coral D, Coudert E, Cusin I, Delbard G, Dornevil D, Roggli PD, Duvaud S, Estreicher A, Famiglietti L, Feuermann M, Gehant S, Farriol-Mathis N, Ferro S, Gasteiger E, Gateau A, Gerritsen V, Gos A, Gruaz-Gumowski N, Hinz U, Hulo C, Hulo N, James J, Jimenez S, Jungo F, Junker V, Kappler T, Keller G, Lachaize C, Lane-Guermonprez L, Langendijk-Genevaux P, Lara V, Lemercier P, Le Saux, Lieberherr D, Lima TdeO, Mangold V, Martin X, Masson P, Michoud K, Moinat M, Morgat A, Mottaz A, Paesano S, Pedruzzi I, Phan I, Pilbout S, Pillet V, Poux S, Pozzato M, Redaschi N, Reynaud S, Rivoire C, Roechert B, Schneider M, Sigrist C, Sonesson K, Staehli S, Stutz A, Sundaram S, Tognolli M, Verbregue L, Veuthey AL, Yip L, Zuletta L, Apweiler R, Alam-Faruque Y, Antunes R, Barrell D, Binns D, Bower L, Browne P, Chan WM, Dimmer E, Eberhardt R, Fedotov A, Foulger R, Garavelli J, Golin R, Horne A, Huntley R, Jacobsen J, Kleen M, Kersey P, Laiho K, Leinonen R, Legge D, Lin Q, Magrane M, Martin MJ, O’Donovan C, Orchard S, O’Rourke J, Patient S, Pruess M, Sitnov A, Stanley E, Corbett M, di Martino G, Donnelly M, Luo J, van Rensburg P, Wu C, Arighi C, Arminski L, Barker W, Chen Y, Hu ZZ, Hua HK, Huang H, Mazumder R, McGarvey P, Natale DA, Nikolskaya A, Petrova N, Suzek BE, Vasudevan S, Vinayaka CR, Yeh LS, Zhang J: The Universal Protein Resource (UniProt) 2009. Nucleic Acids Res. 2009, 37: D169-D174. [http://www.ncbi.nlm.nih.gov/pubmed/18836194]
Iseli C, Jongeneel CV, Bucher P: ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 1999, 138-148. [http://www.ncbi.nlm.nih.gov/pubmed/10786296]
Ghedin E, Wang S, Spiro D, Caler E, Zhao Q, Crabtree J, Allen JE, Delcher AL, Guiliano DB, Miranda-Saavedra D, Angiuoli SV, Creasy T, Amedeo P, Haas B, El-Sayed NM, Wortman JR, Feldblyum T, Tallon L, Schatz M, Shumway M, Koo H, Salzberg SL, Schobel S, Pertea M, Pop M, White O, Barton GJ, Carlow CK, Crawford MJ, Daub J, Dimmic MW, Estes CF, Foster JM, Ganatra M, Gregory WF, Johnson NM, Jin J, Komuniecki R, Korf I, Kumar S, Laney S, Li BW, Li W, Lindblom TH, Lustigman S, Ma D, Maina CV, Martin DM, McCarter JP, McReynolds L, Mitreva M, Nutman TB, Parkinson J, Peregrin-Alvarez JM, Poole C, Ren Q, Saunders L, Sluder AE, Smith K, Stanke M, Unnasch TR, Ware J, Wei AD, Weil G, Williams DJ, Zhang Y, Williams SA, Fraser-Liggett C, Slatko B, Blaxter ML, Scott AL: Draft genome of the filarial nematode parasite Brugia malayi. Science. 2007, 317: 1756-1760. [Http://www.ncbi.nlm.nih.gov/pubmed/17885136]
Schmid RMB: annot8r: GO, EC and KEGG annotation of EST datasets. BMC Bioinformatics. 2008, 9: 180-[http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2324097]
Petersen TN, Brunak S, von Heijne G, Nielsen H: SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011, 8: 785-786. [http://www.ncbi.nlm.nih.gov/pubmed/21959131]
Zdobnov EM, Apweiler R: InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001, 17: 847-848. [http://www.ncbi.nlm.nih.gov/pubmed/11590104]
Kasprzyk A: BioMart: driving a paradigm change in biological data management. Database (Oxford). 2011, 2011: bar049-[http://www.ncbi.nlm.nih.gov/pubmed/22083790]
Durinck S, Spellman PT, Birney E, Huber W: Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009, 4: 1184-1191. [http://www.ncbi.nlm.nih.gov/pubmed/19617889]
Ning Z, Cox AJ, Mullikin JC: SSAHA: a fast search method for large DNA databases. Genome Res. 2001, 11: 1725-1729. [http://www.ncbi.nlm.nih.gov/pmc/articles/PMC311141]
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis GR, Durbin R: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009, 25 (16): 2078-2079. 10.1093/bioinformatics/btp352.
Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L: VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009, 25: 2283-2285. [http://www.ncbi.nlm.nih.gov/pubmed/19542151]
Chen H, Boutros PC: VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011, 12: 35-[http://www.ncbi.nlm.nih.gov/pubmed/21269502]
Falcon S, Gentleman R: Using GOstats to test gene lists for GO term association. Bioinformatics. 2007, 23: 257-258. [http://www.ncbi.nlm.nih.gov/pubmed/17098774]
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth G, TSherry ST, McVean GT, Durbin T, The 1000 Genomes Project: The variant call format and VCF tools. Bioinformatics. 2011, 27: 2156-2158. [http://www.ncbi.nlm.nih.gov/pubmed/21653522]
Alho JS, Valimaki K, Merila J: Rhh: an R extension for estimating multilocus heterozygosity and heterozygosity-heterozygosity correlation. Mol Ecol Resour. 2010, 10: 720-722. [http://www.ncbi.nlm.nih.gov/pubmed/21565077]
Amos W, Wilmer JW, Fullard K, Burg TM, Croxall JP, Bloch D, Coulson T: The influence of parental relatedness on reproductive success. Proc Biol Sci. 2001, 268: 2021-2027. [http://www.ncbi.nlm.nih.gov/pubmed/11571049]
Aparicio JM, Ortego J, Cordero PJ: What should we weigh to estimate heterozygosity, alleles or loci?. Mol Ecol. 2006, 15: 4659-4665. [http://www.ncbi.nlm.nih.gov/pubmed/17107491]
ColtMan W, G PJ, A SJ, ton J M P: Parasite-mediated selection against inbred Soay sheep in a free-living, island population. Evolution. 1999, 81: 1259-1267. [http://www.jstor.org/stable/2640828]
Morgan M, Pagès H: Rsamtools: Import aligned BAM file format sequences into R / Bioconductor. [http://bioconductor.org/packages/release/bioc/html/Rsamtools.html] [R package version 1.4.3]
Audic S, Claverie JM: The significance of digital gene expression profiles. Genome Res. 1997, 7: 986-995. [http://www.ncbi.nlm.nih.gov/pubmed/9331369]
Romualdi C, Bortoluzzi S, D’Alessi F, Danieli GA: IDEG6: a web tool for detection of differentially expressed genes in multiple tag sampling experiments. Physiol Genomics. 2003, 12: 159-162. [http://www.ncbi.nlm.nih.gov/pubmed/12429865]
Pages H, Carlson M, Falcon S, Li N: AnnotationDbi: Annotation Database Interface. [R package version 1.16.10]
Alexa A, Rahnenfuhrer J: topGO: topGO: Enrichment analysis for Gene Ontology. 2010, [R package version 2.6.0]
R Development Core Team: R: A Language and Environment for Statistical Computing. 2009, Vienna: R Foundation for Statistical Computing, [http://www.R-project.org]
Wickham H: ggplot2: Elegant Graphics for Data Analysis. 2009, New York: Springer, [http://had.co.nz/ggplot2/book]
Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J: Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003, 421 (6920): 231-237. [http://www.ncbi.nlm.nih.gov/pubmed/12529635]
Harris TW, Antoshechkin I, Bieri T, Blasiar D, Chan J, Chen WJ, De La Cruz N, Davis P, Duesbury M, Fang R, Fernandes J, Han M, Kishore R, Lee R, Muller HM, Nakamura C, Ozersky P, Petcherski A, Rangarajan A, Rogers A, Schindelman G, Schwarz EM, Tuli MA, Van Auken K, Wang D, Wang X, Williams G, Yook K, Durbin R, Stein LD, Spieth J, Sternberg PW: WormBase: a comprehensive resource for nematode research. Nucleic Acids Res. 2010, 38 (Database issue): D463-D467. [http://www.ncbi.nlm.nih.gov/pubmed/19910365]
Schwartz TS, Tae H, Yang Y, Mockaitis K, Van Hemert JL, Proulx SR, Choi JH, Bronikowski AM: A garter snake transcriptome: pyrosequencing, de novo assembly, and sex-specific differences. BMC Genomics. 2010, 11: 694-[http://www.ncbi.nlm.nih.gov/pubmed/21138572]
Hale MC, Jackson JR, Dewoody JA: Discovery and evaluation of candidate sex-determining genes and xenobiotics in the gonads of lake sturgeon (Acipenser fulvescens). Genetica. 2010, 138: 745-756. [http://www.ncbi.nlm.nih.gov/pubmed/20386959]
Papanicolaou A, Stierli R, Ffrench-Constant RH, Heckel DG: Next generation transcriptomes for next generation genomes using est2assembly. BMC Bioinformatics. 2009, 10: 447-[http://www.ncbi.nlm.nih.gov/pubmed/20034392]
Emmersen J, Rudd S, Mewes HW, Tetko IV: Separation of sequences from host-pathogen interface using triplet nucleotide frequencies. Fungal Genet Biol. 2007, 44: 231-241. 10.1016/j.fgb.2006.11.010.
Gregory R, Darby AC, Irving H, Coulibaly MB, Hughes M, Koekemoer LL, Coetzee M, Ranson H, Hemingway J, Hall N, Wondji CS: A de novo expression profiling of anopheles funestus, malaria vector in Africa, using 454 pyrosequencing. PLoS ONE. 2011, 6: e17418-[http://www.ncbi.nlm.nih.gov/pubmed/21364769]
Kunstner A, Wolf JB, Backstrom N, Whitney O, Balakrishnan CN, Day L, Edwards SV, Janes DE, Schlinger BA, Wilson RK, Jarvis ED, Warren WC, Ellegren H: Comparative genomics based on massive parallel transcriptome sequencing reveals patterns of substitution and selection across 10 bird species. Mol Ecol. 2010, 19 (Suppl 1): 266-276. [http://www.ncbi.nlm.nih.gov/pubmed/20331785]
The C elegans Sequencing Consortium: Genome sequence of the nematode C elegans: a platform for investigating biology. Science. 1998, 282: 2012-2018. [http://www.ncbi.nlm.nih.gov/pubmed/9851916]
Parkinson J, Mitreva M, Whitton C, Thomson M, Daub J, Martin J, Schmid R, Hall N, Barrell B, R H W, McCarter J, Blaxter M: A transcriptomic analysis of the phylum Nematoda. Nat Genet. 2004, 36 (12): 1259-1267. 10.1038/ng1472.
Wasmuth J, Schmid R, Hedley A, Blaxter M: On the Extent and Origins of Genic Novelty in the Phylum Nematoda. PLoS Neglected Tropical Diseases. 2008, 2 (7): e258-[http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2432500]
Parkinson J, Anthony A, Wasmuth J, Schmid R, Hedley A, Blaxter M: PartiGene–constructing partial genomes. Bioinformatics. 2004, 20 (9): 1398-1404. [http://bioinformatics.oxfordjournals.org/cgi/content/abstract/20/9/1398]
Harcus Y, Parkinson J, Fernandez C, Daub J, Selkirk M, Blaxter M, Maizels R: Signal sequence analysis of expressed sequence tags from the nematode Nippostrongylus brasiliensis and the evolution of secreted proteins in parasites. Genome Biology. 2004, 5 (6): R39-[http://genomebiology.com/2004/5/6/R39]
Balzer S, Malde K, Jonassen I: Systematic exploration of error sources in pyrosequencing flowgram data. Bioinformatics. 2011, 27: i304-309. [http://www.ncbi.nlm.nih.gov/pubmed/21685085]
Yang H, Chen X, Wong WH: Completely phased genome sequencing through chromosome sorting. Proc Natl Acad Sci USA. 2011, 108: 12-17. [http://www.ncbi.nlm.nih.gov/pubmed/21169219]
Adey A, Morrison H, Asan X, Xun X, Kitzman J, Turner E, Stackhouse B, MacKenzie A, Caruccio N, Zhang X, Shendure J: Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010, 11 (12): R119-[http://genomebiology.com/content/11/12/R119]
Kryazhimskiy S, Plotkin JB: The population genetics of dN/dS. PLoS Genet. 2008, 4: e1000304-[http://www.ncbi.nlm.nih.gov/pubmed/19081788]
Novaes E, Drost DR, Farmerie WG, Pappas GJ, Grattapaglia D, Sederoff RR, Kirst M: High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics. 2008, 9: 312-[http://www.ncbi.nlm.nih.gov/pubmed/18590545]
O’Neil ST, Dzurisin JD, Carmichael RD, Lobo NF, Emrich SJ, Hellmann JJ: Population-level transcriptome sequencing of nonmodel organisms Erynnis propertius and Papilio zelicaon. BMC Genomics. 2010, 11: 310-[http://www.ncbi.nlm.nih.gov/pubmed/20478048]
Swanson WJ, Wong A, Wolfner MF, Aquadro CF: Evolutionary expressed sequence tag analysis of Drosophila female reproductive tracts identifies genes subjected to positive selection. Genetics. 2004, 168: 1457-1465. [http://www.ncbi.nlm.nih.gov/pubmed/15579698]
Wang Z, Abubucker S, Martin J, Wilson RK, Hawdon J, Mitreva M: Characterizing Ancylostoma caninum transcriptome and exploring nematode parasitic adaptation. BMC Genomics. 2010, 11: 307-[http://www.ncbi.nlm.nih.gov/pubmed/20470405]
Knopf K, Madriles Helm A, Lucius R, Bleiss W, Taraschewski H: Migratory response of European eel (Anguilla anguilla) phagocytes to the eel swimbladder nematode Anguillicola crassus. Parasitol Res. 2008, 102 (6): 1311-1316. [http://www.ncbi.nlm.nih.gov/pubmed/18311570]
Molnár K: Formation of parasitic nodules in the swimbladder and intestinal walls of the eel Anguilla anguilla due to infections with larval stages of Anguillicola crassus. Dis Aquat Organ. 1994, 20 (3): 163-170.
Danecek Pandothers: The variant call format and VCFtools. Bioinformatics. 2011, 27: 2156-2158. [http://www.ncbi.nlm.nih.gov/pubmed/21653522]
Johnstone IL: Cuticle collagen genes. Expression in Caenorhabditis elegans. Trends Genet. 2000, 16: 21-27. [http://www.ncbi.nlm.nih.gov/pubmed/10637627]
Middleton B: The oxoacyl-coenzyme A thiolases of animal tissues. Biochem J. 1973, 132: 717-730. [http://www.ncbi.nlm.nih.gov/pubmed/4721607]
Cutter AD, Ward S: Sexual and temporal dynamics of molecular evolution in C. elegans development. Mol Biol Evol. 2005, 22: 178-188. [http://www.ncbi.nlm.nih.gov/pubmed/15371532]
Eberhard WG: Evolutionary conflicts of interest: are female sexual decisions different?. Am Nat. 2005, 165 (Suppl 5): 19-25. [http://www.ncbi.nlm.nih.gov/pubmed/15795858]
Swanson WJ, Clark AG, Waldrip-Dail HM, Wolfner MF, Aquadro CF: Evolutionary EST analysis identifies rapidly evolving male reproductive proteins in Drosophila. Proc Natl Acad Sci USA. 2001, 98: 7375-7379. [http://www.ncbi.nlm.nih.gov/pubmed/11404480]
Acknowledgements
This work has been made possible through a grant provided to EH by Volkswagen Foundation, “Förderinitiative Evolutionsbiologie”. The GenePool Genomics Facility is core funded by The School of Biological Sciences, University of Edinburgh, the Darwin Trust of Edinburgh, the UK Natural Environment Research Council (award reference R8/H10/56) and the UK Medical Research Council (award reference G0900740). We are grateful to Yun-San Han for his help collecting samples in Taiwan and to Karim Gharbi for overseeing the project within the GenePool. Timothée Cezard, Sujai Kumar and Graham Thomas gave essential analytic and informatic support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EH, HT and MB conceived and designed the experiments. EH carried out bioinformatic analyses. SB assisted in bioinformatic analyses. AM prepared sequencing libraries. HT provided close supervision throughout. EH and MB interpreted results and prepared the manuscript. All authors have read and approved the final manuscript.
Electronic supplementary material
12864_2012_7097_MOESM1_ESM.pdf
Additional file 1: Additional text. Supporting_information.pdf. This document contains the 3 additional figures referenced in the main text and an additional text describing the assembly process and evaluation of assembly quality in further detail. This text contains additional 7 figures (additional figures 4-10) and 3 tables. (PDF 307 KB)
12864_2012_7097_MOESM2_ESM.zip
Additional file 2: Additional data. A.crassus_transcriptome_contig_data.csv. All data computed on the contig level, as described in the manuscript and additional text including sequences (raw, coding, imputed, protein). (ZIP 16 MB)
12864_2012_7097_MOESM3_ESM.csv
Additional file 3: A.crassus_transcriptome_screened_SNPs.csv. High quality SNPs. The contig, the base relative to the start of the contig (base), the reference base-call (from), the alternative base-call (to), the number of reads supporting the reference (nfrom) and the alternative (nto), the percentage of the alternate allele (perc), whether the SNP is in the region of an ORF (inORF), the position in the Frame (inFrame) and the effect of the SNP (effect; synonymous, non-synonymous or nonsense) are given. (CSV 302 KB)
12864_2012_7097_MOESM4_ESM.pdf
Additional file 4: Additional figure 11 (a-i). Subgraphs of GO induced by the top 10 terms identified as enriched in different sets of genes. Subgraphs of the GO ontology categories induced by the top 10 terms identified as enriched in different sets of genes. Boxes indicate the 10 most significant terms. Box colour represents the relative significance, ranging from dark red (most significant) to light yellow (least significant). In each node the category-identifier, a (eventually truncated) description of the term, the significance for enrichment and the number of DE / total number of annotated gene is given. Black arrows indicate an “is-a” relationship. GO ontology category and the set of genes analysed for the enrichment are indicated in each figure. (PDF 337 KB)
12864_2012_7097_MOESM5_ESM.csv
Additional file 5: A.crassus_transcriptome_sexDE.csv. Contigs differentially expressed between male and female worms. Normalised counts and the natural logarithm of fold changes are given. (CSV 4 KB)
12864_2012_7097_MOESM6_ESM.csv
Additional file 6: A.crassus_transcriptome_originDE.csv. Contigs differentially expressed between European and Taiwanese worms. Normalised counts and the natural logarithm of fold changes are given. (CSV 3 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Heitlinger, E., Bridgett, S., Montazam, A. et al. The transcriptome of the invasive eel swimbladder nematode parasite Anguillicola crassus. BMC Genomics 14, 87 (2013). https://doi.org/10.1186/1471-2164-14-87
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-87