
Abstract
The flat periwinkles, Littorina fabalis and L. obtusata, comprise two sister gastropod species that have an enormous potential to elucidate the mechanisms involved in ecological speciation in the marine realm. However, the molecular resources currently available for these species are still scarce. In order to circumvent this limitation, we used RNA-seq data to characterize the transcriptome of four individuals from each species sampled in different locations across the Iberian Peninsula. Four de novo transcriptome assemblies were generated, as well as a pseudo-reference using the L. saxatilis reference transcriptome as backbone. After transcripts’ annotation, variant calling resulted in the identification of 19,072 to 45,340 putatively species-diagnostic SNPs. The discriminatory power of a subset of these SNPs was validated by implementing an independent genotyping assay to characterize reference populations, resulting in an accurate classification of individuals into each species and in the identification of hybrids between the two. These data comprise valuable genomic resources for a wide range of evolutionary and conservation studies in flat periwinkles and related taxa.
Measurement(s) | RNA • sequence_assembly • sequence feature annotation • SNP |
Technology Type(s) | RNA sequencing • sequence assembly process • sequence annotation • single-nucleotide polymorphism analysis |
Factor Type(s) | geographic location • species |
Sample Characteristic - Organism | Littorina fabalis • Littorina obtusata |
Sample Characteristic - Environment | marine biome |
Sample Characteristic - Location | Iberian Peninsula |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.11800293
Similar content being viewed by others


Background & Summary
Marine gastropods of the genus Littorina are among the most interesting models for studying adaptation and speciation1,2,3. For example, the recent genomic resources made available for the rough periwinkle (L. saxatilis) have provided important knowledge about the genomic architecture of adaptation and of parallel ecotype evolution4,5,6. Although less studied, the flat periwinkles, L. fabalis and L. obtusata, have recently gained recognition as a model system to inform us about the late stages of ecological speciation and how different reproductive barriers accumulate and interact across the speciation continuum7,8,9. These two sister-species usually dwell on brown algae (Fig. 1), presenting a largely overlapping distribution across the European Atlantic shores10. In some countries (e.g. Spain and United Kingdom), their different ecological requirements result in vertical zonation, with L. obtusata mainly inhabiting the mid to upper intertidal, whereas L. fabalis is more commonly found in the lower part of the shore11. Despite clear morphological and ecological differences, recent studies suggest that they are not fully reproductively isolated9,12, with a remarkable incidence of hybrids in Cabo do Mundo (Portugal)7,8. However, these studies were based on a relatively small number of markers, precluding a more detailed characterization of the type of hybrids. In order to circumvent these limitations, here we use RNA-seq to increase the genomic resources available for this system. We present the first transcriptome for flat periwinkles, identify putative fixed SNP differences between the two species and demonstrate the potential of this information to detect hybrids between them. These resources will facilitate additional evolutionary studies, including the role of adaptive introgression and reinforcement in the diversification of flat periwinkles, producer-herbivore interactions, as well as the identification of genomic regions involved in key traits that diverged between rough and flat periwinkles (e.g. adaptation to live on a rocky substrate versus macroalgae, respectively). Finally, together with the first transcriptome available within the genus Littorina (L. littorea), these data will also allow for comparative immunology studies and contribute to understanding the evolutionary dynamics of host/parasite immune conflicts13,14,15.

Flat periwinkles dwelling on Fucus spp. in the rocky intertidal of Galicia, Spain.
Methods
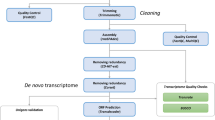
A description of the workflow of our approach is represented in a flowchart (Fig. 2).

Flowchart of our pipeline. Overview of the two main approaches employed to analyse the RNA-seq data. In orange the processes common to both pipelines.
Sampling locations and sample processing
Samples for transcriptome sequencing were collected from different locations in Northwestern Iberian Peninsula, from Abelleira (Galicia, Spain) in the North to Mindelo (Portugal) in the South, during 2014. Individuals collected in each sampling site (N = 12 to 27) were transported alive to the laboratory (ECIMAT, University of Vigo, Spain) within 24 hours after collection. Individuals were sexed under a dissection microscope. Because the two species can be distinguished based on male genitalia morphology10, only males were retained. These were placed in separate aquaria (one per site and species) with running marine water and aeration during 11 to 12 days for acclimation. The maintenance of the samples in similar environmental conditions before RNA extraction was performed to ensure that differences in expression between individuals from the two species were not specifically related to their condition at collection time. The only source of nutrients during this period was the running water, to minimize sequence contamination with DNA from other organisms present in their digestive tract. At the end of the acclimation period, samples were flash frozen in liquid nitrogen and kept at ‐80 °C until further analyses. Four males per species, each from a different location, were then selected for sequencing (Table 1; Fig. 3). Forty-eight additional samples were used to test the SNPs developed in this study. These consisted of 16 L. obtusata from Redondela and 16 L. fabalis from Canido used as reference populations, as well as 16 individuals from Cabo do Mundo, where hybrids have been detected before (Table 1; Fig. 3). In order to certify that individuals were accurately assigned to one of the species, only males were analysed in the reference populations. However, in Cabo do Mundo, where admixture was previously inferred based on microsatellites, two nuclear introns and mtDNA7,8,9, both males and females were included.

Map of the sampling locations. Samples were collected from the Northwestern Iberia (a). Zoom in of the sampling area is shown in (b). L. obtusata sampling sites are shown in blue, L. fabalis sampling sites are shown in orange, and the site where hybrids were previously described is marked with both colours. Circles represent sampling sites for transcriptome sequencing, while squares represent sampling sites for SNP genotyping/validation.
RNA extraction
For each snail, shell and hepatopancreas were first removed (Table 2), and total RNA was extracted from all remaining tissues using the TRIzol® reagent (Invitrogen) according to manufacturer’s instructions. Residual DNA was subsequently eliminated with the TURBO DNA-free kit (Ambion) according to the manufacturer as well. RNA was purified with the RNeasy MinElute Cleanup Kit (Qiagen).
Library preparation and sequencing
Starting with 1 μg of RNA from each sample, library building (with individual barcodes) was performed with the TruSeq RNA Sample Preparation Kit v2 (Illumina) following the manufacturer’s protocol and aiming for insert sizes of ~200 bp. The final libraries were pooled altogether in equimolar ratios before sequencing. Sequencing was performed in two different runs (using the same pool), each consisting of 4/5 of a lane in paired-end mode (first run 2 × 100 bp and second 2 × 125 bp) on an Illumina HiSeq 1500 in CIBIO/InBIO laboratories.
Quality control of sequence data
The quality of the raw reads16 was assessed with FastQC v0.11.517. Subsequently, reads were clipped to remove Illumina adapters and trimmed for quality with TRIMMOMATIC v0.3618 with the following steps: 1. Remove the adapters; 2. Cut the initial 10 bases at the start of the reads; 3. Trimming when average quality of the nucleotides within a 4 bp window was below 15; 4. Remove bases at the start and end of the reads if quality was below 20; and 5. Discard reads shorter than 25 bp. Read quality was re-evaluated with FastQC at the end of this process19.
Transcriptome assembly and annotation
In order to identify highly discriminant SNPs between L. fabalis and L. obtusata, reference transcriptomes were generated using two main approaches. The first one (Fig. 2) was based on the recently available reference transcriptome for L. saxatilis (deposited at DDBJ/ENA/GenBank under the accession GHPE00000000.1), a member of the sister clade of flat periwinkles closest relatives. It consisted of applying the Pseudo-it pipeline developed by Sarver et al.20 to obtain a pseudo-reference minimizing mapping biases towards any of the species. More specifically, this pipeline allows the generation of a pseudo-reference by replacing the original nucleotides in the reference (L. saxatilis) transcript sequences (e.g. A) by those that represent a fixed difference among the eight flat periwinkle individuals (e.g. T) or by using the IUPAC ambiguity code (e.g. R) when an alternative allele is fixed among the four L. fabalis (T) and the four L. obtusata (C) sequenced in this study. This was done through an iterative process with multiple steps, including: mapping reads to the reference using bwa mem v0.7.1521, removing duplicates with Picard v2.8.2 (http://broadinstitute.github.io/picard), identifying insertions and deletions following realignment and calling haplotypes with Genome Analysis Toolkit - GATK v3.722, and injecting the filtered variants in the original reference making use of the FastaAlternateReferenceMaker within GATK. In line with the authors’ recommendations, five iterations were implemented to guarantee a full incorporation of species variability. Further processing (filtering, indexing, merging and sorting) was done with SAMtools v1.3.123.
The second approach consisted of reconstructing species-specific de novo transcriptome assemblies. Four de novo assemblies were generated: two using the reads from the four individuals sequenced for each species (Fab4ind and Obt4ind), and the other two using only the reads from the individual with the highest coverage for each species (fSAM-G03R12 and oMIN-G09R07 for L. fabalis and L. obtusata, respectively). All these assemblies were performed using TRINITY v2.2.024 with default parameters, using the cleaned paired reads. The quality and completeness of the produced assemblies was evaluated with Transrate v1.0.325. This tool defines an optimal cut-off for each assembly, enabling the removal of possible chimeras and poorly supported contigs by mapping back the cleaned reads against the generated reference. Only the best-supported contigs (“good transcripts”) were retained for further analyses. In order to reduce redundancy, an extra step was performed with CD-HIT-EST v4.6.426 to keep only one transcript among those with more than 95% similarity. CD-HIT-EST was used after Transrate for all de novo transcriptomes, except for L. fabalis based on multiple individuals probably due to its high complexity. Finally, TransDecoder v3.0.027 was employed to predict candidate coding regions, discarding possible non-coding RNA and DNA contamination.
The final transcriptome comprises those transcripts with predicted open reading frames and with homologs in the Littorina saxatilis transcriptome reference plus those showing homology with PFAM common protein domains28. Raw and curated versions of each transcriptome are available in Figshare19. Annotation of transcripts was carried out using Conditional Reciprocal Best BLAST (crb-blast) v0.6.929 against the L. saxatilis reference transcriptome and the Swiss-Prot database30 and is available in Figshare19.
SNP calling
SNP calling was independently performed for L. fabalis and L. obtusata16. First, reads from all individuals were aligned to the pseudo-reference and to the respective species-specific de novo references with BWA-MEM v0.7.1521 using default parameters. Read group information was added to each sequencing lane-sample pair. SAMtools v1.3.123 was used for converting the resulting alignments (SAM format) into binary files (BAM format), sorting and fixing mate-pair information. Duplicate reads were removed with Picard v1.140 using MarkDuplicates. Read realignment was performed with RealignerTargetCreator and IndelRealigner tools within GATK v3.722. The pseudo-reference was chosen for downstream analyses based on mapping statistics (Table 3). However, SNPcall was also performed using the references obtained with the other assemblies19. Raw variant calling was carried out using bcftools mpileup and bcftools calls31, keeping both variant and non-variant sites and defining a maximum read coverage of 250. The resulting raw VCF files are available in Figshare19.
Genetic variation and population structure
Genetic variability across individuals and species was first assessed by a Principal Component Analysis (PCA) using PLINK v1.90b3.4532, which was plotted with the ggplot2 R package33. This analysis was based on a subset of SNPs that were called in all eight individuals, with a minimum coverage of 20 and a minimum minor allele count of 2 (i.e. singletons excluded to avoid errors) obtained with VCFtools34. Furthermore, only one random biallelic SNP per transcript was retained, reducing the effect of linkage disequilibrium. Population structure analysis was performed with fastStructure v1.035 based on the same SNP subset, with 10 cross-validation tests for each of three replicate runs, and considering the number of genetic clusters (K) between 2 and 8. The best number of clusters was inferred with the chooseK python script of fastStructure.
Putative fixed single nucleotide differences between species were identified using the python tools created by Simon Martin (https://github.com/simonhmartin/genomics_general), after filtering the raw VCF file for minimum SNP quality of 30 (Phred-scaled probability that the polymorphism exists), minimum coverage of 5 per sample and minimum number of calls of 8 (1 per individual). A more conservative set of putatively fixed SNP differences was also created by applying a more stringent filter of a minimum coverage of 20 per sample19.
Gene ontology enrichment
A Gene Ontology enrichment analysis was performed to assess if the annotated subset of transcripts/genes with putatively fixed differences between L. obtusata and L. fabalis were enriched for any functional category. This analysis was performed using the more conservative set of candidate fixed SNPs with the online tool gProfiler36. The background panel consisted of all annotated genes with at least one SNP. Over-represented GO categories were identified using a hypergeometric test with a significance threshold of p < 0.05 after Benjamini-Hochberg correction37.
Selection of informative SNPs for validation
A genotyping assay was designed with the goal of validating a subset of the identified SNPs and assessing their informativeness for species discrimination. Sequences of 101 bp with a fixed nucleotide difference in the central position were randomly selected from the list of annotated contigs/genes containing putative diagnostic SNPs (less conservative set). Sequences containing at least one polymorphism across 50 bp to each side of the target SNP were discarded to reduce the probability of missing genotypes or null alleles. Finally, to ensure that the 101 nucleotides were located in the same exon avoiding fragments that span introns, all the selected fragments were blasted against the L. saxatilis genome38 and only those with a single hit of 101 bp continuous alignment with a maximum of 1 bp difference were retained for developing the assay.
SNP genotyping and informativeness
A final set of 42 putatively diagnostic SNPs was tested with an independent genotyping assay at the National Genomics Infrastructure (NGI), SNP&SEQ Technology Platform (www.genotyping.se) at Uppsala University, Sweden. This consisted of using multiplexed primer extension (iPLEX) and a MassARRAY analyzer39 from Agena Bioscience for allele detection by mass spectrometry, and subsequent conversion to a genotype using Typer (Agena Bioscience). Forty-eight samples from the Iberian Peninsula were genotyped (Fig. 3, Table 1). Hardy-Weinberg equilibrium (HWE) was estimated for each locus and reference population combination. Since there was no variability for most SNPs within the reference populations, linkage disequilibrium (LD) between locus pairs was evaluated across all populations. Both HWE and LD (genotypic disequilibrium) were evaluated through exact probability tests in Genepop v4.240,41, using a Markov chain with default parameters values. A Bonferroni correction for multiple tests was applied. After removing markers in LD or not conforming to HWE, the ancestry of each individual (L. fabalis, L. obtusata, or admixed) was inferred using STRUCTURE v2.3.442,43,44, running 10 replicates for K = 2, each consisting of 1,000,000 iterations after a 100,000 burn-in. Since all replicates resulted in a similar outcome, the one with the highest likelihood was plotted with DISTRUCT v1.145.
Data Records
The core dataset of this work comprises the transcriptomic data of flat periwinkles. The raw data (eight SRA experiments) were deposited in NCBI Sequence Read Archive, with the BioProject accession number PRJNA55698416 (Table 1). FASTQ files were divided by species (L. obtusata - Lobt and L. fabalis - Lfab) and biosample-specimen (oMOI, oCAN, oBAI, oMIN, fSAM, fABE, fTIR and fMUR). For each biosample, four files were submitted, corresponding to two different Illumina HiSeq lanes and one for each of the paired reads. All intermediate and final files produced during the previous steps are available on Figshare19, including: pre- and post-cleaning FASTQC quality reports (FastQC_quality_reports_raw and FastQC_quality_reports_clean, the pseudo reference transcriptome (LfabLobt_5it.pseudoref.FINAL.fa), the raw trinity assembly results (Lobt4ind_rawAssembly.fasta, Lfab4ind_rawAssembly.fasta, Lobt_oMIN_ G09R07_rawAssembly.fasta and Lfab_fSAM_G03R012_rawAssembly.fasta), curated versions of the transcriptome assemblies (Lobt4ind_curatedAssembly.fasta, Lfab4ind_curatedAssembly.fasta, Lobt_oMIN_G09R07_curatedAssembly.fasta and Lfab_fSAM_G03R012_curatedAssembly.fasta), annotated pseudoreference transcriptome based on Swiss-Prot and the L. saxatilis transcriptome (LfabLobt_5it.pseudoref.annotated.fa) and combined crb-blast annotation file based on Swiss-Prot and the L. saxatilis transcriptome (Annotation_PseudoRef_SP_and_LsaxTranscriptome.txt), transcriptomes crb-blast annotation files based on Swiss-Prot and the L. saxatilis transcriptome, separately (Annotation.7z), raw SNP call VCF files (SNPcall_rawPseudoRef.vcf.gz, SNPcall_rawLobt4ind.vcf, SNPcall_rawLfab4ind.vcf.gz, SNPcall_rawLob_oMIN_ G09R07.vcf.gz, SNPcall_rawLfab_fSAm_G03R012.vcf.gz), filtered VCF file (SNPcall_Biallelic_NoMissing_PseudoRef_min20.vcf), list of putatively diagnostic SNPs (FixedSNPs_PseudoRef_min5.vcf and FixedSNPs_PseudoRef_min20.vcf), and genotypes of the samples used for SNP validation analysed with Structure (SNPvalidation_genotypes_for_Structure_Analyses.xlsx). The L. saxatilis transcriptome and genome sequence data used as reference in this study are publically available at GenBank (GHPE00000000.1)46.
Technical Validation
RNA integrity
RNA quality was evaluated after electrophoresis on the Bioanalyser 2,100 (Agilent Technologies) with the RNA 6000 Nano Chip Kit (Agilent Technologies). RNA quantity was measured with Qubit using the RNA BR Assay Kit (Life Technologies).
RNA-Seq data quality
A total of 736 million reads were generated (368,266,256 paired-end reads), with an average of ~92,000 reads per sample (Table 3). From these, ~705 million reads (96%) were kept after removing the adapters and trimming for quality. These post-cleaning reads passed the minimum quality standards of FastQC17.
Pseudo-reference information
The pseudo-reference transcriptome is composed of 37,873 contigs (containing a total of 82,360,655 bp) with a mean contig length and N50 of 2,139 bp and 2,963 bp, respectively (Table 4). Annotation using a conditional reciprocal best blast hit approach against the Swiss-Prot database coding sequences resulted in 10,968 annotated transcripts (29%), while 91% of the transcripts had 1to1 orthologs in the L. saxatilis reference transcriptome.
Species-specific transcriptome assembly and annotation
The L. fabalis assembly based on the “cleaned” reads from 4 individuals of this species initially resulted in 396,047 contigs with a mean length of 612 bp, a N50 of 832 bp and a total size of 242,362,528 bp; whereas the L. obtusata transcriptome is 251,985,325 bp long, distributed across 349,459 contigs with a mean length of 721 bp and N50 of 1,133 bp (Table 5). The assemblies based on a single individual from each species resulted in lower values for all parameters when compared to those based in all four individuals: 177,022 contigs with N50 of 715 bp for L. fabalis; and 208,362 contigs with N50 of 1,159 bp for L. obtusata (Table 5).
Filtering of these contigs based on CD-HIT-EST and TransRate optimal assembly score (0.08 for L. obtusata four individuals, 0.19 for L. fabalis four individuals, 0.12 one individual L. obtusata and 0.09 one individual L. fabalis), resulted in the retention of 48.5% (L. obtusata one individual) to 75.3% (L. fabalis one individual) of the initial number of contigs. Final curated flat periwinkle assemblies based on four individuals are composed of 53,214 and 32,433 predicted open reading frame (ORF) transcripts with mean lengths of 818 bp and 707 bp, and a N50 length of 1,080 and 861, for L. fabalis and L. obtusata respectively. The single individual assemblies are composed of 31,279 (fSAM) and 24,047 (oMIN) ORF, with mean lengths of 685 bp and 740 bp, and a N50 length of 801 and 912, respectively (Table 5).
Regarding the annotation of the assemblies based on data from four individuals, a total of 38,439 transcripts in the L. fabalis and 23,237 in L. obtusata were annotated (72% in both species), of which 10,592 and 6,275 were uniquely annotated to the Swiss-Prot database, while 20,246 and 15,690 had homologs in the L. saxatilis reference transcriptome. The annotation of the assemblies based on a single individual resulted in 23,947 annotated transcripts for L. fabalis (fSAM) and 19,228 for L. obtusata (oMIN)19.
Mapping, SNP calling, genetic differentiation and gene ontology enrichment
In total, 253,969,198 reads (mean = 36%) were mapped to the pseudo-reference transcriptome (Table 3). The relatively small number of mapped reads can be due to the large number of transcripts filtered out during the assembly, and also possibly due to exogenous DNA. After filtering, a total of 90,826 SNPs, present in all samples, were inferred (SNPcall_Biallelic_NoMissing_PseudoRef_min20.vcf). Principal Component Analysis was based on the filtered dataset, from which 7,061 SNPs were randomly sampled from independent transcripts. The first component of the PCA explains 64% of the total variance and separates the individuals from different species, while the second component, explains 8% of the variance and essentially separates the L. fabalis individuals according to geographical distance: two from Ria de Vigo (fSAM-G03R12 and fTIR-G10R14) and two from Ria de Muros (fABE-G04R13 and fMUR-G11R15) (Fig. 4). The Bayesian assignment analysis with fastStructure, with K = 2 as the number of clusters that maximized the marginal likelihood and better explained the structure in the data, confirms the classification of the four individuals from each species, without any evidence of admixture (Fig. 5).

Principal Component Analysis (PCA) for the eight flat periwinkle samples sequenced for the transcriptome based on a total of 7,061 SNPs randomly sampled from independent transcripts.

Admixture plot showing the membership of each individual sequenced for the transcriptome to the two genetic clusters. Sample codes are the same as in Table 1. No signatures of admixture were found in these individuals. The analysis was based on 7,061 random SNPs from different transcripts.
Putative species-diagnostic SNPs were identified from the raw SNP call using the python tools created by Simon Martin and two different filtering criteria. In the more stringent filter (minimum coverage of 20), 19,072 SNPs were inferred as putatively species-diagnostic (i.e. alternative alleles fixed within the group of four individuals from each species) whereas in the less stringent filter (minimum coverage of 5), 45,340 were identified as putatively species-diagnostic SNPs (FixedSNPs_PseudoRef_min5.vcf and FixedSNPs_PseudoRef_min20.vcf, respectively19).
Among the genes with fixed differences between species no enrichment for a particular GO term was found. However, since RNA was extracted basically from the whole individual (not from individual tissues) and many of the transcripts may not be complete, this result should be interpreted with caution.
SNP genotyping validation and informativeness
From the 42 putatively diagnostic SNPs selected for validation, two failed the quality control tests for primer design. Among the remaining SNPs, one failed during genotyping, one revealed no variability and three others revealed more than two alleles. All these SNPS were excluded resulting in a final set of 35 SNPs. All loci were genotyped for at least 91.7% of the individuals, whereas all individuals were genotyped for at least 94.3% of the loci. No conflicts were found among genotyping replicates (215), resulting in 100% reproducibility. Among these 35 loci, 26 (74.3%) SNPs are potentially diagnostic between L. fabalis and L. obtusata, i.e. fixed differences between the putatively pure populations. The non-diagnostic set consisted of two (5.7%) nearly diagnostic SNPs (one allele with a frequency ≥0.95 in one species and 0 in the other), five (14.2%) informative SNPs (one allele with a frequency ≥0.70 in one species and 0 to 0.03 in the other), and two others (5.7%) with very little or no discriminatory power between the two species defined morphologically according to male genitalia.
No significant Hardy-Weinberg deviations for any of the 35 loci were detected in the reference samples. However, LD remained significant between eight pairs of loci across the three populations. This resulted in the exclusion of six additional SNPs to avoid using non-independent information in subsequent analysis of ancestry. The STRUCTURE analysis for K = 2 based on these 29 SNPs confirmed the pure genetic ancestry of the reference L. fabalis and L. obtusata individuals and the occurrence of admixture in Cabo do Mundo, demonstrating the usefulness of these markers for species discrimination and detection of hybridization between flat periwinkles (Fig. 6).

Structure plot showing the membership of each individual genotyped for a subset of putatively diagnostic SNPs to each genetic cluster. Each column represents one individual. Signatures of admixture were only found in Cabo do Mundo, whereas individuals from Redondela (L. obtusata) and Canido (L. fabalis) represent two different genetic clusters.
Usage Notes
The two species-specific transcriptomes and the SNPs made available in this study are the first genomic/transcriptomic resources made publicly available for flat periwinkles. This information will be useful for a wide range of studies, including evolutionarily-oriented projects concerning adaptation, parallel evolution, comparative immunology, producer-herbivore interactions, hybridization and speciation; as well as for characterization of population genetic diversity and differentiation across the species’ distribution ranges to help implementing appropriate management and conservation measures. However, it is important to emphasise that the discriminatory power of the SNPs here developed was based on individuals from the Iberian Peninsula. Although we expect that most of these SNPs will also be diagnostic in other regions, their power to discriminate between L. fabalis and L. obtusata should first be tested in other parts of the species distribution range before they are applied outside Iberia.
Code availability
1. FASTQC v0.11.517, options: default
2. Trimmomatic v0.3618, options: PE ILLUMINACLIP:Adapters.fa:2:30:10 HEADCROP:10 SLIDINGWINDOW:4:15 LEADING:20 TRAILING:20 MINLEN:25
3. Pseudo-it v120, options: –PE1 R1.fastq.gz –PE2 R2.fastq.gz –iupac 5 REF NAME
4. Trinity v2.2.024, default
5. Transrate v1.0.325, options: –left –right –reference
6. CD-HIT-EST v4.6.426, options: -c 0.95 -M 0
7. TransDecoder.LongOrfs v3.0.027, default
8. hmmscan v v3.1b247, options: default
9. TransDecoder.Predict v3.0.027, options: –retain_pfam_hits–retain_blastp_hits –single_best_only
10. crb-blast v0.6.929, options: -e 1.0e-05
11. BWA v0.7.1521, options: mem -M -R ‘@RG\tID:\tLB:\tPL:\tSM:’
12. Samtools v1.3.123, options: sort; fixmate, index
13. Picard v1.140 (“Picard Toolkit.” 2019. Broad Institute, GitHub Repository: http://broadinstitute.github.io/picard/), options: MarkDuplicates REMOVE_DUPLICATES = true ASSUME_SORTED = true
14. GATK v3.722, options: RealignerTargetCreator; IndelRealigner
15. BCFtools v1.631, options: mpileup -f REF.fa -Ou -a DP -b list|bcftools call -m -f GQ,GP
16. Plink v1.90b3.4532, options: –bp-space 10000 –pca
17. VCFtools v. 0.1.1434, vcftools –gzvcf target.vcf.gz–max-missing 1.0 –min-alleles 2 –max-alleles 2 –mac 2 –remove-indels–recode–stdout|bgzip -c > Filtered.biallelic.vcf.gz
18. fastStructure v1.035, options: –full –cv = 10
19. Simon Martin’s scripts (Simon Martin, GitHub Repository: https://github.com/simonhmartin/genomics_general accessed on July 2018), options: parseVCF.py –skipIndels –minQual 30 –gtf flag = DP min = 20 (or 5); filterGenotypes.py –fixedDiffs–minCalls 8
20. Genepop v4.240,41, options: 1. Hardy Weinberg Exact Tests and 2. Linkage disequilibrium, default
21. STRUCTURE v2.3.442,43,44, options: burnin = 100,000, numreps = 1,000,000, usepopinfo = 0; inferalpha = 1; maxpops = 2.
References
Johannesson, K. Evolution in Littorina: Ecology matters. J. Sea Res. 49, 107–117 (2003).
Rolán-Alvarez, E., Austin, C. J. & Boulding, E. G. The contribution of the genus littorina to the field of evolutionary ecology. Oceanograf. Mar. Biol. 53, 157–214 (2015).
Ravinet, M. Notes from a snail island: Littorinid evolution and adaptation. Mol. Ecol. 27, 2781–2789 (2018).
Westram, A. M. et al. Clines on the seashore: The genomic architecture underlying rapid divergence in the face of gene flow. Evol. Lett. 2, 297–309 (2018).
Faria, R. et al. Multiple chromosomal rearrangements in a hybrid zone between Littorina saxatilis ecotypes. Mol. Ecol. 18, 1375–1393 (2018).
Morales, H. E. et al. Genomic architecture of parallel ecological divergence: beyond a single environmental contrast. Sci. Adv. 5, eaav9963 (2019).
Carvalho, J., Sotelo, G., Galindo, J. & Faria, R. Genetic characterization of flat periwinkles (Littorinidae) from the Iberian Peninsula reveals interspecific hybridization and different degrees of differentiation. Biol. J. Linn. Soc. 118, 503–529 (2016).
Costa, D. et al. Hybridization patterns between two marine snails, Littorina fabalis and L. obtusata. Ecol. Evol. 10, 1158–1179 (2020).
Sotelo, G. et al. Phylogeographic history of flat periwinkles, Littorina fabalis and L. obtusata. BMC Evol. Biol. 30, https://doi.org/10.1186/s12862-019-1561-6 (2020).
Reid, D. G. Systematics and Evolution of Littorina (London: The Ray Society, 1996).
Williams, G. The comparative ecology of the flat periwinkles, Littorina obtusata (L.) and L. mariae Sacchi et Rastelli. Field Stud. 7, 469–482 (2012).
Kemppainen, P., Panova, M., Hollander, J. & Johannesson, K. Complete lack of mitochondrial divergence between two species of NE Atlantic marine intertidal gastropods. J. Evol. Biol. 22, 2000–2011 (2009).
Gorbushin, A. M. & Borisova, E. A. Lectin-like molecules in transcriptome of Littorina littorea hemocytes. Dev. Comp. Immunol. 48, 210–220 (2015).
Gorbushin, A. M. Immune repertoire in the transcriptome of Littorina littorea reveals new trends in lophotrochozoan proto-complement evolution. Dev. Comp. Immunol. 84, 250–263 (2018).
Gorbushin, A. M. Immune response of a caenogastropod host: A case study of Littorina littorea and its digenean parasites. Dev. Comp. Immunol. 101, 103465 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP216581 (2019).
Andrews, S. FastQC: a quality control tool for high throughput sequence data, http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Marques, J. P. et al. Transcriptomic resources for evolutionary studies in flat periwinkles and related species: catching the tide. figshare. https://doi.org/10.6084/m9.figshare.c.4621634.v3 (2020).
Sarver, B. A. J. et al. Phylogenomic Insights into Mouse Evolution Using a Pseudoreference Approach. Genome Biol. Evol. 9, 726–739 (2017).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at ArXiv: 1303, 3997v2 (2013).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Smith-Unna, R., Boursnell, C., Patro, R., Hibberd, J. & Kelly, S. TransRate: reference free quality assessment of de novo transcriptome assemblies. Genome Res. 26, 1134–1144 (2016).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat. Protoc. 8, 1494–1512 (2013).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Aubry, S., Kelly, S., Kümpers, B. M. C., Smith-Unna, R. D. & Hibberd, J. M. Deep Evolutionary Comparison of Gene Expression Identifies Parallel Recruitment of Trans-Factors in Two Independent Origins of C4 Photosynthesis. Plos Genet. 10, e1004365 (2014).
The UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 (2019).
Danecek, P., Schiffels, S. & Durbin, R. Multiallelic calling model in bcftools (-m). samtools.github.io/bcftools/call-m.pdf.
Purcell, S. et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York, 2009.
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Raj, A., Stephens, M. & Pritchard, J. K. fastSTRUCTURE: Variational Inference of Population Structure in Large SNP Data Sets. Genetics 197, 573–589 (2014).
Reimand, J. et al. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 44, W83–W89 (2016).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple. Testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300 (1995).
Westram, A. M. et al. Data from: Clines on the seashore: the genomic architecture underlying rapid divergence in the face of gene flow. Dryad. https://doi.org/10.5061/dryad.bp25b65 (2018).
Gabriel, S., Ziaugra, L. & Tabbaa, D. SNP genotyping using the sequenom massARRAY iPLEX Platform. Curr. Protoc. Hum. Genet. 60, 2.12.1–2.12.18 (2009).
Raymond, M. & Rousset, F. GENEPOP (Version 1.2): Population Genetics Software for Exact Tests and Ecumenicism. J. Hered. 68, 248–249 (1995).
Rousset, F. GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 8, 103–106 (2008).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 7, 574–578 (2007).
Hubisz, M. J., Falush, D., Stephens, M. & Pritchard, J. K. Inferring weak populati on structure with the assistance of sample group information. Mol. Ecol. Resour. 9, 1322–1332 (2009).
Rosenberg, N. A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138 (2004).
Chaube, P., Larsson, T., Panova, M., Johannesson, K. & Butlin, R. TSA: Littorina saxatilis, transcriptome shotgun assembly. GenBank https://identifiers.org/ncbi/insdc:GHPE00000000.1 (2019).
Wheeler, T. J. & Eddy, S. R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489 (2013).
Acknowledgements
This study was supported by: European Regional Development Fund (FCOMP-01-0124-FEDER-014272), FCT – Foundation for Science and Technology (PTDC/BIA-EVF/113805/2009), and for JH the Swedish Research Council (2015-05001). RF was financed by FCT (SFRH/BPD/89313/2012) by the European Union’s Horizon 2020 research and innovation programme, under the Marie Sklodowska-Curie grant agreement number 706376; GS was financed by Volkswagen Stiftung (50500776); and JPM was financed by a PhD fellowship from FCT (SFRH/BD/115089/2016). RKB was financed by the European Research Council. We would like to thank Isabel Pintor for the flat periwinkles’ photo and Peter Stadler and Jens Steuck for access to a server at the Bioinformatics Group of the Department of Computer Science, University of Leipzig. Finally, we are extremely thankful to Tomas Larsson and Kerstin Johannesson, for performing the L. saxatilis genome assembly and giving access to it, respectively.
Author information
Authors and Affiliations
Contributions
J.P.M., G.S., K.N. and R.F. conceived and designed the study. D.C. and G.S. collected the samples. J.G. performed the RNA extractions. G.S. and S.A. built the libraries and prepared the samples for sequencing. J.P.M. set up the bioinformatics pipeline. P.C. performed the L. saxatilis transcriptome assembly together with R.B., whereas M.P. led the assembly of the L. saxatilis reference genome. J.P.M. and R.F. analysed the data. J.H. contributed with SNP data. J.P.M., G.S. and R.F. wrote the manuscript with the contribution of R.B. All authors contributed to the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Marques, J.P., Sotelo, G., Galindo, J. et al. Transcriptomic resources for evolutionary studies in flat periwinkles and related species. Sci Data 7, 73 (2020). https://doi.org/10.1038/s41597-020-0408-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-0408-8
- Springer Nature Limited
This article is cited by
-
Neuronal gene expression in two generations of the marine parasitic worm, Cryptocotyle lingua
Communications Biology (2023)