
Abstract
Background
Obesity, excess fat tissue in the body, can underlie a variety of medical complaints including heart disease, stroke and cancer. The pig is an excellent model organism for the study of various human disorders, including obesity, as well as being the foremost agricultural species. In order to identify genetic variants associated with fatness, we used a selective genomic approach sampling DNA from animals at the extreme ends of the fat and lean spectrum using estimated breeding values derived from a total population size of over 70,000 animals. DNA from 3 breeds (Sire Line Large White, Duroc and a white Pietrain composite line (Titan)) was used to interrogate the Illumina Porcine SNP60 Genotyping Beadchip in order to identify significant associations in terms of single nucleotide polymorphisms (SNPs) and copy number variants (CNVs).
Results
By sampling animals at each end of the fat/lean EBV (estimate breeding value) spectrum the whole population could be assessed using less than 300 animals, without losing statistical power. Indeed, several significant SNPs (at the 5% genome wide significance level) were discovered, 4 of these linked to genes with ontologies that had previously been correlated with fatness (NTS, FABP6, SST and NR3C2). Quantitative analysis of the data identified putative CNV regions containing genes whose ontology suggested fatness related functions (MCHR1, PPARα, SLC5A1 and SLC5A4).
Conclusions
Selective genotyping of EBVs at either end of the phenotypic spectrum proved to be a cost effective means of identifying SNPs and CNVs associated with fatness and with estimated major effects in a large population of animals.
Similar content being viewed by others

Background
The study of the process through which pigs convert food comparatively into fat and lean tissue (i.e. the control of fatness) has many potential applications and implications. From an agricultural perspective, pork is the primary source of meat protein worldwide (43%) and global consumption has doubled over the last ten years (World Health Organisation, 2012). The increasing global population and the constant increase in meat consumption in developing countries, especially East and Southeast Asia, bring an unprecedented challenge to the pig breeding industry who are charged, in part, with feeding this growing number of people [1]. Furthermore, market forces demand the tailoring of meats to specific populations and cultures. Pig breeding companies constantly focus their efforts on either producing lean or fat animals or specifically targeted traits e.g. intramuscular fat in their products [2]. While fatness is inexorably linked to diet, genetic factors undoubtedly have an influence and marker assisted selection regimes, aimed at increasing or decreasing growth of fat or lean tissues selectively, require further sophistication. Pork producers aim to improve genetically advanced breeding stock by improving both the food conversion ratio (FCR) and the residual feed intake of each slaughter pig, as well as producing higher yielding carcasses with significant improvement in lean meat percentage [3]. Such sophistication could be advanced further through a deeper understanding of the genetic control of fatness.
The pig is generally considered an excellent model organism for the study of many aspects of human physiology and disease states including cancer, diabetes, [4] maternal aggression [5] and obesity [6–8]. Obesity, excess fat accumulating in the tissues [9], can lead to a variety of illnesses including coronary heart disease, stroke and cancer [10]. Study of the role of genetic variation in the fatness of pigs therefore can have biomedical implications for the understanding and control of one of the biggest killers in the Western world, through the identification of orthologous genes and their variants.
It addition to chromosomal level genomic changes, normal genetic variation at the sequence level includes insertions and deletions (indels) [11], single nucleotide polymorphisms (SNPs) and copy number variants (CNVs). SNPs are the most frequent genetic variation between humans [12]. SNPs are usually biallellic and the least abundant allele (or minor allele frequency (MAF)) usually occurs at in at least 1% of the population [13]. SNPs are commonly found in non-coding regions, however generally do not reside within genes [14]. SNPs act as markers linked to phenotypically relevant loci and they therefore function as powerful tools in non-hypothesis driven research [15]. Certain SNPs have been causally linked with monogenic traits (1 gene/1 trait). Examples of such traits include achondroplasia [16] and sickle cell anaemia [17] as well as variants located near the MC4R gene, which are known to influence fat mass, weight and risk of obesity, with mutations of this gene being a cause of monogenic severe childhood onset diabetes). It is the association of SNPs with polygenic and multifactorial traits however, that has received the most attention in recent years. That is, post-genomic technologies such as SNP microarrays have permitted genome wide association studies (GWAS) for thousands of traits in humans. Such studies include research into complex diseases in humans such as breast cancer [18], type 2 diabetes (T2D) [19, 20], Crohn’s disease [21, 22], Parkinson’s disease [23], coeliac disease [24] and obesity. Some examples of obesity GWAS in humans include research into early onset extreme obesity. One particular study provided proof of principle for the concept that GWAS are useful in detecting genes relevant to complex phenotypes. In this case a human SNP array comprising of 440,794 SNPs from 487 extremely obese young German samples and 442 healthy lean German controls. Fifteen significant SNPs were determined, 6 of which were associated with the FTO gene, suggesting that it strongly contributes to early onset obesity [25]. To date however, the studies of SNPs associated with fatness in pigs have been limited in comparison to the number of human studies [26–28]. Indeed, there are few GWAS in the pig in relation to phenotypic traits. Popularly studied traits in the pig, apart from fatness include boar taint [29–31], body composition and structural soundness [32, 33], however such studies are also still in their infancy; this perhaps reflects the fact that a SNP microarray (chip) for the pig has only been developed within the last 4 years. Although these studies have provided much insight into the molecular biology of many traits, more are necessary in order to fill gaps in our knowledge of fatness, as it is a multifactorial, complex trait. The agricultural sector stands to benefit financially from such research and there are potentially biomedical applications if orthologous human genes can be associated with obesity, especially if GWAS can be performed in a low-cost manner by means such as use of selective genotyping. Selective genotyping, i.e. use of individuals at the extreme ends of a phenotypic spectrum provides an effective means of performing GWAS on a large population by sampling small numbers of animals and has its theoretical basis in the early 1990s [34]. Most recently applied to identify SNPs associated with back fat thickness in pigs used for Italian dried ham [35].
CNV regions (CNVRs) are segments of DNA (ranging from 1 kilobase (kb) to several megabases (Mb) in size [36]) that vary in copy-number by comparison with reference genomes [36]. It is thought that 12% of the human genome contains CNVRs [37] that are heritable in normal individuals. Around 0.4% of genome content in people who are unrelated is thought to vary in copy number [38]. Specific algorithms for CNV detection from SNP chip raw data include ‘PennCNV’, ‘GADA’ [39], ‘CONAN’ [40], ‘cnvPartition’ and ‘QuantiSNP’, the last 2 of which have been used in this study. Comparative analyses of the above have suggested a preference for QuantiSNP [41]. The study of CNVs has been pioneered in humans and catalogues of human CNVs are now available [42–44]. Significant associations of CNVs with human disease are numerous as CNVs are thought to be able to influence gene expression and may also play a role in affecting metabolic traits. These include susceptibility to HIV1 [45], a role in auto immune disease and lupus glomerulonephritis [46]. CNVs are strongly correlated with gene, repeat and segmental duplication content [47] and play a significant role in the development of complex traits. A recent study showed that 19 CNVs are significantly associated with the mechanisms for the control of a number of metabolic traits in mice. Indeed mouse chromosomes 1, 4 and 17 all have CNVRs in regulatory regions influencing body weight [48]. Wang et al. [49] used PennCNV on SNP chip data in humans to study obese individuals with ‘never overweight’ controls. The authors determined that in a study of over 800 individuals, large CNVs and rare deletions were associated with the risk of moderate obesity [49]. Moreover, CNVs contained candidate obesity genes including a 3.3 Mb deletion disrupting NAP1L5 (nucleosome assembly protein 1-like 5) as well as a 2.1 Mb deletion disrupting UCP1 (uncoupling protein 1) and IL15 (interleukin 15). Such studies for the determination of CNVRs in pigs however are, to date, limited to only 6. The first focussed on chromosomes 4, 7, 14 and 17, in which 37 CNVRs were identified [50], with the second identifying 49 CNVRs genome wide [51]. The third, published in 2012, used a sample size of 474 pigs, across 3 pure-bred lines, Yorkshire, Landrace and Songliao Black as well as a cross-bred line, Duroc-Erualian. PennCNV was the chosen platform, with 382 CNVs identified, genome wide, from information from the Illumina SNP60 platform [52]. Chen and colleagues also used PennCNV to analyse porcine CNVs; they found 565 CNVRs containing a total of 1315 CNVs, from 18 populations. Hotspots for these CNVs included areas on chromosomes 6, 11, 13, 14 and 17, with the Duroc breed having the most CNVs found per individual [53]. A study in 2012 using array comparative genomic hybridisation (CGH) on 12 pigs from several different breeds (including Large White and Duroc) highlighted 259 CNVRs [54], whilst Wang and colleagues used PennCNV to glean CNV information from data derived from the Illumina Porcine SNP60 Beadchip, finding 382 CNVs in a total number of 474 pigs [52].
Studies in the pig have not before however, studied the effect of CNVs on specific traits, with few using QuantiSNP or cnvPartition.
Given the above, it is clear that, despite the need to understand the genetic control of fatness in pigs, both from an agricultural and biomedical standpoint, very few genes have hitherto been associated with fatness and/or leanness in this species. The availability of the porcine 60 K SNP chip (Illumina) allows us to ask whether there are SNPs and/or CNVRs significantly associated with defined fatness phenotypes in different pig breeds. To date, we are aware of only one GWAS and no CNV searches that have addressed this question. Fontanesi and colleagues [35] looking at a single breed and finding gene ontology terms associated with nervous system development and regulation. With this in mind, in this study, we performed a GWAS using selective genotyping to sample a population of over 70,000 animals from 3 breeds identifying both significant SNPs and extracting quantitative information from the raw data to identify also CNVs.
Results
SNP analysis
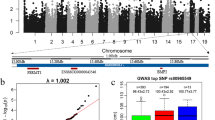
Selective genotyping permitted the generation of statistically significant results using Estimated Breeding Values (EBVs) from animals in the upper and lower 5th percentiles. After SNP chip interrogation, a total of 33,080 SNPs were eliminated from the analysis from the original 64,232 due to either the Minor Allele Frequency (MAF) being <0.05, SNPs being located on the sex chromosomes or those that deviated from Hardy Weinberg Equilibrium (HWE). 31,152 SNPs were taken further for analysis. The results shown in Figure 1 (A, SLLW, B Duroc and C Titan) are the Manhattan plots obtained for the 3 breeds. This figure displays the results from an additive model with significant SNPs at the 5% genome wide level; (p value greater than 0.000019952 (i.e. –log10(p) equal to 5.7)) (see Methods). An example of a chromosome that had significant clustered SNPs present in chromosome 7 (SLLW breed) is shown in Figure 2. Clustering of other SNPs near a SNP of interest reinforces its significance. For the 3 breeds, Duroc, Titan and SLLW, a total of 50, 10 and 12 SNPs (respectively) were considered to be significant, all of these being breed specific. The most significant SNPs were located on chromosomes 7 and 15 (SLLW), 5 and 15 (Duroc) and 9 and 13 (Titan). Following gene ontology, genes that either had an association with fatness (4 genes) or those that resided within a gene (17 genes) are shown in Table 1. Table 2 shows SNPs that were deemed as significant in this study, but without genes upstream or downstream that were associated with fatness. Significant SNPs that were located either up or downstream from genes and implicated in the fatness phenotype in the Duroc breed, were MARC00776967 downstream from NTS on chromosome 5 (neurotensin, involved in maintenance of gut structure and function, and in the regulation of fat metabolism), ASGA0073794 upstream from FABP6 on chromosome 16 (FABPs roles include fatty acid uptake, transport, and metabolism), INRA0040972 upstream from SST on chromosome 13 (somatostatin, an important regulator of the endocrine system through its interactions with pituitary growth hormone, thyroid stimulating hormone, and most hormones of the gastrointestinal tract) and ASGA0102890 upstream from NR3C2 located on chromosome 8 (which encodes the mineralocorticoid receptor, involved in metabolism). 12 other significant SNPs identified in the Duroc breed, resided in the genes TECTA, EPAS1, TMPRSS4, ADAM32, MX2, HSF5, MPZL3, CAMK1D, DOCK5, CCDC39, DENND2D and TEX14. Only 1 significant SNP (ALGA0027483) was found to be located within an intronic region of a gene for the Titan breed. This gene was SPAG17, known to be involved in maintenance of the structural integrity of the central apparatus of the sperm axoneme. There were no genes implicated with fatness found in the Titan breed. Significant SNPs in the SLLW breed were identified in 3 chromosomes, 7, 15 and 16. Whilst there were no genes identified either up or downstream from significant SNPs, that could have been directly implicated with a fatness phenotype, other significant SNPs that resided within genes included ALGA0039880 in the gene TINAG (tubulointerstitial nephritis antigen), INRA0024695 and ALGA0040094 both in the gene DST (dystonin) and DIAS0000554 in the gene GLO1 (glyoxalase I). The only gene that had a SNP located within a coding region in any of the 3 breeds was GLO1. After analysis using the Ensembl variant effect predictor, the amino acid at position 41 displayed a synonymous substitution, with the residue leucine not being modified after the SNP change. All significance levels for all SNPs in all breeds are depicted in Figure 1. It was interesting to note that the Duroc breed had many more SNPs associated with a fatness phenotype than any of the other breeds analysed with these SNPs having a higher level of significance also.

Graphical interpretation of SLLW (A), Duroc (B) and Titan (C) SNP results for Rib Fat for all chromosomes. Legend: Graphical interpretation of SNP results for Rib Fat for all chromosomes. Red dots represent SNPs on odd numbered chromosomes while the green dots are from even numbered chromosomes. The dotted lines represent the 5% (bottom line) and 1% (top line) confidence thresholds.

Graphical microarray outputs from chromosome 7 in the SLLW breed. Legend: Graphical microarray outputs from chromosome 7 in the SLLW breed. Each of the dots represents a SNP plotted against its chromosome position (bp) and significance value (-log10(p)). The circled SNPs represent SNPs discovered within the following genes: TINAG, DST and GLO1.
CNV Analysis
In terms of CNV Analysis, 1 example of the output from QuantiSNP is shown in Figure 3 for Titan chromosome 1. All red lines on this figure depict CNVs discovered in ‘fat’ (upper 5th percentile) pigs and green lines depict those found in ‘lean’ (lower 5th percentile) pigs. In order to distinguish between levels of significance (determined by Log Bayes Factors), a darker line (either red or green, as shown in the key) indicates a high Log Bayes Factor (>30), and therefore a higher level of significance, while a paler line indicates samples with a Log Bayes Factor of between 10 and 30 (therefore with a lower significance value). Lines above the x-axis in the middle of the figure, indicate gains in copy number whilst lines below are losses. The chromosome position in Mb is shown below the graph. The start and ends of the putative CNVs are defined on the chip and through reference to the published porcine genome sequence. The total number of CNVs detected was higher from QuantiSNP (216) than from cnvPartition (27). Of the total 243 CNVs detected in 5 or more animals (from either CNV calling approach), 202 were unique to QuantiSNP (83%), 14 were unique to cnvPartition and 27 were detected by both (11%). Generally, there were 3 times more losses found by both programs than gains. In order to determine whether regions of the genome differ in DNA copy number in statistically different numbers of ‘fat’ and ‘lean’ animals, Mann Whitney U tests (2 tailed) were performed; samples with a p value of ≤0.05 were considered significant. The statistical results for significant CNVRs can be found in Table 3, that shows CNVs present in more than 5 animals, for both CNV detecting algorithms. The estimated copy number and the number of animals in which the CNVs were found is shown. Each CNV was assigned a CNV ID; 1.S2 represents a CNV on chromosome 1, from the SLLW breed, assigned the number 2. 2 CNVRs contained genes that their ontology suggested could play a role in fatness, shown in Table 4. These CNVs were 5.D2 (chromosome 5, Duroc) and 14.D1 (chromosome 14, Duroc). 98% of the CNVs overlapped coding sequence. In the SLLW breed, cnvPartition results covered 6.11% of the genome, while QuantiSNP determined a 11.26% coverage. cnvPartition and QuantiSNP results for the Duroc breed indicated genome coverage of 5.03% and 10.93% respectively, with the percentage coverage in the Titan breed being 4.22% (cnvPartition) and 0.62% (QuantiSNP). The distribution of the CNVs can be found in the Additional file 1: Figure S1. Here, the positions of the CNVs are noted with respect to a standard porcine ideogram (ignoring whether these animals are fat or lean). It is depicted whether the CNVs (found in 5 or more animals) are losses or gains by the position, left or right of the chromosome respectively, in which breed they were observed (colour code) and in how many animals each CNV was observed (number inside the coloured ellipse). Results from both QuantiSNP and cnvPartition are given side by side.

QuantiSNP output for Titan chromosome 1. Legend: QuantiSNP output for Titan chromosome 1. Red lines depict CNVs discovered in fat pigs and green lines depict those found in lean pigs. To distinguish between levels of significance (determined by Log Bayes Factors), a darker line (either red or green, as shown in the key) indicates a high Log Bayes Factor (>30), and therefore a higher level of significance, while a paler line indicates samples with a Log Bayes Factor of between 10 and 30 (therefore with a lower significance value). Lines above the x-axis in the middle of the figure, indicate gains in copy number whilst lines below are losses. The chromosome position in Mb is shown below the graph and the y axis is scaled by copy number count.
Discussion
The results presented demonstrate the applicability of selective genotyping when using a GWAS approach. Unlike the most recent similar study [35] the breeds under investigation are ones marketed extensively worldwide and clearly display breed specific differences. For GWAS data to be used in practical applications, it is essential that the SNPs discovered as associated with fatness and leanness (or indeed any other commercially relevant trait) are segregating in the population in question. To this end, in any association study, the benefits must outweigh the costs; here we have provided evidence of a low-cost approach to GWAS by using EBV data for animals in the upper and lower 5th percentiles. It is also essential that the relevant samples are archived and readily accessed. Storage of large numbers of blood or DNA samples in freezers is expensive and space consuming. Here we demonstrate that amplifying DNA from archived blood spots can overcome this problem. Finally, we demonstrate that CNV information can be derived from the raw SNP chip data, providing the opportunity for studies of DNA copy number and its association with commercially and biologically relevant traits.
SNP discovery and gene ontology
In the current study, we identified a total of 12 SNPs in the SLLW breed, 50 in Duroc breed and 12 in Titan breed significantly associated with fatness or leanness as defined by EBVs for back fat. The reasons why Duroc had so many more than the other two (indeed over twice as many as the other two put together) is not clear. We are not aware that Duroc is any more genetically diverse – one possible explanation is a technical one in that the chip itself was made from a Duroc pig and the results may reflect ascertainment bias. SNPs were identified that were either contained within, or 0.5 Mb up or downstream of genes whose ontology terms strongly implicated them in a fatness phenotype as follows: NTS (neurotensin), is involved in energy homeostasis, a complex physiological process most probably related to fatness [55]. Genes such as NTS have been shown to be involved in both central and peripheral signals affecting feed intake [56, 57]. SST (somatostatin) was first isolated from the hypothalamus of sheep as a 14 amino acid peptide in 1973 [58]; SST plays a vital role in the regulation of growth and development in vertebrates, particularly muscle growth. It is known to be one of the most important genes involved in both the regulation of animal growth, metabolism and development through its negative role on growth, as it acts as an inhibitor of growth hormone release [59], inhibition of cell proliferation as well as affecting nutrient absorption in the alimentary canal [60, 61]. A study focussing on a polymorphism in SST and its association with growth traits in Chinese cattle was published earlier this year that has been correlated with improving the establishment of meat production performance by breeding of new beef cattle [58]. Fatty acid binding protein 6 (FABP6), which was mapped to chromosome 16 in 2007 [62], was also found to be significant in the Duroc population in this study. It has been shown that FABP6 is associated with type II diabetes therefore suggesting an association of variants between fatness and type II diabetes susceptibility [63], as well as the role of FABPs in fat absorption and in the development of metabolic syndrome [64]. GLO1 (SSC17) was a gene of particular interest highlighted throughout this study due to the fact that it was the only candidate gene isolated in this GWAS that had a SNP residing within the gene itself. GLO1 is a candidate gene that is thought to be involved with fatness; a study that focussed on a QTL for carbohydrate and total energy intake on chromosome 17 in mice showed that genes (including GLO1) involved in fat metabolism were decreased in carbohydrate preferring mice [65, 66]. A GWAS performed in 2009 discovered specific alleles that interestingly were associated with both increased back fat and better leg action [67]. These genes included MTHFR, WNT2, APOE, BMP8, GNRHR and OXTR.
The results presented have also been compared to a recent GWAS performed in a specific sire line large white breed used in dried Italian ham production [35]. The genes which Fontanesi and colleagues found to be associated with significant SNPs by gene ontology, did not overlap with ones found in this study, however interesting insights were made, possibly indicating that neuronal genes may play a role in fat deposition in the pig. This concurs with one of the genes we found to be associated with fatness, neurotensin, widely distributed throughout the central nervous system that may be a neuromodulator or neurotransmitter.
Technical issues pertaining to GWAS
There are several factors to consider when interpreting GWAS for example, in replication of such studies, the consistency of the results vary [68]. Some genes are reproducibly found in follow-up studies, such as genes related to diabetes including the peroxisome proliferator-activated receptor-γ (PPARG). This was of particular interest as the peroxisome proliferator activated receptor alpha, was found in a CNV which was significant in this study. Replication problems involved in replication of GWAS have been widely reported [69–72] also mentioning that small sample sizes can be problematic. Inconsistent results have been reported in obesity studies [69], which suggests that many results may be population dependent.
One possible criticism of the results presented here is the relatively small sample size used. We would argue that this might have been more of a problem had we considered the data as a binary trait (i.e. either ‘fat’ or ‘lean’). We believe that there is no loss of power (when compared to analysing the entire population of over 70,000 animals, from which the EBVs were derived) by adopting selective genotyping, indeed we suggest that use of this approach (i.e. using EBVs from animals in the tails of the distribution), in fact, retains most of the power of the calculations. If the EBVs are calculated from a larger number of individuals then it removes a source of environmental variation and potentially provides a more accurate estimate of the genetic effect (taking into account information from other animals in the population that were not genotyped). As Darvasi and colleagues showed, selective genotyping is very effective to retain the power of the experiment while reducing the cost of genotyping [34]. There is no evidence that selective genotyping would bias results, thereby increasing the number of false positives, providing that an appropriate significance threshold is calculated to account for multiple testing. A similar approach was recently published in this journal by Fontanesi and colleagues [35] have demonstrated that selective genotyping is very effective to retain the power of the experiment while reducing the cost of genotyping. Given this information, we were able to derive data from a 70,000+ animals, thereby avoiding a high false positive rate. Admittedly we might miss QTL with relatively small effects, however part of the point of the exercise was to discover traits that were most biologically significant and thus commercially relevant.
CNV analysis
The CNV information presented is, one of few such studies in pigs and has identified 243 candidate CNVs. It is worthy of note however that further studies involving deep sequencing, array CGH and or qPCR would be required to confirm the extent to which the data presented here represent physical changes on DNA copy number. Nonetheless we have demonstrated that use of SNP chips can identify “putative” CNVs that warrant further investigations. Our attempts to confirm some of the results are mentioned below. Only 2 putative CNVs that were discovered contained genes that, as implied by their ontology terms, might be involved in fatness. Both programs identified CNVR14.D1 whereas QuantiSNP only called CNVR5.D2. In CNVR5.D2, a copy number loss (in comparison to the reference genome) was observed in both fat and lean animals, however the number of animals that displayed a loss was significantly greater in lean animals (40) compared to fat animals (23). This is particularly interesting, due to the fact that 1 of the genes located within this putative CNVR was MCHR1, known to regulate feeding behaviour. It has been shown that mice lacking MCHR1 eat less and are therefore leaner [73, 74]. PPARα, involved in metabolic control of the expression of genes encoding fatty acid oxidation enzymes [75], is also located in this putative CNVR. CNVR14.D1, again displayed a greater number of samples with a loss in copy number in ‘lean’ compared to ‘fat’ animals (28 vs 12). Genes contained within this CNVR include sodium/glucose transporters and co-transporters, responsible for glucose absorption across the brush border of gastrointestinal tract cells; similar gene families have also been identified in other pig CNVR studies [52, 53]. When comparing the data produced from this study to other published work, there were 7 losses identified by QuantiSNP that were also found by Fadista et al. [50]. Seven overlapping CNVRs were also found when comparing our data to Ramayo-Caldas et al. [51], however there were discrepancies in calling whether these were gains or losses.
A total of 83% of the 243 putative CNVs identified were unique to QuantiSNP, 6% were unique to cnvPartition and 11% were detected by both programs. One possible reason for these discrepancies is that each of the programs made use of a different algorithm in order to detect putative CNVs. cnvPartition, the plug-in available for Illumina’s Genome Studio, produces 1 of 14 possible outputs assuming 5 copy number states; a homozygous deletion, heterozygous deletion, dizygous (normal state), trizygous (1 extra copy) and finally tetrazygous (2 more copies than the normal state). This algorithm models log r ratios (LRRs) and b allele frequencies (BAFs) as a bivariate Gaussian distribution. In contrast, QuantiSNP is based on a Hidden-Bayes Objective Markov Model (HB-OMM) that considers the number of copies in both the hidden and observed states. QuantiSNP uses a filtering process by which any CNVs called with a Log Bayes Factor of less than ten were removed whereas cnvPartition does not. Taking this into consideration it is surprising that QuantiSNP called more CNV regions than cnvPartition, however this could suggest that the algorithm used by the cnvPartition software is more accurate at calling CNVs. Generally, there were more losses found by both programs than gains. A paper published this year discussed new freely available software called ParseCNV. This is a CNV call association software that uses CNV information to create SNP statistics from information in the PennCNV format [76]; this would be an interesting future study to perform on this data.
qPCR was attempted in order to verify the results produced from both cnvPartition and QuantiSNP, however, after numerous attempts, results did not give adequate consistency in order to calibrate the system and therefore make it possible to verify the putative CNV calls by independent means. As discussed in a study published in 2010, there are several reasons as to why CNVR prediction varies between qPCR analysis and in silico analysis of data. The 4 x sequence depth of the Sus scrofa genome build 9 and low probe density of the Porcine SNP60 BeadChip makes it difficult to determine the genuine boundaries of CNVRs. This can therefore lead to an over-estimation of the real size of the region. SNPs and indels also have the ability to affect the hybridisation of qPCR primers and true CNVR boundaries may be polymorphic between analysed animals [51]. Array CGH could also be used as an alternative platform to derive CNV information from and compared to data produced in this study. There have been several such studies performed [37, 77, 78] including one in pigs [51]. Whilst this would be an interesting comparison, it is possible that similar amplification problems might be experienced.
Conclusions
The combination of a cost effecting selective genotyping-based GWAS, data from 3 widely consumed pig breeds, the derivation of data from archived blood spots and the simultaneous mining of both SNP and CNV data are the unique aspects of this study. The discovery of novel SNPs and CNVs associated with fatness are, potentially of value to the pig breeding industry and shed light on the aetiology of fatness in mammals (including humans). However they demonstrate the phenomenon of breed-specificity and thus highlight the need for the study of multiple populations to verify genotype - phenotype correlations.
Methods
Sample acquisition
An Aloka 500 Ultrasound scanner, coupled with AUSkey fat and muscle depth system software was able to provide an accurate representation of average fat depth (aFd) and average muscle depth (aMd) for 18757 Duroc pigs, 26992 Sire line large white and 27537 Titan (Pietrain) animals (this measurement is directly correlated with the total fat in the carcass [79]). Raw data was then converted to EBVs correcting the rib fat depth to the fixed animal weight of 91 kg. EBVs for each of the 70,000+ animals were obtained from the standard evaluation scheme employed by JSR using best linear unbiased prediction (BLUP) analysis. All samples were from males, with a similar genetic background and reared under identical conditions (stocking, density, feed, space etc.), in order to prevent other potential contributing factors that could influence any conclusions drawn. For selective genotyping, animals in the upper 5th percentile and the lower 5th percentile of the EBV range for fatness were taken forward for further investigation. All DNA samples were extracted from blood spots stored on FTA Whatman Cards™ that were sourced from the JSR Genetics (Driffield UK) archive. Of the 288 samples used, 96 were from each of the 3 aforementioned breeds; 48 from each group either in the upper or lower 5th percentile and their EBV values used for subsequent calculation. DNA isolation, amplification and SNP chip interrogation was performed using the method previously developed in house [80], with several alterations to the manufacturer’s instructions, including 2 punches being removed as opposed to 1, heating of the cards and washes being performed using water instead of FTA purification reagent and Tris EDTA. Extracted samples were amplified via Whole Genome Amplification (WGA) using the Sigma-Aldrich WGA2 kit, in order to produce an appropriate amount of DNA for microarray analysis, following manufacturer’s instructions. This fragmentation based WGA produces short 400-600 bp overlapping fragments that are primed with defined 3′and 5′ ends and amplified via linear amplification followed by geometrical amplification, therefore generating a thorough coverage of the genome.
Genotyping
SNP analysis
Illumina Porcine SNP60 Genotyping BeadChips were interrogated with WGA amplified DNA from each porcine sample, according to manufacturer’s instructions, in an approach similar to that described by our own group [81, 82] at Cambridge Genomic Services, Department of Pathology, University of Cambridge. All DNA concentrations were adjusted to a concentration of 50 ng/μl, in a final volume of 5 μl per sample. Raw fluorescence data were captured and normalized, using internal and external controls, and stored as image files. Following scanning, image data were transferred to the GenomeStudio Software framework V2010.1 and converted from fluorescence data to genotypic data based on the manufacturer’s design algorithms and the call rates produced by the Illumina software were determined. Significance analysis of the SNP chip data was performed, assuming an additive model and the raw data was corrected to the fixed animal weight of 91 kg, and then converted to EBVs using the PEST software. The model used herd/sex/season as a fixed effect with litter fitted as a random effect. The genotype scores for a given SNP were 1, 2 and 3 for genotypes AA, AB, and BB, respectively. For a given SNP only records were used when the genotype was known, any animals for which records were unknown were removed from the study. Analysis of variance was performed to obtain the P values. Due to multiple testing, Bonferroni corrections were implemented in order to determine the appropriate significance value. Due to multiple testing, an empirical genome wide significance threshold was calculated using permutation analysis, where SNPs are not considered to all be independent. Genotypes for all individuals were permuted and the GWAS analysis was calculated in all SNPs with the smallest p value being used to calculate the distribution. The permutation analysis was repeated 10,000 times and the value for the top 5% was deemed to be the significance threshold. SNPs that deviated from Hardy-Weinberg Equilibrium (HWE) were also removed, so results were not skewed. Significant SNPs were consequently investigated to determine whether they were located within, or close to a gene, using the Ensembl output for orthologous regions. This involved data mining using a combination of Ensembl (http://www.ensembl.org) and NCBI to interpret where the SNP resided (pig genome assembly version 9.2). A window of 1 Mb was examined (0.5 Mb upstream and 0.5 Mb downstream from the SNP was considered). If a SNP resided within a gene this was also noted. Subsequently, gene function was determined and SNPs were then placed into 2 groups, those in which their function suggested a pathway in which the control of fatness might be implicated, and those that were not. In order to determine the location and function of a SNP of interest within a gene, the variant effect predictor, a tool available from Ensembl was used (http://www.ensembl.org/info/website/upload/var.html).
CNV analysis
In order to derive CNV information from the existing SNP data, 2 analytical tools for the determination of copy number variation using SNP genotyping data were used, QuantiSNP and cnvPartition. QuantiSNP uses an Objective Bayes Hidden-Markov Model (OB-HMM) to estimate probabilities of CNV/aneuploidy detection [83]. QuantiSNP uses both LRRs and BAFs in order to call CNVs with corrections for differences in GC content also being employed in order to correct signal strength [84]. cnvPartition calculates copy number variants along with scores of confidence, therefore indicating the locations of CNV regions (http://www.illumina.com) using both BAFs and LRRs for each of 14 genotypes (double deletion, A, B, AA, AB, BB, AAA, AAB, ABB, BBB, AAAA, AAAB, ABBB, BBBB) as a simple bivariate Gaussian distribution. Samples with a call rate below 0.9 were removed, and the confidence thresholds and number of probes needed to determine a CNV event were altered accordingly; increasing the threshold improved the clarity of the output, while using a high probe count increased the stringency. 3 outputs were produced, 1 per breed with CNV fold change being represented in different colours for 5 groups (0–0.5, 0.5-1.5, 1.5-2.5, 2.5-3.5 and 3.5-4.5). Data derived from QuantiSNP graphs and cnvPartition outputs were subsequently collated. Only CNVs present in more than 5 animals for both QuantiSNP and cnvPartition were considered. The chromosome, the start and end of each of the CNVRs, the estimated copy number of the sequence and the number of animals in which each was observed (2 copies was considered to be typical as pigs are diploid organisms) were recorded. Mann Whitney U tests (2 tailed) were performed the data to determine if there were any significant differences between samples in the upper and lower 5th percentiles with p values of ≤0.05 being considered statistically significant.
Author’s contributions
KF performed the majority of the experiments in the paper and co-wrote the manuscript. RPW analysed the raw SNP chip data. JB analysed the raw data from cnvPartition. EC, CR and NA performed the microarray experiments and generated the call rate data. SW coordinated all the samples for the project as well as the phenotyping. GAW and DKG conceived the project, supervised all aspects of it and co-wrote the manuscript. All authors read and approved the final manuscript.
Abbreviations
- aFd:
-
average fat depth
- aMd:
-
average muscle depth
- BAF:
-
B allele frequency
- BLUP:
-
Best linear unbiased prediction
- CNV:
-
Copy number variant
- CNVR:
-
Copy number variable region
- EBV:
-
Estimated Breeding Value
- FDR:
-
False discovery rate
- FCR:
-
Food conversion ratio
- GWA:
-
Genome wide association
- GWAS:
-
Genome wide association study
- HWE:
-
Hardy Weinberg equilibrium
- HSA:
-
Homo sapiens
- LRR:
-
Log r ratio
- MAF:
-
Minor allele frequency
- SNP:
-
Single nucleotide polymorphism
- SSC:
-
Sus scrofa
- T2D:
-
Type II diabetes
- WGA:
-
Whole genome amplification.
References
Ngapo T, Martin JF, Dransfield E: International preferences for pork appearance: I Consumer choices. Food Qual Prefer. 2007, 18 (1): 26-36. 10.1016/j.foodqual.2005.07.001.
Webb J: New Opportunities for Genetic Change in Pigs. Advances in Pork Production. 2000, 11: 83-95.
Mrode R, Kennedy B: Genetic variation in measures of food efficiency in pigs and their genetic relationships with growth rate and backfat. Animal Production-Glasgow. 1993, 56: 225-232. 10.1017/S0003356100021309.
Miller E, Ullrey D: The pig as a model for human nutrition. Annu Rev Nutr. 1987, 7 (1): 361-382. 10.1146/annurev.nu.07.070187.002045.
Quilter C, Gilbert C, Oliver G, Jafer O, Furlong R, Blott S, Wilson A, Sargent C, Mileham A, Affara N: Gene expression profiling in porcine maternal infanticide: a model for puerperal psychosis. Am J Med Genet B Neuropsychiatr Genet. 2008, 147 (7): 1126-1137.
Bidanel JP, Milan D, Iannuccelli N, Amigues Y, Boscher MY, Bourgeois F, Caritez JC, Gruand J, Le Roy P, Lagant H, Quintanilla R, Renard C, Gellin J, Ollivier L, Chevalet C: Detection of quantitative trait loci for growth and fatness in pigs. Genet Sel Evol. 2001, 33: 289-309. 10.1186/1297-9686-33-3-289.
Houpt K, Houpt T, Pond W: The pig as a model for the study of obesity and of control of food intake: a review. Yale J Biol Med. 1979, 52 (3): 307-
Andersson L: Genes and obesity. Ann Med. 1996, 28 (1): 5-7. 10.3109/07853899608999066.
Naggert J, Harris T, North M: The genetics of obesity. Curr Opin Genet Dev. 1997, 7 (3): 398-404. 10.1016/S0959-437X(97)80155-4.
Colditz GA: Economic costs of obesity and inactivity. Med Sci Sports Exerc. 1999, 31 (11): 663-667.
Kondrashov AS, Rogozin IB: Context of deletions and insertions in human coding sequences. Hum Mutat. 2004, 23 (2): 177-185. 10.1002/humu.10312.
Lander ES, Schork NJ: Genetic Dissection of Complex Traits. Focus. 2006, 4 (3): 442-458.
Brookes AJ: The essence of SNPs. Gene. 1999, 234 (2): 177-186. 10.1016/S0378-1119(99)00219-X.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A: Finding the missing heritability of complex diseases. Nature. 2009, 461 (7265): 747-753. 10.1038/nature08494.
Fan B, Du ZQ, Gorbach DM, Rothschild MF: Development and application of high-density SNP arrays in genomic studies of domestic animals. Asian-Aust J Anim Sci. 2010, 23 (7): 833-847. 10.5713/ajas.2010.r.03.
Traeger-Synodinos J: Real-time PCR for prenatal and preimplantation genetic diagnosis of monogenic diseases. Mol Aspects Med. 2006, 27 (2): 176-191.
Driss A, Asare K, Hibbert J, Gee B, Adamkiewicz T, Stiles J: Sickle cell disease in the post genomic era: a monogenic disease with a polygenic phenotype. Genomics insights. 2009, 2009 (2): 23-
Easton DF, Pooley KA, Dunning AM, Pharoah PDP, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R: Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007, 447 (7148): 1087-1093. 10.1038/nature05887.
Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PIW, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ: Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007, 316 (5829): 1331-
Frayling TM: Genome–wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet. 2007, 8 (9): 657-662.
Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R: Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010, 42 (12): 1118-1125. 10.1038/ng.717.
Franke A, Balschun T, Karlsen TH, Hedderich J, May S, Lu T, Schuldt D, Nikolaus S, Rosenstiel P, Krawczak M: Replication of signals from recent studies of Crohn's disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet. 2008, 40 (6): 713-715. 10.1038/ng.148.
Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG: Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009, 41 (12): 1308-1312. 10.1038/ng.487.
van Heel DA, Franke L, Hunt KA, Gwilliam R, Zhernakova A, Inouye M, Wapenaar MC, Barnardo MCNM, Bethel G, Holmes GKT: A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet. 2007, 39 (7): 827-829. 10.1038/ng2058.
Hinney A, Nguyen TT, Scherag A, Friedel S, Bronner G, Muller TD, Grallert H, Illig T, Wichmann HE, Rief W: Genome Wide Association (GWA) Study for Early Onset Extreme Obesity Supports the Role of Fat Mass and Obesity Associated Gene (FTO) Variants. PLoS ONE. 2007, 2 (12): e1361-10.1371/journal.pone.0001361.
Fontanesi L, Scotti E, Buttazzoni L, Davoli R, Russo V: The porcine fat mass and obesity associated (FTO) gene is associated with fat deposition in Italian Duroc pigs. Anim Genet. 2008, 40 (1): 90-93.
Fan B, Du ZQ, Rothschild MF: The fat mass and obesity-associated (FTO) gene is associated with intramuscular fat content and growth rate in the pig. Anim Biotechnol. 2009, 20 (2): 58-70. 10.1080/10495390902800792.
Kim KS, Larsen N, Short T, Plastow G, Rothschlid MF: A missense variant of the porcine melanocortin-4 receptor (MC4R) gene is associated with fatness, growth, and feed intake traits. Mamm Genome. 2000, 11: 131-135. 10.1007/s003350010025.
Duijvesteijn N, Knol EF, Merks JWM, Crooijmans RPMA, Groenen MAM, Bovenhuis H, Harlizius B: A genome-wide association study on androstenone levels in pigs reveals a cluster of candidate genes on chromosome 6. BMC Genet. 2010, 11 (1): 42-
Skinner T, Doran E, McGivan J, Haley C, Archibald A: Cloning and mapping of the porcine cytochrome‒p450 2E1 gene and its association with skatole levels in the domestic pig. Anim Genet. 2005, 36 (5): 417-422. 10.1111/j.1365-2052.2005.01342.x.
Grindflek E, Lien S, Hamland H, Hansen MHS, Kent M, van Son M, Meuwissen THE: Large scale genome-wide association and LDLA mapping study identifies QTLs for boar taint and related sex steroids. BMC Genomics. 2011, 12 (1): 362-10.1186/1471-2164-12-362.
Fan B, Onteru SK, Du ZQ, Garrick DJ, Stalder KJ, Rothschild MF, Sorensen TIA: Genome-Wide Association Study Identifies Loci for Body Composition and Structural Soundness Traits in Pigs. PloS one. 2011, 6 (2): e14726-10.1371/journal.pone.0014726.
Fan B, Onteru SK, Mote BE, Serenius T, Stalder KJ, Rothschild MF: Large-scale association study for structural soundness and leg locomotion traits in the pig. Genet Sel Evol. 2009, 41: 14-10.1186/1297-9686-41-14.
Darvasi A, Soller M: Selective genotyping for determination of linkage between a marker locus and a quantitative trait locus. Theor Appl Genet. 1992, 85 (2): 353-359.
Fontanesi L, Schiavo G, Galimberti G, Calò D, Scotti E, Martelli P, Buttazzoni L, Casadio R, Russo V: A genome wide association study for backfat thickness in Italian Large White pigs highlights new regions affecting fat deposition including neuronal genes. BMC Genomics. 2012, 13 (1): 583-10.1186/1471-2164-13-583.
Feuk L, Carson AR, Scherer SW: Structural variation in the human genome. Nat Rev Genet. 2006, 7 (2): 85-97.
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W: Global variation in copy number in the human genome. Nature. 2006, 444 (7118): 444-10.1038/nature05329.
Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F: Mapping and sequencing of structural variation from eight human genomes. Nature. 2008, 453 (7191): 56-64. 10.1038/nature06862.
Pique-Regi R, Ortega A, Asgharzadeh S: Joint estimation of copy number variation and reference intensities on multiple DNA arrays using GADA. Bioinformatics. 2009, 25 (10): 1223-10.1093/bioinformatics/btp119.
Forer L, Schönherr S, Weissensteiner H, Haider F, Kluckner T, Gieger C, Wichmann HE, Specht G, Kronenberg F, Kloss-Brandstätter A: CONAN: copy number variation analysis software for genome-wide association studies. BMC bioinformatics. 2010, 11 (1): 318-10.1186/1471-2105-11-318.
Dellinger AE, Saw SM, Goh LK, Seielstad M, Young TL, Li YJ: Comparative analyses of seven algorithms for copy number variant identification from single nucleotide polymorphism arrays. Nucleic Acids Res. 2010, 38 (9): e105-10.1093/nar/gkq040.
Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P: Origins and functional impact of copy number variation in the human genome. Nature. 2009, 464 (7289): 704-712.
Pang AW, MacDonald JR, Pinto D, Wei J, Rafiq MA, Conrad DF, Park H, Hurles ME, Lee C, Venter JC: Towards a comprehensive structural variation map of an individual human genome. Genome Biol. 2010, 11 (5): R52-10.1186/gb-2010-11-5-r52.
Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, Krauss RM, Myers RM, Ridker PM, Chasman DI: Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009, 84 (2): 148-161. 10.1016/j.ajhg.2008.12.014.
Nakajima T, Kaur G, Mehra N, Kimura A: HIV-1/AIDS susceptibility and copy number variation in CCL3L1, a gene encoding a natural ligand for HIV-1 co-receptor CCR5. Cytogenet Genome Res. 2008, 123: 156-160. 10.1159/000184703.
McCarroll SA, Altshuler DM: Copy-number variation and association studies of human disease. Nat Genet. 2007, 39: S37-S42. 10.1038/ng2080.
Cooper GM, Nickerson DA, Eichler EE: Mutational and selective effects on copy-number variants in the human genome. Nat Genet. 2007, 39: S22-S29. 10.1038/ng2054.
Orozco LD, Cokus SJ, Ghazalpour A, Ingram-Drake L, Wang S, van Nas A, Che N, Araujo JA, Pellegrini M, Lusis AJ: Copy number variation influences gene expression and metabolic traits in mice. Hum Mol Genet. 2009, 18 (21): 4118-4129. 10.1093/hmg/ddp360.
Wang K, Li WD, Glessner JT, Grant SFA, Hakonarson H, Price RA: Large copy-number variations are enriched in cases with moderate to extreme obesity. Diabetes. 2010, 59 (10): 2690-10.2337/db10-0192.
Fadista J, Nygaard M, Holm LE, Thomsen B, Bendixen C: A Snapshot of CNVs in the Pig Genome. PLoS ONE. 2008, 3 (12): e3196-
Ramayo-Caldas Y, Castelló A, Pena RN, Alves E, Mercadé A, Souza CA, Fernández AI, Perez-Enciso M, Folch JM: Copy number variation in the porcine genome inferred from a 60 k SNP BeadChip. BMC genomics. 2010, 11 (1): 593-10.1186/1471-2164-11-593.
Wang J, Jiang J, Fu W, Jiang L, Ding X, Liu JF, Zhang Q: A genome-wide detection of copy number variations using SNP genotyping arrays in swine. BMC genomics. 2012, 13 (1): 273-10.1186/1471-2164-13-273.
Chen C, Qiao R, Wei R, Guo Y, Ai H, Ma J, Ren J, Huang L: A comprehensive survey of copy number variation in 18 diverse pig populations and identification of candidate copy number variable genes associated with complex traits. BMC genomics. 2012, 13 (1): 733-10.1186/1471-2164-13-733.
Li Y, Mei S, Zhang X, Peng X, Liu G, Tao H, Wu H, Jiang S, Xiong Y, Li F: Identification of genome-wide copy number variations among diverse pig breeds by array CGH. BMC genomics. 2012, 13 (1): 725-10.1186/1471-2164-13-725.
Woods SC: Signals that influence food intake and body weight. Physiology & behavior. 2005, 86 (5): 709-716. 10.1016/j.physbeh.2005.08.060.
Gerken T, Girard CA, Tung YCL, Webby CJ, Saudek V, Hewitson KS, Yeo GSH, McDonough MA, Cunliffe S, McNeill LA: The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007, 318 (5855): 1469-10.1126/science.1151710.
Fredriksson R, Hägglund M, Olszewski PK, Stephansson O, Jacobsson JA, Olszewska AM, Levine AS, Lindblom J, Schiöth HB: The obesity gene, FTO, is of ancient origin, up-regulated during food deprivation and expressed in neurons of feeding-related nuclei of the brain. Endocrinology. 2008, 149 (5): 2062-10.1210/en.2007-1457.
Gao L, Zan L, Wang H, Hao R, Zhong X: Polymorphism of somatostatin gene and its association with growth traits in Chinese cattle. Genet Mol Res. 2011, 10 (2): 703-711. 10.4238/vol10-2gmr1029.
Very N, Sheridan M: The role of somatostatins in the regulation of growth in fish. Fish Physiol Biochem. 2002, 27 (3): 217-226.
Yamada Y, Stoffel M, Espinosa R: Human somatostatin receptor genes: localization to human chromosomes 14, 17, and 22 and identification of simple tandem repeat polymorphisms. Genomics. 1993, 15 (2): 449-452. 10.1006/geno.1993.1088.
Kubota A, Yamada Y, Kagimoto S, Shimatsu A, Imamura M, Tsuda K, Imura H, Seino S, Seino Y: Identification of somatostatin receptor subtypes and an implication for the efficacy of somatostatin analogue SMS 201–995 in treatment of human endocrine tumors. J Clin Invest. 1994, 93 (3): 1321-10.1172/JCI117090.
Szczerbal I, Chmurzynska A, Switonski M: Cytogenetic mapping of eight genes encoding fatty acid binding proteins (FABPs) in the pig genome. Cytogenet Genome Res. 2007, 118: 63-66. 10.1159/000106442.
Fisher E, Nitz I, Lindner I, Rubin D, Boeing H, Möhlig M, Hampe J, Schreiber S, Schrezenmeir J, Döring F: Candidate gene association study of type 2 diabetes in a nested case control study of the EPIC Potsdam cohort–Role of fat assimilation. Mol Nutr Food Res. 2007, 51 (2): 185-191. 10.1002/mnfr.200600162.
Chmurzyska A: The multigene family of fatty acid-binding proteins (FABPs): function, structure and polymorphism. J Appl Genet. 2006, 47 (1): 39-48. 10.1007/BF03194597.
Kumar K, Smith Richards B: Transcriptional profiling of chromosome 17 quantitative trait Loci for carbohydrate and total calorie intake in a mouse congenic strain reveals candidate genes and pathways. J Nutrigenet Nutrigenomics. 2008, 1 (4): 155-171. 10.1159/000113657.
Kumar KG, Poole AC, York B, Volaufova J, Zuberi A, Richards BKS: Quantitative trait loci for carbohydrate and total energy intake on mouse chromosome 17: congenic strain confirmation and candidate gene analyses (Glo1, Glp1r). Am J Physiol Regul Integr Comp Physiol. 2007, 292 (1): R207-
Fan B, Onteru S, Nikkilä M, Stalder K, Rothschild M: Identification of genetic markers associated with fatness and leg weakness traits in the pig. Anim Genet. 2009, 40 (6): 967-970. 10.1111/j.1365-2052.2009.01932.x.
Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G, Hirschhorn JN, Abecasis GÇR, Altshuler D, Bailey-Wilson JE: Replicating genotype-phenotype associations. Nature. 2007, 447 (7145): 655-660. 10.1038/447655a.
Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K: A comprehensive review of genetic association studies. Genet Med. 2002, 4 (2): 45-10.1097/00125817-200203000-00002.
Ioannidis JPA, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG: Replication validity of genetic association studies. Nat Genet. 2001, 29 (3): 306-309. 10.1038/ng749.
Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN: Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet. 2003, 33 (2): 177-182. 10.1038/ng1071.
Colhoun HM, McKeigue PM, Smith GD: Problems of reporting genetic associations with complex outcomes. Lancet. 2003, 361 (9360): 865-872. 10.1016/S0140-6736(03)12715-8.
Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E: Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature. 1998, 396 (6712): 670-673. 10.1038/25341.
Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen AS, Guan XM, Jiang MM, Feng Y, Camacho RE: Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci. 2002, 99 (5): 3240-10.1073/pnas.052706899.
Barger PM, Kelly DP: PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc Med. 2000, 10 (6): 238-10.1016/S1050-1738(00)00077-3.
Glessner JT, Li J, Hakonarson H: ParseCNV integrative copy number variation association software with quality tracking. Nucleic Acids Res. 2013, 41 (5): e64-10.1093/nar/gks1346.
Fadista J, Thomsen B, Holm LE, Bendixen C: Copy number variation in the bovine genome. BMC genomics. 2010, 11 (1): 284-10.1186/1471-2164-11-284.
De Smith AJ, Tsalenko A, Sampas N, Scheffer A, Yamada NA, Tsang P, Ben-Dor A, Yakhini Z, Ellis RJ, Bruhn L: Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum Mol Genet. 2007, 16 (23): 2783-10.1093/hmg/ddm208.
Liu Y, Stouffer JR: Pork carcass evaluation with an automated and computerized ultrasonic system. J Anim Sci. 1995, 73 (1): 29-38.
Fowler KE, Reitter CP, Walling GA, Griffin DK: Novel approach for deriving genome wide SNP analysis data from archived blood spots. BMC Research Notes. 2012, 5 (1): 503-10.1186/1756-0500-5-503.
Handyside AH, Harton GL, Mariani B, Thornhill AR, Affara N, Shaw MA, Griffin DK: Karyomapping: a universal method for genome wide analysis of genetic disease based on mapping crossovers between parental haplotypes. J Med Genet. 2010, 47 (10): 651-10.1136/jmg.2009.069971.
Gabriel AS, Thornhill AR, Gordon A, Brown A, Taylor J, Bennett K, Handyside AH, Griffin DK: Array CGH on human first polar bodies suggests that non-disjunction is not the predominant mechanism leading to aneuploidy. Am J Med Genet. 2011, 48 (7): 433-
Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, Bassett AS, Seller A, Holmes CC, Ragoussis J: QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007, 35 (6): 2013-2015. 10.1093/nar/gkm076.
Cronin S, Blauw HM, Veldink JH, van Es MA, Ophoff RA, Bradley DG, van den Berg LH, Hardiman O: Analysis of genome-wide copy number variation in Irish and Dutch ALS populations. Hum Mol Genet. 2008, 17 (21): 3392-10.1093/hmg/ddn233.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
GAW and SW are employees of JSR genetics, who could, potentially, benefit financially from the results of this study.
Electronic supplementary material
12864_2013_7160_MOESM1_ESM.pdf
Additional file 1: Figure S1: Chromosome position of putative CNVs ascertained by quantiSNP and CNV partition. Left hand chromosome denotes result from CNV partition, right hand chromosome from quantSNP. Each putative CNVR is depicted as an elliptoid shape, colour coded for each breed as indicated. The numbers within the shape indicate the number of animals in with each putative CNV was found. If to the left of each chromosome a potential loss compared to the reference genome is apparent, a potential gain if to the right. (PDF 520 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fowler, K.E., Pong-Wong, R., Bauer, J. et al. Genome wide analysis reveals single nucleotide polymorphisms associated with fatness and putative novel copy number variants in three pig breeds. BMC Genomics 14, 784 (2013). https://doi.org/10.1186/1471-2164-14-784
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-784