
Abstract
Background
In the alpha subclass of proteobacteria iron homeostasis is controlled by diverse iron responsive regulators. Caulobacter crescentus, an important freshwater α-proteobacterium, uses the ferric uptake repressor (Fur) for such purpose. However, the impact of the iron availability on the C. crescentus transcriptome and an overall perspective of the regulatory networks involved remain unknown.
Results
In this work we report the identification of iron-responsive and Fur-regulated genes in C. crescentus using microarray-based global transcriptional analyses. We identified 42 genes that were strongly upregulated both by mutation of fur and by iron limitation condition. Among them, there are genes involved in iron uptake (four TonB-dependent receptor gene clusters, and feoAB), riboflavin biosynthesis and genes encoding hypothetical proteins. Most of these genes are associated with predicted Fur binding sites, implicating them as direct targets of Fur-mediated repression. These data were validated by β-galactosidase and EMSA assays for two operons encoding putative transporters. The role of Fur as a positive regulator is also evident, given that 27 genes were downregulated both by mutation of fur and under low-iron condition. As expected, this group includes many genes involved in energy metabolism, mostly iron-using enzymes. Surprisingly, included in this group are also TonB-dependent receptors genes and the genes fixK, fixT and ftrB encoding an oxygen signaling network required for growth during hypoxia. Bioinformatics analyses suggest that positive regulation by Fur is mainly indirect. In addition to the Fur modulon, iron limitation altered expression of 113 more genes, including induction of genes involved in Fe-S cluster assembly, oxidative stress and heat shock response, as well as repression of genes implicated in amino acid metabolism, chemotaxis and motility.
Conclusions
Using a global transcriptional approach, we determined the C. crescentus iron stimulon. Many but not all of iron responsive genes were directly or indirectly controlled by Fur. The iron limitation stimulon overlaps with other regulatory systems, such as the RpoH and FixK regulons. Altogether, our results showed that adaptation of C. crescentus to iron limitation not only involves increasing the transcription of iron-acquisition systems and decreasing the production of iron-using proteins, but also includes novel genes and regulatory mechanisms.
Similar content being viewed by others


Background
Iron is an essential micronutrient required for almost all organisms, functioning as a cofactor for proteins that are involved in a number of fundamental metabolic and enzymatic functions. Despite its high abundance, iron is a limiting nutrient in most biological systems due to its poor solubility under physiological conditions or because it is tightly sequestered by high-affinity proteins, such as transferrin and lactoferrin in eukaryotic hosts [1, 2]. On the other hand, high iron levels can generate toxic hydroxyl radicals by the Fenton reaction [3]. Thus, organisms have evolved multiple strategies to maintain accurate control over intracellular iron levels.
In most bacteria, iron homeostasis is mediated by Fur (ferric uptake regulator), an iron-sensing repressor protein, that controls the expression of genes involved in iron uptake, storage and usage. Under iron sufficiency, Fe2+-Fur (holo-Fur) binds at operator sites (Fur boxes) in the promoters of multiple iron-responsive genes, and represses their transcription [4]. In a few bacterial species, Fur seems to have a broader scope of regulation, acting also as a direct transcriptional activator [5–7] or as an apo-regulator (apo-Fur) [8, 9]. However, the most common Fur-mediated activation mechanism occurs indirectly via small regulatory RNAs (sRNA), such as RyhB in Escherichia coli[10], PrrF1 and PrrF2 in Pseudomonas aeruginosa[11], NrrF in Neisseria meningitidis[12] and FsrA in Bacillus subtilis[13]. In all these cases, the sRNAs inhibit the production of non-essential iron-using proteins under iron limitation, allowing relocation of the intracellular iron for essential proteins [14].
The Fur protein is the most widely found and best-studied iron-responsive regulator in bacteria from diverse taxonomic groups, such as subdivisions γ, β, δ and ϵ of proteobacteria and bacilli [4]. However, in α-proteobacteria iron regulation is still little studied and appears to be mediated by regulators different from Fur. Direct experimental data, available mostly to Rhizobiales, indicate that RirA and Irr are the master regulators of iron homeostasis while a Fur-like protein, named Mur, regulates only a manganese transporter [15, 16]. It has been suggested, based on bioinformatics and phylogenetic analyses, that RirA and Irr emerged as the main iron regulators in the common ancestor of the Rhizobiales and Rhodobacterales, whereas in more basal lineages of α-proteobacteria (Caulobacterales, Rhodospirillales and Sphingomonadales), Fur remained as the global iron regulator [17]. This in silico prediction was recently confirmed by experimental data for at least two α-proteobacteria, Caulobacter crescentus[6] and the magnetotactic bacterium Magnetospirillum gryphiswaldense[18, 19].
We have previously demonstrated, using an in silico approach combined with experimental data, that Fur controls iron homeostasis in C. crescentus by regulating many iron-responsive genes, and protect this freshwater oligotrophic bacterium from oxidative stress [6]. However, the response of C. crescentus to iron limitation and a comprehensive investigation of its Fur regulon remain to be determined on a global scale. In this work, we performed DNA microarray analysis to determine the transcriptional response of C. crescentus to iron availability, using wild-type cells growing under iron-replete versus iron-limiting conditions. We also used transcriptional profiling, comparing wild-type versus fur-mutant strains, to find novel members of the C. crescentus Fur regulon.
Results and discussion
Effect of iron and Fur on the C. crescentus transcriptome
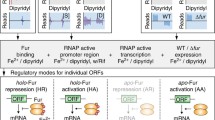
Whole genome transcriptional profiling using DNA microarrays were performed to identify iron-responsive and Fur-regulated genes in C. crescentus. Two sets of microarray experiments were conducted in duplicate using RNA samples prepared from two independent biological cultures. First, to define the C. crescentus iron limitation stimulon, we compared the transcriptome of wild-type cells treated with 100 μM FeSO4 (iron sufficiency) with that of wild-type cells treated with 100 μM 2,2-dipyridyl for 2 hours (iron limitation), an experimental condition previously established to study gene expression of iron-responsive genes in C. crescentus[6]. Iron limitation altered expression of 182 genes of which 108 were upregulated and 74 downregulated (Figure 1). Second, to identify Fur-regulated genes, we compared the transcriptome of wild-type cells with that of a fur mutant both cultivated in iron sufficiency. The expression of 121 genes was found to be significantly changed by the fur mutation (58 upregulated genes and 63 downregulated genes) (Figure 1).

Overview of iron-responsive and Fur-regulated genes in C. crescentus identified by microarray analyses. The Venn diagrams were constructed using the number of up- and down-regulated genes from experiments comparing wild type cells exposed to iron-limiting versus iron-replete conditions or comparing fur mutant strain versus wild type strain both in iron-replete condition. The complete set of the genes belonging to each group is listed in Tables 1, 2, 3, 4 and Additional file 1: Table S1. The upstream region of these genes (−200 to +50 bp relative to the start codon) were searched for sequence motifs using the MEME tool. A 19-pb palindromic motif, corresponding to the Fur binding site, was exclusively found in the group of genes regulated by both iron and Fur.
The up- and down-regulated genes, identified in these two microarray experiments, were compared to identify genes regulated by both iron limitation and fur mutation or genes affected by only one of these conditions (Figure 1). We found 42 genes upregulated both under iron limitation and in the fur mutant (Fe2+-Fur repressed genes) and 27 genes that were found to be downregulated on both these conditions (Fe2+-Fur activated genes), indicating that Fur has a major role on controlling expression of iron-responsive genes in C. crescentus. We also found many genes regulated exclusively in response to iron limitation, namely 66 upregulated genes and 47 downregulated genes, suggesting that the C. crescentus iron limitation stimulon is controlled by additional regulatory mechanisms.
Lastly, a group of genes showed differential expression in the fur mutant (16 up- and 36 downregulated genes) independent of iron availability (Figure 1; Additional file 1: Table S1). We were unable to determine whether these transcriptional changes are secondary effects or are mediated directly by Fur in an iron-independent manner. Nevertheless, the most upregulated genes in the fur mutant are the genes involved in transport (CC0859-60-61) and catabolism (CC1296, CC1298, CC1299 and CC1302) of myo-inositol in C. crescentus, belonging to the IolR regulon [20]. As expected, the level of fur mRNA (CC0057) was severely reduced in the fur mutant (7.4 fold). Interestingly, the sodB gene (CC3557) encoding an iron/manganese superoxide dismutase was 2.2-fold downregulated in the fur mutant (Additional file 1: Table S1), although its iron-dependent regulation verified in other bacteria [21] was not observed in our microarrays.
The repertoire of iron-responsive and Fur-regulated genes in C. crescentus
The genes regulated by both iron limitation and Fur are those showed in Tables 1 and 2 (in Figure 2A and 2B, genes indicated in blue). In addition to those genes, we observed that expression of some genes assumed as significantly up or downregulated under only one of the tested conditions changed to levels very close to our cutoff criterion (twofold change) in the other condition (Tables 3, 4 and Additional file 1: Table S1 and in Figure 2, genes indicated in orange). Thus, it is probable that these genes are also responsive to both iron levels and Fur, especially if one considers that most of them are in putative operons with genes whose expression was significantly changed under both iron limitation and fur mutation. Therefore, these genes were discussed here as part of the repertory of iron-responsive and Fur-regulated genes.

Genomic organization of the C. crescentus Fur regulon. The chromosomal clusters of the iron-responsive and Fur-regulated genes are organized in functional categories grouped in separate panels. Genes are also grouped as upregulated (A) or downregulated (B) under both iron limitation and fur mutation. The arrows indicate each open reading frame and their orientation on the chromosome. Differentially expressed genes are indicated in blue. Genes that have been experimentally shown to be directly regulated by Fur [6] are indicated in red. Selected genes that were either iron or Fur regulated (Tables 3, 4 and Additional file 1: Table S1) and showed expression change very close to our cutoff criterion on the other condition are shown in orange. Vertical blocks indicate the location of the Fur binding sites detected by the MEME search described in Figure 1, where sites predicted in silico are white and experimentally validated by EMSA are black [6].
The genes upregulated by both iron limitation and fur mutation (Fe2+-Fur repressed genes) were grouped into functional categories and according to their transcriptional organization in the chromosome (Table 1; Figure 2A). Many of these genes are organized in large clusters that contain at least one gene predicted to be involved in transport, implicating them in iron-acquisition associated functions (Figure 2A). These include four gene clusters containing TonB-dependent receptors, which are outer membrane proteins probably involved in Fe3+-siderophore acquisition (CC0028-27-26, CC0139, CC2194-95-96-97 and CC2928-27-26), the operon encoding the ferrous iron transporter FeoAB (CC0711-12) as well as two gene clusters encoding predicted ABC transporters (CC3692-93-94-95-96 and CC0683-84) and two gene clusters encoding hypothetical proteins that are putative transporters (CC2193-92-91 and CC3059-60-61-62-63) (these last two operons are discussed below). Although none of these putative transporters have been characterized yet, their high derepression by both iron limitation and fur mutation (Table 1) indicates that they could play a major role in the adaptation of C. crescentus to low-iron conditions. Unexpectedly, it has been shown, using hyper-saturated transposon mutagenesis, that feoAB is an essential operon in C. crescentus even for growth on rich media (iron sufficiency) [22], highlighting the vital role of iron acquisition in this bacterium.
In addition to these putative iron acquisition systems, a riboflavin biosynthesis operon (CC0885-86-87-88-89) as well as the bfd gene (CC3263) encoding a ferredoxin associated with bacterioferritin were upregulated by both iron limitation and fur mutation (Table 1; Figure 2A). It has been reported for Helicobacter pylori and Campylobacter jejuni that the production of riboflavin is also regulated by iron and Fur and secreted riboflavin has a role in Fe3+ reduction and hence in iron acquisition [23, 24]. Genes involved in oxidative stress response (CC0220), RNA processing (CC0835), transcriptional regulation (CC0884) and ammonia assimilation (CC1968-69) were also Fe2+-Fur repressed. A tight connection between iron homeostasis and nitrogen metabolism has been reported for the nitrogen-fixing cyanobacterium Anabaena sp. [25].
Finally, seven genes encoding hypothetical proteins were also upregulated by both iron limitation and fur mutation, of which two genes are of particular interest (CC0681 and CC0682). A previous report, based on tiled microarray analysis, suggested the existence of two candidate small regulatory RNAs (sRNAs) located in the intergenic regions between CC0680-CC0681 and C00681-CC0682, but attempts to validate these sRNAs by Northern blot allowed the detection of only a large transcript comprising all this region [26]. Considering that the putative operon CC0682-sRNA1-CC0681-sRNA2 was found to be Fe2+-Fur repressed in our microarray analyses (Table 1, Figure 2A) we are tempted to speculate that it could be processed under iron limitation, generating two sRNAs and two mRNAs translated to small proteins. These components could mediate the iron sparing response in C. crescentus, similarly to what was observed in Bacillus subtilis in which a sRNA (FsrA) and three small basic proteins (FbpA, FbpB e FbpC) act in conjunction to repress the expression of iron-rich proteins [13].
Additionally to these Fe2+-Fur repressed genes, our microarray analyses allowed us to identify the genes positively regulated by Fe2+-Fur, in other words, the genes that were downregulated by both iron limitation and fur mutation (Table 2; Figure 2B). As expected, many of these genes encode iron-containing enzymes. These included succinate dehydrogenase (sdh operon, CC3529-28-27-26-25), NADH ubiquinone oxidoreductase (nuo operon, CC1956-55-54-53-52-51-50), cytochromes (CC0762, CC1401 and CC2115), cytochrome P450 enzyme (CC2494), glutamate synthase (CC3607), a hypothetical protein predicted as catalase and a hypothetical protein with a ferritin-like domain (CC0556-57). This mechanism of repressing iron-rich enzymes to prioritize iron usage when this metal is scarce, sometimes referred as iron sparing response, has been described in many bacteria, such as E. coli[10, 21, 27], P. aeruginosa[11] and B. subtilis[13, 28].
Unexpectedly, a large number of genes encoding proteins involved in transport were also downregulated by both iron limitation and fur mutation (Table 2; Figure 2B). Among these, there are transporters belonging to the major facilitator superfamily (MFS) (CC1628, CC2485-86), porins (CC0925 and CC1409) and many TonB-dependent receptors. At least six of these genes (CC3336, CC3161, CC3461, CC0991, CC2804 and CC2485) are also highly induced by carbon limitation [29] and are positively regulated by CfrA, a sRNA that regulates adaptation to carbon starvation in C. crescentus[30]. Although the reason for these genes to be repressed by iron limitation and induced by carbon starvation is still not clear, it is reasonable to suppose that these TonB-dependent receptors are required for uptake of carbohydrates instead of Fe3+-siderophore complexes, since it has recently been shown that novel substrates, such as nickel and different carbohydrates, are transported via TonB-dependent receptors [31].
Importantly, three genes (fixK, fixT and ftrB) encoding regulatory proteins that specify an oxygen signaling network required for C. crescentus growth under hypoxia [32] were found to be downregulated by both iron limitation and fur mutation (Table 2; Figure 2B). The C. crescentus Fix signaling system consists of the sensor histidine kinase FixL (a heme-binding oxygen sensor), its cognate response regulator FixJ, the transcriptional regulator FixK, and the kinase inhibitor FixT (the core FixLJ–FixK–FixT), besides the downstream regulators FtrA and FtrB [32]. Consistent with downregulation of fixK, many hypoxia-dependent FixK-activated genes containing a FixK binding site [32], were also downregulated by both iron limitation and fur mutation, including CC1409 (ompW), CC1410 (ftrB), CC0762 (cydA), CC1401 (ccoN), CC0753 (fixT), CC2115 and CC0277 (Table 2; Figure 2B). Therefore, the FixK-dependent hypoxia stress response seems to be positively regulated by Fe2+-Fur under iron sufficiency and repressed in iron limitation condition, similarly to what was described for the anaerobic regulator Fnr in E. coli[21] and Salmonella enterica serovar Typhimurium[33]. The regulatory link between oxygen and iron availability could be mediated by the histidine kinase FixL that senses oxygen through its heme-containing amino-terminal PAS domain [32].
To further discriminate whether regulation by Fur was direct or indirect, we conducted in silico searches in the upstream region of all up- and down-regulated genes identified in the microarray experiments (Figure 1). MEME-based analyses, including all genes together or each group of genes separately, identified a motif very similar to the Fur binding site previously detected in C. crescentus[6]. These Fur binding sites were detected only for genes regulated by both iron and Fur (Figure 1). As indicated in Figure 2, sixteen Fur binding sites were identified in the group of the genes upregulated by both iron limitation and fur mutation, indicating that most of these genes (37 out of 47 genes) are direct target for Fur-mediated repression. In contrast, only three Fur binding sites were detected in the group of the genes downregulated by both iron limitation and fur mutation, suggesting that Fur indirectly mediates positive regulation of many genes, in addition to the direct positive regulation previously demonstrated [6].
Fur-independent regulation of C. crescentus iron-responsive genes
In addition to the Fur modulon iron limitation also affected the C. crescentus transcriptome in a Fur-independent manner, given that 66 genes were upregulated (Table 3) and 47 genes were downregulated (Table 4) during growth in iron-limitation condition that were not affected by the fur mutation (Figure 1).
Among the genes strongly upregulated exclusively in response to iron limitation there is a large gene cluster (CC1866-65-64-63-62-61-60-59-58-57), which encodes the transcriptional repressor IscR (CC1866) and enzymes of the Suf system of Fe-S cluster biogenesis (Table 3). E. coli possesses two operons implicated in Fe-S cluster assembly, iscRSUA-hscBA-fdx, encoding the housekeeping Fe-S cluster biogenesis pathway and sufABCDSE, which synthesize Fe-S clusters under iron limitation or oxidative stress conditions [34,35), whereas C. crescentus appears to have only one operon that contains a combination of isc (CC1866-65, iscRS) and suf (CC1864-62-61-60, sufBCDS) genes. In E. coli both isc and suf operons are induced by iron limitation and oxidative stress, but while the isc genes are regulated by IscR, the suf genes are under control of OxyR and Fur [21, 34–36]. In C. crescentus upregulation of this large operon by iron limitation is Fur-independent and we postulate that it could be mediated by IscR via an IscR binding site previously predicted upstream of the CC1866 gene [17]. Because IscR senses damage to the Fe-S clusters of the cell, it is possible that iron limitation is generating some kind of stress in C. crescentus which is able to damage Fe-S clusters.
In agreement with this assumption, many of the genes upregulated exclusively by iron limitation are related to various stress responses (Table 3) and were found to be induced when C. crescentus was submitted to heavy metal stress [37]. Among the genes induced by both iron limitation and heavy metal stress (mainly cadmium stress), there are those related to oxidative stress defense (CC0141, CC0994, CC1316), detoxification efflux pumps (CC3195, CC3197), DNA repair (CC2590) and nucleotide biosynthesis (CC0260, CC3492) (Table 3). Interestingly, 12 heat shock genes, encoding chaperones, proteases and small heat shock proteins, were also upregulated by iron limitation, as well as some genes encoding peptidases containing metals as cofactors (Table 3), what is consistent with previous observations in Shewanella oneidensis[38]. Induction of these genes might be directly mediated by the heat shock sigma factor RpoH (σ32), for the reason that the own rpoH gene (CC3098) is upregulated in iron limitation (Table 3). Moreover, a predicted σ32-binding motif (m_6 motif), which has been identified upstream of cadmium-induced genes [39], was found here upstream of nearly half (15 sites upstream of 30 genes/operons) of the 63 genes upregulated in iron limitation (Table 3), indicating induction of the RpoH regulon by iron limitation. The C. crescentus rpoH gene is transcribed from two promoters, a σ70-dependent P1 promoter and a heat shock autoregulated σ32-dependent P2 promoter [40]. It remains to be determined how these different signals (cadmium stress and iron limitation) could increase transcription of rpoH in C. crescentus, activating its regulon.
When the genes downregulated exclusively in iron limitation are grouped into functional categories, the most prominent groups of genes are involved in amino acid metabolism, chemotaxis and motility, and energy metabolism (Table 4). Among the enzymes of amino acid biosynthesis pathways repressed by iron limitation there are many involved in methionine biosynthesis, such as methionine synthases (CC0482, CC2137, CC2138), adenosylmethionine synthtase (CC0050), S-adenosyl-L-homocysteine hydrolase (CC0257) and methylenetetrahydrofolate reductase (CC2140), which is required to produce 5-methyltetrahydrofolate as methyl-group donor for methionine synthesis. Pathways of protein catabolism were also repressed by iron limitation as revealed by downregulation of many genes encoding peptidases (CC0167, CC0984, CC1048, CC2480 and CC3246) (Table 4). Furthermore, some genes for flagella assembly (CC0901-02, CC1456) and chemotaxis (CC0430-31-32-33, CC1399 and CC2847) were downregulated in iron limitation. Repression of motility and chemotaxis genes by iron limitation has been described in Sinorhizobium meliloti[41] and Acinetobacter baumannii[42]. Finally, some known Fe2+-Fur activated genes [6, 13] were downregulated in iron limitation, but not in the fur mutant in this work. Of these, there are genes encoding the Fe-S clusters-containing enzymes aconitate hydratase (CC3667), NADH ubiquinone oxidoreductase (nuo genes CC1946, CC1944-43-42), glutamate synthase (CC3606) and dihydroxy-acid dehydratase (CC3044) (Table 4). In some cases, at least part of the operons (nuo and CC3607) was downregulated by both iron limitation and fur mutation (Figure 2B). A possible explanation is that the Fe2+-Fur activated genes showed modest differential expression (approximately 2 fold) (Table 2), thus small experimental fluctuations could exclude some genes based on our cutoff criteria for differential expression in the microarray analyses.
Comparing our microarray data with other large-scale transcriptomic studies performed under iron-limiting condition in bacteria from diverse taxonomic groups [21, 28, 38, 41, 42], we observed that, in spite of the multiplicity of regulatory mechanisms, the core of iron-regulated genes is extremely conserved, including mainly those related to transport, use and storage of this metal. Some responses seems to be confined to few bacteria, such as upregulation of the heat shock response, also described in S. oneidensis[38] and downregulation of chemotaxis and motility, observed in S. meliloti[41] and A. baumannii[42]. However, our study expands the range of genes involved in iron homeostasis when we consider physiological processes unique to the C. crescentus lifestyle, such as adaptation to growing in oligotrophic environments and under different oxygen tensions. In fact, many TonB-dependent receptors, predicted to be required for sugar transport, and the hypoxia FixK regulon were surprisingly downregulated by both iron limitation and fur mutation.
Verification of iron- and Fur-dependent expression of the CC2193 and CC3059 operons
Nearly all of the genes previously identified as members of the C. crescentus Fur regulon [6] were found to be differentially expressed by microarray analyses (Figure 2, red arrows), validating the experimental procedure. To further confirm our microarray data, we selected genes located in two clusters that encode putative transporters for validation by β-galactosidase activity assays and EMSA. The first cluster (CC2193-92-91) encodes a hypothetical protein containing an EF hand motif (CC2193), a putative glutathione peroxidase (CC2192) and a hypothetical protein (CC2191). The CC2193 gene appears to have been incorrectly annotated in the CB15 strain given that in the chromosome of the C. crescentus NA1000 strain, recently sequenced [43], two open reading frames were annotated in this region, CCNA02274 (encoding a shorter EF hand protein) and CCNA02275, encoding a hypothetical protein with a domain of unknown function (DUF4198). The second cluster (CC3059-60-61-62-63) contains three genes encoding a putative transporter (CC3059-60-61), and two genes involved in iron-related functions (sulfite reductase iron-flavoprotein and Fe-S cluster repair protein) (Figure 2; Table 1). Interestingly, the genes of these two clusters most highly upregulated in iron limitation and fur mutant (CC2193-corresponding to CCNA02275 in NA1000, and CC3061) (Table 1) encode two paralogous proteins belonging to the widespread Pfam family DUF4198. Although the proteins of this family are widely distributed in various groups of bacteria (750 sequences in 486 species, Pfam database February 2013), nothing is known about their function or regulation.
The promoter regions of CC2193 and CC3059 were cloned in a lacZ reporter plasmid and the constructions were introduced into the wild type and fur mutant strains. Beta-galactosidase activity assays indicated that the expression of these two genes was induced under iron limitation and derepressed in the fur mutant strain, validating the microarray data (Figure 3A, Table 1). To verify whether Fur acts as repressor by directly binding upstream of the CC2193-92-91 and CC3059-60-61-62-63 gene clusters (Figure 2) we performed gel mobility shift assays using purified Fur protein (Figure 3B). Fur bound to the probes corresponding to the promoter region of CC2193 (data not shown,[6]) as well as to that of CC3059, validating the Fur binding sites found overlapping the −35 and −10 promoter elements of these two genes (Figure 3B). These data illustrate the high performance of the microarray analysis to identify unknown genes with potential functions in iron acquisition and new members of the Fur regulon. For instance, the high derepression of CC2193 and CC3059 by iron limitation and fur mutation suggests that these two members of a widespread family of proteins with unknown function (DUF4198) could be involved in transport or signaling in response to iron limitation.

The CC2193 and CC3059 operons are members of the C. crescentus Fur regulon. (A) Promoter activities of the CC2193 and CC3059 operons in response to iron and Fur. Wild type (NA1000) and fur mutant (∆fur) strains containing plasmids pLAC2193 or pLAC3059 were grown in PYE medium and treated with 100 μM FeSO4 (Fe) or 100 μM 2,2-dipyridyl (DP) for two hours. The β-galactosidase activity generated by these lacZ fusions was determined. The experiments were performed in duplicate from three independent biological cultures. (B) Fur binds directly to the promoter of the CC3059 operon. EMSAs were performed using the purified His-Fur protein and a probe containing the promoter region of CC3059. The 32P-labeled probe was incubated with increasing concentrations of protein (0, 50, 200, 500 and 1000 nM) (left). A competition assay using 250 nM Fur and the labeled CC3059 probe was performed, where binding of Fur was challenged with a 30-fold excess of unlabeled DNA fragments of the same region (SE) or the 16S rRNA coding region (SI) as competitors (right). Below is shown the promoter region of the CC3059 operon, indicating the previously identified transcriptional start site (+1) and conserved −35 and −10 sequences of Caulobacter σ70 promoters (TTGAC-16 bp-G/CCTANA) [39]. The initiation codon (GTG) is underlined. The Fur binding site predicted in silico is shaded.
Conclusions
Using DNA microarray analyses, we have defined the global transcriptional response of Caulobacter crescentus to iron availability, providing an overview of the physiological strategies that this oligotrophic α-proteobacterium employs for survival in iron limiting conditions (Figure 4). Our data reveal that the iron stimulon in C. crescentus is larger than the Fur regulon previously identified [6], involving a more complex regulatory network. Among the responses mediated by Fur it is worth pointing out the upregulation of genes involved in iron acquisition systems and biosynthesis of riboflavin in iron limiting condition, as well as the downregulation of genes encoding many iron-using enzymes involved in energy metabolism (Figure 4). Fur binding site prediction suggests that Fur acts mainly as a direct transcriptional repressor, whereas positive regulation could be mediated either directly by Fur in a few cases or indirectly for most genes. In many cases this indirect effect was provoked by downregulation of the hypoxia regulator FixK, causing decreased expression of FixK-activated genes in iron limitation condition (Figure 4). Other genes could be indirectly activated by Fur via an unidentified iron-responsive sRNA. While the Fe2+-Fur mediated repression of some genes encoding TonB-dependent receptors confirmed our previous data [6], the Fe2+-Fur mediated activation of many other TonB-dependent receptors putatively associated with sugar transport was unexpected. Fur-independent regulation of C. crescentus iron-responsive genes was also observed, indicating an overlap with other regulatory pathways (Figure 4). It is worth mentioning that iron limitation caused upregulation of the heat shock sigma factor RpoH with consequent activation of its regulon, and upregulation of the IscR regulon, whose genes are involved in Fe-S cluster biogenesis. Since most of these iron-responsive genes identified in this work have not been experimentally investigated in C. crescentus, they are good targets for future studies.

Schematic representation of the main changes in gene expression and cell processes under iron-limiting conditions. Upregulated genes and pathways are shown in red, downregulated are shown in green. Large arrows indicate activation and blunt-head lines indicate repression by the respective transcription regulator (Fur is represented as iron-bound). Thin arrows indicate enzyme reactions. Traced arrow indicates activation of the enzyme GlnB by GlnD via uridylylation. L-hCys: L-homocysteine, SAM: S-adenosylmethionine, SAhC: S-adenosylhomocysteine.
Methods
Bacterial strains and growth conditions
Caulobacter crescentus, also known as Caulobacter vibrioides[44], strains NA1000 (wild-type) [45] and SP0057 (fur mutant) [6] were grown aerobically at 30°C in peptone-yeast extract (PYE) medium [46]. Iron-replete and iron-limiting conditions were achieved by supplementing PYE medium with 100 μM FeSO4 and 100 μM 2,2-dipyridyl (DP) (Sigma), respectively. Plasmids were introduced into C. crescentus by conjugation with Escherichia coli strain S17-1. E. coli was grown at 37°C in LB medium supplemented with ampicillin (100 μg ml-1) or tetracycline (12.5 μg ml-1) as required. His-Fur protein was purified after overexpression in E. coli DH5α as described [6].
Microarray analysis
For the DNA microarray experiments, overnight C. crescentus cultures were diluted to an optical density at 600 nm (OD600) of 0.1 in 35 ml of PYE medium. Cells were grown up to midlog phase (OD600 ~ 0.5) and the cultures were divided and treated with either 100 μM FeSO4 (iron sufficiency) or 100 μM DP (iron limitation). The incubation was continued for two hours prior to RNA isolation as previously described [6]. Total RNA was extracted using Trizol Reagent (Invitrogen), according to the manufacturer’s instructions. RNA samples were treated with RNase-free DNase I (Fermentas) to digest residual chromosomal DNA and then precipitated using sodium acetate/ethanol prior to spectrophotometric quantification and visualization on formaldehyde-agarose gels. RNA samples were isolated from two independent bacterial cultures for each strain or condition analyzed as biological replicates. Amino allyl modified cDNA was generated by reverse transcription from 20 μg of total RNA and labeled with either Cy3 or Cy5 mono-reactive fluorescent dyes using the FairPlay III Microarray Labeling System (Stratagene). Labeled cDNA samples were hybridized to a custom-designed DNA oligo microarray (Agilent) (each gene is covered by 9–11 probes located −300 to +200 relative to the translational start site) using a protocol previously described [47, 48]. The arrays were scanned for the Cy3 and Cy5 fluorescent signals with an Agilent High Resolution Microarray Scanner. Data extraction and normalization was performed with the Feature Extraction Software 9.0 (Agilent). A gene was considered as upregulated or downregulated if it showed 2-fold change relative to the control considering at least three out of four last probes (that are downstream of the translational start site) in both biological replicates. The values for the relative expression of each gene were obtained as the average of the four last probes. The microarray data have been deposited in the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE45653.
Fur binding site detection
Bioinformatics analyses were performed using the Multiple Em for Motif Elicitation (MEME) tool [49] to identify motifs within the promoter regions of iron regulated genes. Putative gene regulatory regions (−200 to +50 bp relative to the start codon) were searched using the following parameters: motifs size from 6 to 50 bp; zero or one motif per sequence; search given strand only; palindromic and nonpalindromic models were tested. Sequence logos were generated using WebLogo [50].
lacZ fusions and ß-Galactosidase assays
DNA fragments covering the promoter regions of CC2193 (193 bp) and CC3059 (183 bp) were PCR-amplified using primer pairs CC2193-fw (5'-TGGATCCCGGCGAGTTTCAGGCGCGAC-3')/CC2193-rv (5'-TAAGCTTACGGATCATTGGACAAACCC-3') and CC3059-fw (5'-TGGATCCAGTTGACGGCGCAATAGGCC-3')/CC3059-rv (5'-TAAGCTTGCGGCGGCGGATTTCACAGG-3'), respectively. These PCR products were cloned into pGEM-T Easy, sequenced and subcloned as BamH I/Hind III fragments into the reporter vector pRKlacZ 290 [51], resulting in plasmids pLAC2193 and pLAC3059. These constructs were introduced into C. crescentus NA1000 and SP0057 strains by conjugation. Cultures were grown in PYE medium up to mid-log phase, divided into two flasks, and treated with either 100 μM FeSO4 or 100 μM DP for two hours. The ß-galactosidase activity from these strains was determined colorimetrically using o-nitrophenyl-ß-D-galactoside (ONPG) as substrate [52].
Electrophoretic mobility shift assay (EMSA)
A probe corresponding the promoter region of CC3059 (the same 183 bp- fragment used in lacZ fusion) was obtained by PCR amplification and was end-labeled with [γ32P]-ATP using T4 polynucleotide kinase (Invitrogen). For competition assay, a 101-bp 16S rRNA intragenic fragment was PCR-amplified using the primers 16SA-fw (5'-CCGCGTGAATGATGAAGGTC-3') and 16SA-rv (5'-GCTGCTGGCACGAAGTTAGC-3'). For EMSA, purified His-Fur protein and labeled DNA probes were incubated in binding buffer exactly as previously described [6].
References
Andrews SC, Robinson AK, Rodriguez-Quinones F: Bacterial iron homeostasis. FEMS Microbiol Rev. 2003, 27: 215-237. 10.1016/S0168-6445(03)00055-X.
Schaible UE, Kaufmann SHE: Iron and microbial infection. Nat Rev Microbiol. 2004, 2: 946-953. 10.1038/nrmicro1046.
Imlay JA: Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem. 2008, 77: 755-776. 10.1146/annurev.biochem.77.061606.161055.
Lee JW, Helmann JD: Functional specialization within the Fur family of metalloregulators. Biometals. 2007, 20: 485-499. 10.1007/s10534-006-9070-7.
Delany I, Rappuoli R, Scarlato V: Fur functions as an activator and as a repressor of putative virulence genes in Neisseria meningitidis. Mol Microbiol. 2004, 52: 1081-1090. 10.1111/j.1365-2958.2004.04030.x.
da Silva Neto JF, Braz VS, Italiani VCS, Marques MV: Fur controls iron homeostasis and oxidative stress defense in the oligotrophic alpha-proteobacterium Caulobacter crescentus. Nucleic Acids Res. 2009, 37: 4812-4825. 10.1093/nar/gkp509.
Yu C, Genco CA: Fur-mediated activation of gene transcription in the human pathogen Neisseria gonorrhoeae. J Bacteriol. 2012, 194: 1730-1742. 10.1128/JB.06176-11.
Carpenter BM, Whitmire JM, Merrell DS: This is not your mother's repressor: the complex role of Fur in pathogenesis. Infect Immun. 2009, 77: 2590-2601. 10.1128/IAI.00116-09.
Butcher J, Sarvan S, Brunzelle JS, Couture JF, Stintzi A: Structure and regulon of Campylobacter jejuni ferric uptake regulator Fur define apo-Fur regulation. Proc Natl Acad Sci USA. 2012, 109: 10047-10052. 10.1073/pnas.1118321109.
Masse E, Gottesman S: A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc Natl Acad Sci USA. 2002, 99: 4620-4625. 10.1073/pnas.032066599.
Wilderman PJ, Sowa NA, FitzGerald DJ, FitzGerald PC, Gottesman S, Ochsner UA, Vasil ML: Identification of tandem duplicate regulatory small RNAs in Pseudomonas aeruginosa involved in iron homeostasis. Proc Natl Acad Sci USA. 2004, 101: 9792-9797. 10.1073/pnas.0403423101.
Mellin JR, Goswami S, Grogan S, Tjaden B, Genco CA: A novel Fur- and iron-regulated small RNA, NrrF, is required for indirect Fur-mediated regulation of the sdhA and sdhC genes in Neisseria meningitidis. J Bacteriol. 2007, 189: 3686-3694. 10.1128/JB.01890-06.
Gaballa A, Antelmann H, Aguilar C, Khakh SK, Song KB, Smaldone GT, Helmann JD: The Bacillus subtilis iron-sparing response is mediated by a Fur-regulated small RNA and three small, basic proteins. Proc Natl Acad Sci USA. 2008, 105: 11927-11932. 10.1073/pnas.0711752105.
Masse E, Salvail H, Desnoyers G, Arguin M: Small RNAs controlling iron metabolism. Curr Opin Microbiol. 2007, 10: 140-145. 10.1016/j.mib.2007.03.013.
Rudolph G, Hennecke H, Fischer HM: Beyond the Fur paradigm: iron-controlled gene expression in rhizobia. FEMS Microbiol Rev. 2006, 30: 631-648. 10.1111/j.1574-6976.2006.00030.x.
Johnston AW, Todd JD, Curson AR, Lei S, Nikolaidou-Katsaridou N, Gelfand MS, Rodionov DA: Living without Fur: the subtlety and complexity of iron-responsive gene regulation in the symbiotic bacterium Rhizobium and other alpha-proteobacteria. Biometals. 2007, 20: 501-511. 10.1007/s10534-007-9085-8.
Rodionov DA, Gelfand MS, Todd JD, Curson AR, Johnston AW: Computational reconstruction of iron- and manganese-responsive transcriptional networks in alpha-proteobacteria. PLoS Comput Biol. 2006, 2: e163-10.1371/journal.pcbi.0020163.
Uebe R, Voigt B, Schweder T, Albrecht D, Katzmann E, Lang C, Böttger L, Matzanke B, Schüler D: Deletion of a fur-like gene affects iron homeostasis and magnetosome formation in Magnetospirillum gryphiswaldense. J Bacteriol. 2010, 192: 4192-4204. 10.1128/JB.00319-10.
Qi L, Li J, Zhang W, Liu J, Rong C, Li Y, Wu L: Fur in Magnetospirillum gryphiswaldense influences magnetosomes formation and directly regulates the genes involved in iron and oxygen metabolism. PLoS ONE. 2012, 7: e29572-10.1371/journal.pone.0029572.
Boutte CC, Srinivasan BS, Flannick JA, Novak AF, Martens AT, Batzoglou S, Viollier PH, Crosson S: Genetic and computational identification of a conserved bacterial metabolic module. PLoS Genet. 2008, 4: e1000310-10.1371/journal.pgen.1000310.
McHugh JP, Rodriguez-Quinones F, Abdul-Tehrani H, Svistunenko DA, Poole RK, Cooper CE, Andrews SC: Global iron-dependent gene regulation in Escherichia coli. A new mechanism for iron homeostasis. J Biol Chem. 2003, 278: 29478-29486. 10.1074/jbc.M303381200.
Christen B, Abeliuk E, Collier JM, Kalogeraki VS, Passarelli B, Coller JA, Fero MJ, McAdams HH, Shapiro L: The essential genome of a bacterium. Mol Syst Biol. 2011, 7: 528-
Worst DJ, Gerrits MM, Vandenbrouke-Grauls CM, Kusters JG: Helicobacter pylori ribBA-mediated riboflavin production is involved in iron acquisition. J Bacteriol. 1998, 180: 1473-1479.
Crossley RA, Gaskin DJ, Holmes K, Mulholland F, Wells JM, Kelly DJ, van Vliet AH, Walton NJ: Riboflavin biosynthesis is associated with assimilatory ferric reduction and iron acquisition by Campylobacter jejuni. Appl Environ Microbiol. 2007, 73: 7819-7825. 10.1128/AEM.01919-07.
López-Gomollón S, Hernández JA, Pellicer S, Angarica VE, Peleato ML, Fillat MF: Cross-talk between iron and nitrogen regulatory networks in Anabaena (Nostoc) sp. PCC 7120: identification of overlapping genes in FurA and NtcA regulons. J Mol Biol. 2007, 374: 267-281. 10.1016/j.jmb.2007.09.010.
Landt SG, Abeliuk E, McGrath PT, Lesley JA, McAdams HH, Shapiro L: Small non-coding RNAs in Caulobacter crescentus. Mol Microbiol. 2008, 68: 600-614. 10.1111/j.1365-2958.2008.06172.x.
Masse E, Vanderpool CK, Gottesman S: Effect of RyhB small RNA on global iron use in Escherichia coli. J Bacteriol. 2005, 187: 6962-6971. 10.1128/JB.187.20.6962-6971.2005.
Baichoo N, Wang T, Ye R, Helmann JD: Global analysis of the Bacillus subtilis Fur regulon and the iron starvation stimulon. Mol Microbiol. 2002, 45: 1613-1629. 10.1046/j.1365-2958.2002.03113.x.
England JC, Perchuk BS, Laub MT, Gober JW: Global regulation of gene expression and cell differentiation in Caulobacter crescentus in response to nutrient availability. J Bacteriol. 2010, 192: 819-833. 10.1128/JB.01240-09.
Landt SG, Lesley JA, Britos L, Shapiro L: CrfA, a small noncoding RNA regulator of adaptation to carbon starvation in Caulobacter crescentus. J Bacteriol. 2010, 192: 4763-4775. 10.1128/JB.00343-10.
Schauer K, Rodionov DA, de Reuse H: New substrates for TonB-dependent transport: do we only see the ‘tip of the iceberg’?. Trends Bioch Sci. 2008, 33: 330-338. 10.1016/j.tibs.2008.04.012.
Crosson S, McGrath PT, Stephens C, McAdams HH, Shapiro L: Conserved modular design of an oxygen sensory/signaling network with species-specific output. Proc Natl Acad Sci USA. 2005, 102: 8018-8023. 10.1073/pnas.0503022102.
Troxell B, Fink RC, Porwollik S, McClelland M, Hassan HM: The Fur regulon in anaerobically grown Salmonella enterica sv. Typhimurium: identification of new Fur targets. BMC Microbiol. 2011, 11: 236-
Outten FW, Djaman O, Storz G: A suf operon requirement for Fe-S cluster assembly during iron starvation in Escherichia coli. Mol Microbiol. 2004, 52: 861-872. 10.1111/j.1365-2958.2004.04025.x.
Py B, Barras F: Building Fe-S proteins: bacterial strategies. Nat Rev Microbiol. 2010, 8: 436-446. 10.1038/nrmicro2356.
Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G: DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J Bacteriol. 2001, 183: 4562-4570. 10.1128/JB.183.15.4562-4570.2001.
Hu P, Brodie EL, Suzuki Y, McAdams HH, Andersen GL: Whole-genome transcriptional analysis of heavy metal stresses in Caulobacter crescentus. J Bacteriol. 2005, 187: 8437-8449. 10.1128/JB.187.24.8437-8449.2005.
Yang Y, Harris DP, Luo F, Xiong W, Joachimiak M, Wu L, Dehal P, Jacobsen J, Yang Z, Palumbo AV, Arkin AP, Zhou J: Snapshot of iron response in Shewanella oneidensis by gene network reconstruction. BMC Genomics. 2009, 10: 131-10.1186/1471-2164-10-131.
McGrath PT, Lee H, Zhang L, Iniesta AA, Hottes AK, Tan MH, Hillson NJ, Hu P, Shapiro L, McAdams HH: High-throughput identification of transcription start sites, conserved promoter motifs and predicted regulons. Nat Biotechnol. 2007, 25: 584-592. 10.1038/nbt1294.
Wu J, Newton A: The Caulobacter heat shock sigma factor gene rpoH is positively autoregulated from a sigma32-dependent promoter. J Bacteriol. 1997, 179: 514-521.
Chao TC, Buhrmester J, Hansmeier N, Pühler A, Weidner S: Role of the regulatory gene rirA in the transcriptional response of Sinorhizobium meliloti to iron limitation. Appl Environ Microbiol. 2005, 71: 5969-5982. 10.1128/AEM.71.10.5969-5982.2005.
Eijkelkamp BA, Hassan KA, Paulsen IT, Brown MH: Investigation of the human pathogen Acinetobacter baumannii under iron limiting conditions. BMC Genomics. 2011, 12: 126-10.1186/1471-2164-12-126.
Marks ME, Castro-Rojas CM, Teiling C, Du L, Kapatral V, Walunas TL, Crosson S: The genetic basis of laboratory adaptation in Caulobacter crescentus. J Bacteriol. 2010, 192: 3678-3688. 10.1128/JB.00255-10.
Abraham WR, Strömpl C, Meyer H, Lindholst S, Moore ER, Christ R, Vancanneyt M, Tindall BJ, Bennasar A, Smit J, Tesar M: Phylogeny and polyphasic taxonomy of Caulobacter species. Proposal of Maricaulis gen. nov. with Maricaulis maris (Poindexter) comb. nov. as the type species, and emended description of the genera Brevundimonas and Caulobacter. Int J Syst Bacteriol. 1999, 49: 1053-1073. 10.1099/00207713-49-3-1053.
Evinger M, Agabian N: Envelope-associated nucleoid from Caulobacter crescentus stalked and swarmer cells. J Bacteriol. 1977, 132: 294-301.
Ely B: Genetics of Caulobacter crescentus. Methods Enzymol. 1991, 204: 372-384.
Lourenço RF, Kohler C, Gomes SL: A two-component system, an anti-sigma factor and two paralogous ECF sigma factors are involved in the control of general stress response in Caulobacter crescentus. Mol Microbiol. 2011, 80: 1598-1612. 10.1111/j.1365-2958.2011.07668.x.
Kohler C, Lourenço RF, Avelar GM, Gomes SL: Extracytoplasmic function (ECF) sigma factor σF is involved in Caulobacter crescentus response to heavy metal stress. BMC Microbiology. 2012, 12: 210-10.1186/1471-2180-12-210.
Bailey TL, Elkan C: Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. 1994, Menlo Park, CA: AAAI Press, 28-36.
Crooks GE, Hon G, Chandonia JM, Brenner SE: WebLogo: a sequence logo generator. Genome Res. 2004, 14: 1188-1190. 10.1101/gr.849004.
Gober JW, Shapiro L: A developmentally regulated Caulobacter flagellar promoter is activated by 3' enhancer and IHF binding elements. Mol Biol Cell. 1992, 3: 913-916. 10.1091/mbc.3.8.913.
Miller JH: Experiments in Molecular Genetics. 1972, Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press
Acknowledgements
We are grateful to Michael T. Laub for making the C. crescentus DNA microarray slides available and Carla Rosenberg lab for assistance with the microarray scanning. This work was supported by grant 470663/2011-1 from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). During the course of this work, JFSN and RFL were supported by postdoctoral fellowships, grants 2007/56306-0 and 2008/52874-6, from São Paulo Research Foundation (FAPESP). MVM is partly supported by CNPq.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interest.
Authors’ contributions
JFSN and MVM planned the experiments; JFSN performed the experimental work and wrote the manuscript; JFSN and RFL analyzed the microarray data; MVM participated in study design and coordination and helped to prepare the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2013_5267_MOESM1_ESM.docx
Additional file 1: Table S1: Differentially expressed genes in the fur mutant but not affected by iron limitation. (DOCX 19 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
da Silva Neto, J.F., Lourenço, R.F. & Marques, M.V. Global transcriptional response of Caulobacter crescentus to iron availability. BMC Genomics 14, 549 (2013). https://doi.org/10.1186/1471-2164-14-549
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-549