
Abstract
Background
Foxtail millet (Setaria italica (L.) P. Beauv.), one of the most ancient domesticated crops, is becoming a model system for studying biofuel crops and comparative genomics in the grasses. However, knowledge on the level of genetic diversity and linkage disequilibrium (LD) is very limited in this crop and its wild ancestor, green foxtail (Setaria viridis (L.) P. Beauv.). Such information would help us to understand the domestication process of cultivated species and will allow further research in these species, including association mapping and identification of agricultural significant genes involved in domestication.
Results
In this study, we surveyed DNA sequence for nine loci across 50 accessions of cultivated foxtail millet and 34 of its wild progenitor. We found a low level of genetic diversity in wild green foxtail (θ = 0.0059), θ means Watterson's estimator of θ. Despite of a 55% loss of its wild diversity, foxtail millet still harbored a considerable level of diversity (θ = 0.0027) when compared to rice and sorghum (θ = 0.0024 and 0.0034, respectively). The level of LD in the domesticated foxtail millet extends to 1 kb, while it decayed rapidly to a negligible level within 150 bp in wild green foxtail. Using coalescent simulation, we estimated the bottleneck severity at k = 0.6095 when ρ/θ = 1. These results indicated that the domestication bottleneck of foxtail millet was more severe than that of maize but slightly less pronounced than that of rice.
Conclusions
The results in this study establish a general framework for the domestication history of foxtail millet. The low level of genetic diversity and the increased level of LD in foxtail millet are mainly caused by a population bottleneck, although gene flow from foxtail millet to green foxtail is another factor that may have shaped the pattern of genetic diversity of these two related gene pools. The knowledge provided in this study will benefit future population based studies in foxtail millet.
Similar content being viewed by others


Background
Plant domestication, which began approximately 10,000 years ago, is the most crucial development in human history [1]. Domesticated crops provide most of our food today, and provide the foundation for human civilization. Yet, only a small fraction of flowering plants were actually domesticated. It will be of great benefit for future crop breeding and improvement if we have a better understanding of the domestication process.
The evolutionary footprints left by domestication at the population level are dispersed throughout the genome. Due to population bottlenecks, a large proportion of genetic diversity is typically lost during the domestication process, as seen by a 25% reduction of diversity in maize and an 80% reduction in rice [2, 3]. In some species like rice and sunflower, the transition of mating system from outcrossing to self-fertilization can further reduce the levels of genetic diversity [4, 5]. Furthermore, some selected loci exhibit a more dramatic loss in genetic diversity [6]. In maize, the reduction in genetic diversity is less than 20% for presumably neutral loci like hm1, hm2, glb1, and sh1, but up to 80% for selected loci like c1, ae1 and tb1[7–11]. Elevated levels of linkage disequilibrium (LD) are also observed in the genome of domesticated plants [3, 6, 12]. This is caused by either the domestication bottleneck and/or reduction of effective recombination rate due to self-fertilization [5]. Additionally, selection can have a similar effect on LD in the target region [13]. Studies that focus on the level and organization of genetic variation in these major crops are essential for our understanding of the process of domestication and are instructive for crop improvement or other research that is based on these population parameters, such as association mapping [2, 12, 14].
To study the patterns of genetic diversity within and between populations, as well as to trace the demographic history of crops and their wild relatives, multilocus surveys of population sequence data have been widely used in recent years [3, 6, 12, 15]. Since selection acts on some but not all genes in genome, a multilocus sample increases the probability that both loci that are under selection and those that are neutrally-evolving will be sampled. Multi-locus sampling is also necessary to understand the demographic history of populations, and enables targets of natural or artificial selection to be more reliably identified. Many studies have been carried out to investigate nucleotide diversity in plants, yet only a few have been focused on their demographic history. Understanding the demographic history of populations will also help in interpretation of population genetic neutrality tests, which are based on the comparison of observed and expected polymorphism patterns under the neutral equilibrium model (NE) [16, 17]. This model assumes random outcrossing and a large stable population size, assumptions which may not be valid in many domesticated crops [16]. Without a reliable knowledge of the demographic history, it is hard to interpret the statistical results of neutrality tests.
Foxtail millet (Setaria italica (L.) P. Beauv) has been a very important cereal since ancient times in Eurasia and has contributed greatly to human civilization both in Asia and Europe [18]. The most recent archaeological evidence demonstrates that foxtail millet is one of the most ancient crops as its domestication in China dates back to 8,700 years ago [19]. With the rapid development of maize and other modern crops, foxtail millet has gradually become a minor crop in the last 80 years, but is nonetheless still widely cultivated in Asia, Europe, North America, Australia and North Africa as grain food or forage [20]. Green foxtail (Setaria viridis (L.) Beauv.), a weed distributed worldwide, is the presumed wild progenitor of domesticated foxtail millet, based on cytological evidence and RAPD, AFLP, and other markers [21, 22]. But the number of domestication centers and the age of domestication of foxtail millet remains controversial [23]. As revealed by earlier studies, genetic diversity is low in foxtail millet [24], yet these analyses were based on genetic markers that only represent a subset of the possible information that can be gained regarding genomic polymorphism [16]. However, research at the sequence level is very limited in foxtail millet and its close relatives. although sequencing of the foxtail millet genome is now nearly completed [25]. This will provide another cereal model system for comparative and functional genomics and model for studying other biofuel crops such as switchgrass (Panicum virgatum), and napiergrass (Pennisetum purpureum). Studies on domestication or other agricultural related characteristics are ongoing[26–28]. In this study, we conduct a multilocus analysis of nucleotide variation in foxtail millet and green foxtail to reveal the pattern of genetic diversity within and between these two species and to establish a population genetic framework for further analysis of the effects of domestication in foxtail millet.
Results
Nucleotide diversity and neutrality test
We collected 50 cultivated foxtail millets and 34 wild green foxtails to represent the broad diversity of these two species (Table 1). Nine loci randomly selected from the genome were used to survey genetic diversity from both sspecies (Table 2& Additional file 1, Fig s1). The alignment length of each locus after excluding gaps and missing data varied from 431 bp to 996 bp. All nine loci contained both coding and noncoding sequences. However, ninety percent of the final alignments were noncoding sequence because the primers were designed to amplify a large proportion of intron fragments. A total of 160 SNPs were found in the nine loci across the 84 accessions, with an average density of 52 bp/SNP. The wild progenitor had more SNPs (147 SNPs with a density of 43 bp/SNP) compared to the cultivars (75 SNPs with a density of 87 bp/SNP). Indel polymorphisms were infrequent across loci and most had a small size of 1 to 3 basepairs. Three large indels were present in ADTY (143 bp), UPL (112 bp) and TIFIIF (43) bp. These indel polymorphisms were excluded from the subsequent analysis.
The nucleotide diversity of the nine loci for each species is summarized in Table 3. For both θ and π, the values for each locus were slightly lower than values for silent sites, presumably due to strong functional constraint in coding regions. Considering individual loci, the most variable gene was MDEH with a mean of 0.01 for θsil across all accessions. Compared to MDEH, PP2C was the least variable site with a value of only 0.0016 for θsil. At the taxon level, the diversity of wild green foxtail (mean θsil 0.0059) was much higher than the domesticated foxtail millet (mean θsil 0.0027). On average, the cultivars lost 55% of the diversity harbored by the wild progenitor during the domestication process. Some loci in cultivars, such as ADTY and PP2C, lost more than 86% and 75% of their diversity in the wild progenitor. Under the assumption of a neutral equilibrium model, the population silent diversity (θsil) and population divergence (Ks) should be correlated with each other across the loci. We calculated the Pearson correlation between them to test if both species were under neutral evolution. The correlation for wild species is high and significant (pearson cor = 0.93, p value = 0.0001), whereas it is not significant for the cultivar (pearson cor = 0.32, p = 0.19). When excluding the most diverged locus ADTY, significant positive correlations were found in both wild (pearson cor = 0.89, p = 0.002) and cultivars species (pearson cor = 0.69, p = 0.03). The significant correlations suggested that most of loci were under neutral evolution in both species, except for ADTY in cultivars. This locus may be under directional selection or influenced by a selective sweep on neighboring loci.
We used a series of neutrality tests to determine the fit of our data to a neutral equilibrium model. Tajima's D and Fu and Li's D* and F* were used to examine the allele frequency spectrum in polymorphism data for each locus. In the wild population, all the loci showed a negative value for D, D* and F*. Two loci, SIGT and MDEH, had a significant negative value for both tests. However, loci in the cultivars exhibited positive values or less negative values than the wild species for both tests except for DACP and ADTY. The results were unsurprising since we expected D to be higher when the species had experienced a recent population bottleneck[29]. The loss of low frequency alleles during the bottleneck process will increase the D for cultivars. On the other hand, a significant negative D value means the locus has an excess of low frequency variants due to population size expansion and/or purifying or directional selection. We also used a multilocus HKA test to determine whether the level of polymorphism and divergence were correlated across the loci. A significant result was found when we used all nine loci (X^2 = 12.26, p < 0.007). As shown in Figure 1, locus ADTY contributed a large proportion to the overall deviation. Further analysis by removing ADTY showed that no significant result was found across the loci (X^2 = 6.92, p < 0.21).

Summary of multilocus HKA test. Blank circles stand for deviations of foxtail millet. Solid circles stand for deviations of green foxtail. Squares stand for deviations of divergence between foxtail millet and green foxtail.
Population divergence
The level of population differentiation of the two related species was examined by the values of Fst and shared, fixed and unique polymorphisms in the two species, as well as the phylogenetic relationship of these accessions. Fst varied from locus to locus with a mean of 0.1536 (Table 4). Although some loci, such as ADTY and UPL, had Fst values over 0.40, the remaining loci were differentiated at a very low level with Fst values between 0.0254-0.1546. No fixed differences were detected between the cultivars and their wild relatives. This was consistent with a low divergence level due to the short history of domestication. However, a high proportion of shared polymorphisms were observed, particularly for the cultivars. The domesticated foxtail millet shared almost 75% of its polymorphism with green foxtail on average, whereas the proportion for green foxtail was 36%. Unique polymorphisms were present in both species, but the number was much less in cultivars compared to the wild species. Due to the nature of the domestication process, we expected that the cultivars had lost much of their diversity compared to its wild progenitor (55%). The unique polymorphisms in the cultivars suggested that new mutations occurred after domestication or that the wild gene pool was insufficiently sampled.
Linkage disequilibrium
The level of linkage disequilibrium measured as squared allele-frequency correlations were plotted against the distance between pairwise SNPs (Figure 2). The regression curves show that the decay of linkage disequilibrium along the distance for the cultivars was much slower than for the wild. The expected value of for wild green foxtail dropped rapidly to 0.1 within 150 bp, whereas a much higher level (> 0.1) extended to 1000 bp in foxtail millet. Comparison of the population recombination rate, ρ, in the two species implied that recombination was more frequent in the wild species (mean = 0.1497) than that in the domesticated species (mean = 0.0329). Together with the data of haplotype number and haplotype diversity (table 3), the cultivar foxtail millet showed increased linkage disequilibrium level compared to the wild progenitor.

Plots of squared allele-frequency correlations along with the distance between pairs of SNP across nine loci.
Bayesian estimate of population mutation rate θ and recombination rate/mutation rate ρ/θ
Under the standard neutral model, we drew the prior distribution of θ and ρ from uniform distribution within intervals 0-0.03 and 0-0.15. Using rejection algorithm, we obtained 1000 samples from simulations. The posterior distribution of θ, ρ and ρ/θ are shown in Figure 3. Compared with the mean values calculated from the sequence data, values estimated by simulations were quite similar for θ in both domesticated (0.0022) and wild species (0.0053). However, the maximum posterior estimates of ρ were not as large as observed data (ρ = 0.001, ρ/θ = 0.43 for the domesticated and ρ = 0.005 and ρ/θ = 0.836 for the wild). Since the sequence used in this study were short, recombination that could be detected in such a short distance would be rare. The method used for calculating recombination rates here may not give correct values. We also found variations among loci for the values of ρ (0-1.1713 for the cultivated and 0-0.2204 for the wild). This method may over-estimate ρ for some loci. We used a grid of values 0, 1, 2, 5, 10 for ρ/θ in the later analysis, since previous studies showed that the level of recombination had influence on posterior estimation of parameters [30].

Posterior distributions of θ, ρ and ρ/θ as estimated using approximate Bayesian approach in foxtail millet (solid line) and green foxtail(dashed line).
Bayesian inference of bottleneck process
Domestication process can be modeled by coalescent simulation using a simple bottleneck model. The model has been described in Maize, Rice, Wheat and other crops, although for species such as Asian rice the domestication scenario may be more complex[2, 3, 6, 15]. In the bottleneck model, it was assumed that the domesticated species experienced a reduction in population size in the initial of domestication state and then the population size increased after the domesticated species were wildly distributed. We can use this model to infer the domestication process based on population structure and other known information. Upon the assumption of the domestication model, a series of coalescent simulations were done to infer the parameters of the bottleneck model. The parameters that need to be estimated were drawn from a uniform distribution with a specific interval (t2 = 5000-15000, d = 100-3000, k = 0.1-10, m12 = 0-100, m21 = 0-100) (table 3). We did simulations for ρ/θ = 0, 1, 2, 5, 10, respectively. As the acceptance rates for simulations of ρ/θ > = 2 were very low, we used a larger interval (60%) for the rejection algorithm in these simulations. For each simulation, we collected 10,000 samples using the rejection algorithm with summary statistics of the wild species. Then the samples were used to fit with summary statistics of the cultivated species. The number of fitted samples used for posterior prediction varied from 702 for ρ/θ = 1 to 2571 for ρ/θ = 2. The posterior distribution of t2, d, M12 and M21 showed no large peak. However, the posterior distribution of the bottleneck intensity k did show a clear peak and depended on the ratio of ρ/θ (Figure 4). The domestication bottleneck was more severe when ρ/θ changed to large values. Since the ratio of ρ/θ estimated by Bayesian estimation in this study was 0.836, the severity of the bottleneck for foxtail millet should be similar to that of ρ/θ = 1 (0.6095). If the ratio of ρ/θ was underestimated, the domestication process would be much more intense. To further estimate the rate of migration between the two species, we employed a Markov Chain Monte Carlo method as implemented in MIMAR [31]. We ran 1.1e7 steps with 1e6 burnin steps and considered convergence was reached when the posterior distribution of two independent chains were similar (Figure 5). The maximum estimated migration from cultivar to wild is 0.3174, whereas the reverse process was 0.1712.

Posterior distributions of population bottleneck severity k estimated using approximate Bayesian approach for a grid of ρ/θ = 0, 1, 2, 5, 10.

Posterior distributions of migrations between foxtail millet and green foxtail using a Markov Chain Monte Carlo (MCMC) method. Two independent runs were run to assess convergence of estimation, where red and black lines stand for the migrations from foxtail millet to green foxtail each generation and blue and green lines stand for the reverse process.
Discussion
Sequence diversity
Previous studies based on isozymes and DNA markers showed a high level of genetic diversity in foxtail millet [32, 33]. However, diversity at the DNA sequence level of foxtail millet was not well documented and comparisons with other crops, especially cereals, was absent. In this study, nine loci were surveyed to investigate the genetic diversity in foxtail millet and its wild progenitor green foxtail at the DNA sequence level. We found a low level of nucleotide variation in both foxtail millet and green foxtail as compared with other domesticated crops and their wild relatives, such as sunflower, barley, maize and rice [3, 6, 12, 34]. The silent nucleotide variation for green foxtail estimated here was 0.0059, which was lower than most of these wild crop relatives but higher than wild soybean. Compared to other species, nucleotide variation indicated by θsil was higher in wild maize Zea mays ssp. parviglumis (0.0247), wild sunflower Helianthus annuus (0.0234), etc [3, 6, 12, 34]. An exception was found in wild soybean Glycine soja, which had a θsil as low as 0.00235 [35]. Further investigation of these wild relatives showed that most wild species with a higher nucleotide variation have an outcrossing mating system, while nucleotide variation was lower for inbreeding species, such as wild barley and wild soybean [34, 36, 37]. Green foxtail, as well as foxtail millet, are self-pollinated plant that have a 0.3% to 4% outcrossing rate in natural conditions [38, 39]. Lower levels of nucleotide variation in these wild species is expected by their mating system, although the samples collected in this study may also influence the estimation. This is because some wild accessions may not represent a local original wild species but a weedy form that derived from the gene flow between the cultivars and their wild relatives. However, the underestimation influenced by sampling may have been very limited because several accessions were collected in the areas where a weedy form was recognized. The low level of genetic diversity of wild green foxtail might be caused mainly by its high inbreeding rate and/or other demographic factors, particularly changes in population size. Consistent with its wild relatives, foxtail millet also showed a lower level of silent nucleotide variation (0.0027). It was much lower than that of maize (0.0149) or sunflower (0.0072), but similar to that of rice (0.0024) and sorghum (0.0034) [3, 6, 12, 40]. The domestication of maize, barley, soybean, and foxtail millet did not involve changes in mating systems. However, the mating system was changed from outcrossing to inbreeding in rice and sunflower [3, 12]. Based on this information, we can conclude that the level of genetic diversity of foxtail millet, which maintained 45% of its wild diversity, was mainly a result of its change in population size during domestication process, followed by mutation accumulated after divergence. To further explore the impact of domestication on genetic diversity, we used Tajima's D test to detect the change of SNP frequency in the species after domestication. As indicated by Tajima' test, D values of most loci in domesticated species were higher than those of the wild species, but only two of them had a positive D value. The increase of D values in the domesticated species is likely because low frequency alleles were preferentially lost during the domestication bottleneck. Detailed site frequency spectra are shown in Figure 6. We detected an excess in both low and high frequency alleles in the domesticated species compared to the wild one. The excess of high frequency variants in the domesticated species was also found in domesticated Asian rice [41]. Taking into account the higher level of LD in the domesticated species, this may mean that artificial selection active during the domestication process may have extended over a long distance. This phenomenon has been observed for tb 1 in maize and waxy in rice, that the influence of directional selection on certain loci had a large effect on regions around them [8, 42].

Derived allele frequency spectra for cultivated foxtail millet and wild green foxtail.
Linkage disequilibrium
Several studies that focus on the domestication of crops indicated that there was an increased level of linkage disequilibrium in domesticated species as compared to their wild relatives [3, 12, 43]. With the bottleneck effect of domestication, the genome-wide level of nonrandom association tends to increase. In some loci that were targeted by artificial selection, LD could extend to a long distance up to 100 kb, such as tb 1 in maize and waxy in rice [8, 42]. In addition to the influence of population size and direction selection, mating system was also shown to affect LD level [16]. In selfing Arabidopsis thaliana, LD can persist for 250 kb. However, in outcrossing maize, LD declined to a very low level of less than 1 kb [43, 44]. Using the same set of sequences, it was shown that the LD level in O. nivara was slightly higher than that in O. rufipogon, which displayed a higher rate of outcrossing compared to O. nivara[3]. In this study, the higher level of LD observed in foxtail millet compared with green foxtail was similar to what has been observed in rice and sunflower [3, 12]. In wild species, which were shown to have a higher outcrossing rate, the LD level declined rapidly within 100-200 bp. In contrast, LD in domesticated foxtail millet extended to 1000 bp. Based on the fact that the mating system did not change after domestication of foxtail millet, and that the influences of directional selection was likely focused on a small proportion of local genomic regions, we suggest that the increased level of LD in the cultivated foxtail millet was mainly due to the change of population size during the domestication process.
Gene flow between the domesticated and the wild
As indicated previously, foxtail millet maintained 45% of its wild diversity. The proportion was similar with that of sorghum (60-70%) and sunflower (40-59%) [12, 40]. However diversity retention was higher in maize (80%) and very low in rice (10-20%) [3, 16]. The retention of wild diversity is a reflection of the bottleneck intensity (such as in rice) or the mating system (such as in maize). We used coalescent simulation to infer the bottleneck severity during the domestication process. The Bayesian estimate of bottleneck severity k was 0.6095 if we set ρ/θ = 1, which is near the maximum estimate of ρ/θ. Compared with the bottleneck severity that modeled in maize (2.45) and in rice (0.2 for japonica and 0.5 for indica) [3, 45], the severity estimated here was compatible with the loss of diversity from its wild relatives. We also incorporated gene flow into a simulation model. The results suggested that there were low levels of gene flow from the cultivated species to wild species, but the reverse process was even smaller. As foxtail millet and green foxtail can have low levels of cross-pollination and the hybridization between them are compatible [46], we expected that gene flow between the two species would be frequent. By intensive human selection, alleles from wild relatives to cultivated foxtail millet were mostly eliminated. But genes introgressed into the wild species might be retained at a higher level, as evidenced by many weedy types morphologically similar to foxtail millet in and around millet fields. This has became a serious problem for field management. Taken together, the population bottleneck and gene flow both contributed to the present pattern of nucleotide diversity of these two species.
Loci under selection in the domestication
Another factor in the domestication process is human selection [16]. We intended to select nearly neutral loci in order to make inferences about the domestication process in this study. However, ADTY had a significant negative D value in the domesticated species. This observation, taken together with an 86% loss of diversity compared to the wild type and a large deviation in the HKA test, suggested that this locus was likely under directional selection during domestication. We used the estimated parameter of bottleneck severity to infer whether this locus was under selection. Two loci were detected to be under selection in this model, ADTY and PP2C (p-value = 0.014 and 0.008). Except for a loss of 75% of its wild diversity, other statistical tests did not show any signal of selection for PP2C, indicating that the simulation may have given a false positive result for this locus due to low diversity in both cultivated and wild species. Furthermore, the model used to detect selection may not be robust enough to give a result with only a few false positive genes. The locus ADTY may be a target gene of human selection or located in a selected region. However, excluding this locus did not affect the calculated parameters and the results of the simulation. The whole genome sequence of foxtail millet will be released very soon; and further work that focuses on whole genome analysis of genes involved in domestication will likely indicate to what extent human selection has acted in the domestication process.
Conclusions
We found a 55% loss of diversity in foxtail millet and an increased level of LD that can be extended to 1 kb. This phenomenon is likely to be caused by the small effective population size due to a population bottleneck during domestication. Low levels of gene flow from foxtail millet to green foxtail may have been another factor that influenced the genetic diversity of these two species.
Methods
Plant materials and DNA sequencing
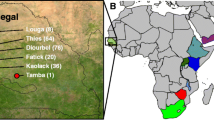
We collected 84 accessions to survey DNA sequence variation in this study, including 50 cultivated foxtail millets and 34 wild green foxtails (Table 1). Accession numbers started with PI or Ames were obtained from the National Germplasm Resources Laboratory, Beltsville, Maryland, USA; others samples were from the Chinese National Germplasm Bank in the Institute of Crop Sciences, Chinese Academy of Agricultural Sciences. The cultivated materials were sampled to represent a broad diversity of foxtail millet, in which all Chinese accessions are landraces. The wild samples were collected throughout the Eurasian continent to cover the distribution range of green foxtail where foxtail millet was claimed to have been domesticated [23, 24]. All the sample seeds were planted in autoclaved soil and fresh leaves were collected to extract genomic DNA using a modified CTAB protocol.
Genomic fragments between 500 and 1500 bp were amplified and sequenced from nine unlinked loci (Table 2 and Additional file 1: figure S1). Based on the high colinearity between the genetic map of foxtail millet and rice [47], we used the rice genome sequence as a reference when selecting the loci so as to have wide coverage of the genome. All EST sequences of Setaria italica were downloaded from the NCBI ftp site http://ftp.ncbi.nlm.nih.gov. The EST set was used to search rice gene models and only those with a single hit were retained for further studies http://rice.plantbiology.msu.edu/. We checked if the homologous region covers an intron in the rice genome and designed primers to amplify the intron.
The nine loci were amplified in the two species using a modified PCR reaction system: 50 ng genomic DNA, 0.2 μmol/l of each primer, 0.2 mM dNTP, 1 U ExTaq DNA polymerase (TaKaRa), 2 μl PCR mix buffer and dH2O to a final volume of 20 μl. After amplification, the products were separated by electrophoresis on a 2% agarose gel. DNA bands were excised, purified, and directly sequenced on an Applied Biosystems 3730 DNA Sequencer. For individual with heterozygous alleles, It is impossible to choose a true allele if it contains more than one heterozygous polymorphism. By directly sequencing the PCR product, the haplotype that was chosen randomly may not be a true haplotype. However both foxtail millet and green foxtail have a low outcross rate, we expected that the influence of heterozygous polymorphism on haplotype inference is very limit. Single base pair changes were further confirmed by PCR and DNA sequencing.
Sequence analysis
The raw sequence trace files were collected and assembled by Phred/Phrap [48, 49]. Alleles of each locus were aligned by ClustalW 1.81 with further manual check [50]. All the alleles containing singletons were subject to a check process, in which we amplified and sequenced the product again to confirm the sequence quality and update the alignment. The alignment files were imported to DnaSP 4.5 with coding regions assigned according to the rice gene annotation [51]. For each locus and species, we calculated the number of segregating sites (S), the population recombination rate (ρ), minimum number of recombination events (Rm), number of haplotypes (Hap), haplotype diversity (Hdiv), average number of nucleotide difference per site between two sequences (π), and the watterson estimator of population mutation rate (θ). To test for neutrality, we calculated Tajima's D [52], and D* and F* of Fu and Li [53] test without outgroup. To access the level of species divergence, we calculated shared, fixed, species-specific S and Fst for the two species. Multilocus HKA test was done by HKA http://lifesci.rutgers.edu/~heylab/ for the nine loci and the results was parsed to R for further analysis [54].
The decay of linkage disequilibrium (LD) with physical distance was described using a nonlinear regression analysis. The expected value of squared allele-frequency correlations (r2) at drift-recombination equilibrium is, E(r2) = 1/(1 + ρ)where ρ is 4Nc and N is the effective population size, c is the recombination rate in Morgans between the 2 markers. Under the assumption of a low mutation rate and finite sample size, the expectation becomes
where n is the sample size of sequences [55]. To introduce the distance between pairs of SNP sites (d) into the formula, we use rho*d to replace ρ, in which rho is the recombination rate per basepair. r2 between pairs of polymorphism was estimated using DnaSP 4.5 for each locus and species. r2 and d were pooled across the loci for each species. The nonlinear regression analysis was performed with the NLS function in the R statistical package http://www.r-project.org.
Coalescent simulation
Coalescent simulation was used to model the process of domestication, as well as to estimate the population mutation rate θ and population recombination rate ρ for each species. The simulations were done using Hudson's ms[56]. In each simulation, we used an rejection-based approximate Bayesian computation approach to obtain a posterior distribution for parameters of interest [57, 58]. Briefly, the initial values of parameters were drawn from a user-specified prior distribution, and, starting with these parameters, the simulation was run under a defined model; For each simulated datasets, several summary statistics were calculated and compared with the observed values. The data was accepted if it was within a defined interval of observed data; and then the parameter set that generated acceptable data were used to obtain a posterior distribution for each parameter. In this study, we used a multilocus approach to assess the acceptability of the data. Summary statistics were calculated for each locus and summarized by mean and/or variance across the loci. The values of mean and/or variance of each summary statistics were compared between simulated and observed data using. The accepted datasets were used for Baysian inference of selceted parameters. Doing this incorporates the variation among the loci into the simulations.
Approximate Bayesian estimate of θ and ρ/θ
To estimate θ and ρ, the simulations were run under the standard neutral model for the wild and domesticated species separately. The means for each four summary statistics (S, π, Hap, Hdiv) were used to access the acceptable of the simulated data. The data was accepted if three of them were within 20% of the observed data [59].
Approximate Bayesian inference of bottleneck parameters
The model used to investigate the bottleneck process was similar as described for maize and rice [3, 6, 45]: N a is the effective population size of the ancestor of the two species.μis the mutation rate for the ancestor. The values of μ were based on the synonymous substitution rate or calculated by θ = 4N μ. The recombination rate ρ together with N a and μ defined the ancestor population. At time t2 generations ago, a new population was derived from the ancestor with a population size of N b and expanded to a population with a size of N p at t1 generations ago. Then, the ancestor population and the newly derived population evolved with constant population size until present. To incorporate gene flow in this model, we defined M12 to be migration rate from wild to cultivar and M21 for the rate for the reverse rate. In this model, the parameters for the wild and domesticated species were calculated by the sequence or estimated using an approximate Bayesian estimate. We defined k to be the ratio of N b and d, d being the duration of the bottleneck in generations (d = t2-t1). Previous studies suggested that N b is positively correlated with d; k is a good indicator for the stringency of the bottleneck [3, 6].
Under this model, we used the mean and variance for each four summary statistics (S, π, Hap, Hdiv) to assess the acceptability of simulated wild species data. The simulation was accepted if both the mean and variance fell within 30% of the observed data. To fit the cultivated data, we use the mean of S, ρ, Hap, and Hdiv to assess the fitness of the simulated data, but we accepted if three of the four fell within 30% of the observed data.
References
Diamond J: Evolution, consequences and future of plant and animal domestication. Nature. 2002, 418 (6898): 700-707. 10.1038/nature01019.
Eyre-Walker A, Gaut RL, Hilton H, Feldman DL, Gaut BS: Investigation of the bottleneck leading to the domestication of maize. Proc Natl Acad Sci USA. 1998, 95 (8): 4441-4446. 10.1073/pnas.95.8.4441.
Zhu Q, Zheng X, Luo J, Gaut BS, Ge S: Multilocus analysis of nucleotide variation of Oryza sativa and its wild relatives: severe bottleneck during domestication of rice. Mol Biol Evol. 2007, 24 (3): 875-888. 10.1093/molbev/msm005.
Pollak E: On the theory of partially inbreeding finite populations. I. Partial selfing. Genetics. 1987, 117 (2): 353-360.
Nordborg M: Linkage disequilibrium, gene trees and selfing: an ancestral recombination graph with partial self-fertilization. Genetics. 2000, 154 (2): 923-929.
Tenaillon MI, U'Ren J, Tenaillon O, Gaut BS: Selection versus demography: a multilocus investigation of the domestication process in maize. Mol Biol Evol. 2004, 21 (7): 1214-1225. 10.1093/molbev/msh102.
Hanson MA, Gaut BS, Stec AO, Fuerstenberg SI, Goodman MM, Coe EH, Doebley JF: Evolution of anthocyanin biosynthesis in maize kernels: the role of regulatory and enzymatic loci. Genetics. 1996, 143 (3): 1395-1407.
Wang RL, Stec A, Hey J, Lukens L, Doebley J: The limits of selection during maize domestication. Nature. 1999, 398 (6724): 236-239. 10.1038/18435.
Whitt SR, Wilson LM, Tenaillon MI, Gaut BS, Buckler ESt: Genetic diversity and selection in the maize starch pathway. Proc Natl Acad Sci USA. 2002, 99 (20): 12959-12962. 10.1073/pnas.202476999.
White SE, Doebley JF: The molecular evolution of terminal ear1, a regulatory gene in the genus Zea. Genetics. 1999, 153 (3): 1455-1462.
Zhang L, Peek AS, Dunams D, Gaut BS: Population genetics of duplicated disease-defense genes, hm1 and hm2, in maize (Zea mays ssp. mays L.) and its wild ancestor (Zea mays ssp. parviglumis). Genetics. 2002, 162 (2): 851-860.
Liu A, Burke JM: Patterns of nucleotide diversity in wild and cultivated sunflower. Genetics. 2006, 173 (1): 321-330. 10.1534/genetics.105.051110.
Clark RM, Linton E, Messing J, Doebley JF: Pattern of diversity in the genomic region near the maize domestication gene tb1. Proc Natl Acad Sci USA. 2004, 101 (3): 700-707. 10.1073/pnas.2237049100.
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ESt: Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet. 2001, 28 (3): 286-289. 10.1038/90135.
Haudry A, Cenci A, Ravel C, Bataillon T, Brunel D, Poncet C, Hochu I, Poirier S, Santoni S, Glemin S, et al: Grinding up wheat: a massive loss of nucleotide diversity since domestication. Mol Biol Evol. 2007, 24 (7): 1506-1517. 10.1093/molbev/msm077.
Wright SI, Gaut BS: Molecular population genetics and the search for adaptive evolution in plants. Mol Biol Evol. 2005, 22 (3): 506-519. 10.1093/molbev/msi035.
Nielsen R: Molecular signatures of natural selection. Annu Rev Genet. 2005, 39: 197-218. 10.1146/annurev.genet.39.073003.112420.
Li Y, Wu S: Traditional maintenance and multiplication of foxtail millet (Setaria italica (L.) P. Beauv.) landraces in China. Euphytica. 1996, 87 (1): 33-38. 10.1007/BF00022961.
Lu H, Zhang J, Liu KB, Wu N, Li Y, Zhou K, Ye M, Zhang T, Zhang H, Yang X, et al: Earliest domestication of common millet (Panicum miliaceum) in East Asia extended to 10,000 years ago. Proc Natl Acad Sci USA. 2009, 106 (18): 7367-7372. 10.1073/pnas.0900158106.
Austin DF: Fox-tail Millets (Setaria: Poaceae)--Abandoned Food in Two Hemispheres. Economic Botany. 2006, 60 (2): 143-158. 10.1663/0013-0001(2006)60[143:FMSPFI]2.0.CO;2.
Fukunaga K, Kato K: Mitochondrial DNA variation in foxtail millet, Setaria italica (L.) P. Beauv. Euphytica. 2003, 129 (1): 7-13. 10.1023/A:1021589019323.
Fukunaga K, Domon E, Kawase M: Ribosomal DNA variation in foxtail millet, Setaria italica (L.) P. Beauv., and a survey of variation from Europe and Asia. Theoretical and Applied Genetics. 1997, 95 (5-6): 751-756. 10.1007/s001220050621.
Fukunaga K, Ichitani K, Kawase M: Phylogenetic analysis of the rDNA intergenic spacer subrepeats and its implication for the domestication history of foxtail millet, Setaria italica. Theor Appl Genet. 2006, 113 (2): 261-269. 10.1007/s00122-006-0291-5.
Li Y, Jia J, Wang Y, S W: Intraspecific and interspecific variation in Setaria revealed by RAPD analysis. Genetic Resources and Crop Evolution. 1998, 45 (3): 279-285. 10.1023/A:1008600123509.
Doust AN, Kellogg EA, Devos KM, Bennetzen JL: Foxtail millet: a sequence-driven grass model system. Plant Physiol. 2009, 149 (1): 137-141. 10.1104/pp.108.129627.
Doust AN, Devos KM, Gadberry MD, Gale MD, Kellogg EA: Genetic control of branching in foxtail millet. Proc Natl Acad Sci USA. 2004, 101 (24): 9045-9050. 10.1073/pnas.0402892101.
Doust AN, Devos KM, Gadberry MD, Gale MD, Kellogg EA: The genetic basis for inflorescence variation between foxtail and green millet (poaceae). Genetics. 2005, 169 (3): 1659-1672. 10.1534/genetics.104.035543.
Doust AN, Kellogg EA: Effect of genotype and environment on branching in weedy green millet (Setaria viridis) and domesticated foxtail millet (Setaria italica) (Poaceae). Mol Ecol. 2006, 15 (5): 1335-1349. 10.1111/j.1365-294X.2005.02791.x.
Tenaillon MI, Sawkins MC, Long AD, Gaut RL, Doebley JF, Gaut BS: Patterns of DNA sequence polymorphism along chromosome 1 of maize (Zea mays ssp. mays L.). Proc Natl Acad Sci USA. 2001, 98 (16): 9161-9166. 10.1073/pnas.151244298.
Thornton K, Andolfatto P: Approximate Bayesian inference reveals evidence for a recent, severe bottleneck in a Netherlands population of Drosophila melanogaster. Genetics. 2006, 172 (3): 1607-1619. 10.1534/genetics.105.048223.
Becquet C, Przeworski M: A new approach to estimate parameters of speciation models with application to apes. Genome Res. 2007, 17 (10): 1505-1519. 10.1101/gr.6409707.
Wang RL, Wendel JF, Dekker JH: Weedy Adaptation in Setaria spp. I. Isozyme Analysis of Genetic Diversity and Population Genetic Structure in Setaria viridis. American Journal of Botany. 1995, 82: 308-317. 10.2307/2445576.
Le Thierry d'Ennequin M, Panaud O, Toupance B, Sarr A: Assessment of genetic relationships between Setaria italica and its wild relative S. viridis using AFLP markers. TAG Theoretical and Applied Genetics. 2000, 100 (7): 1061-1066. 10.1007/s001220051387.
Morrell PL, Toleno DM, Lundy KE, Clegg MT: Low levels of linkage disequilibrium in wild barley (Hordeum vulgare ssp. spontaneum) despite high rates of self-fertilization. Proc Natl Acad Sci USA. 2005, 102 (7): 2442-2447. 10.1073/pnas.0409804102.
Hyten DL, Song Q, Zhu Y, Choi IY, Nelson RL, Costa JM, Specht JE, Shoemaker RC, Cregan PB: Impacts of genetic bottlenecks on soybean genome diversity. Proc Natl Acad Sci USA. 2006, 103 (45): 16666-16671. 10.1073/pnas.0604379103.
Kuroda Y, Kaga A, Tomooka N, Vaughan DA: Gene Flow and Genetic Structure of Wild Soybean (Glycine soja) in Japan. Crop Science. 2008, 48 (3): 1071-1079. 10.2135/cropsci2007.09.0496.
Fujita R, Ohara M, Okazaki K, Shimamoto Y: The Extent of Natural Cross-Pollination in Wild Soybean (Glycine soja). Journal of Heredity. 1997, 88 (2): 124-128.
Li H, Li C, Pao W: Cytological and genetical studies of the interspecific cross of the cultivated foxtail millet, Setaria-Italica (L) Beauv, and the green foxtail millet, S-Viridis L. J Am Soc Agron. 1945, 9: 32-54.
Till-Bottraud I, Reboud X, Brabant P, Lefranc M, Rherissi B, Vedel F, Darmency H: Outcrossing and hybridization in wild and cultivated foxtail millets: consequences for the release of transgenic crops. TAG Theoretical and Applied Genetics. 1992, 83 (8): 940-946.
Hamblin MT, Casa AM, Sun H, Murray SC, Paterson AH, Aquadro CF, Kresovich S: Challenges of detecting directional selection after a bottleneck: lessons from Sorghum bicolor. Genetics. 2006, 173 (2): 953-964. 10.1534/genetics.105.054312.
Caicedo AL, Williamson SH, Hernandez RD, Boyko A, Fledel-Alon A, York TL, Polato NR, Olsen KM, Nielsen R, McCouch SR, et al: Genome-wide patterns of nucleotide polymorphism in domesticated rice. PLoS Genet. 2007, 3 (9): 1745-1756. 10.1371/journal.pgen.0030163.
Olsen KM, Caicedo AL, Polato N, McClung A, McCouch S, Purugganan MD: Selection Under Domestication: Evidence for a Sweep in the Rice Waxy Genomic Region. Genetics. 2006, 173 (2): 975-983. 10.1534/genetics.106.056473.
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ESt: Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci USA. 2001, 98 (20): 11479-11484. 10.1073/pnas.201394398.
Nordborg M, Innan H: Molecular population genetics. Current Opinion in Plant Biology. 2002 (2): 69-73.
Wright SI, Bi IV, Schroeder SG, Yamasaki M, Doebley JF, McMullen MD, Gaut BS: The effects of artificial selection on the maize genome. Science. 2005, 308 (5726): 1310-1314. 10.1126/science.1107891.
Darmency H, Zangre GR, Pernes J: The wild-weed-crop complex in Setaria: a hybridization study. Genetica. 1987, 75 (2): 103-107. 10.1007/BF00055253.
Devos KM, Wang ZM, Beales J, Sasaki T, Gale MD: Comparative genetic maps of foxtail millet (Setaria italica) and rice (Oryza sativa). Theoretical and Applied Genetics. 1998, 96 (1): 63-68. 10.1007/s001220050709.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8 (3): 175-185.
Ewing B, Green P: Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8 (3): 186-194.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Research. 1994, 22: 4673-4680. 10.1093/nar/22.22.4673.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19 (18): 2496-2497. 10.1093/bioinformatics/btg359.
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989, 123 (3): 585-595.
Fu YX, Li WH: Statistical tests of neutrality of mutations. Genetics. 1993, 133 (3): 693-709.
Hudson RR, Kreitman M, Aguade M: A test of neutral molecular evolution based on nucleotide data. Genetics. 1987, 116 (1): 153-159.
Hill WG, Weir BS: Variances and covariances of squared linkage disequilibria in finite populations. Theor Popul Biol. 1988, 33 (54-78):
Hudson RR: Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002, 18 (2): 337-338. 10.1093/bioinformatics/18.2.337.
Beaumont MA, Zhang W, Balding DJ: Approximate Bayesian computation in population genetics. Genetics. 2002, 162 (4): 2025-2035.
Marjoram P, Tavare S: Modern computational approaches for analysing molecular genetic variation data. Nat Rev Genet. 2006, 7 (10): 759-770. 10.1038/nrg1961.
Weiss G, von Haeseler A: Inference of population history using a likelihood approach. Genetics. 1998, 149 (3): 1539-1546.
Acknowledgements
This paper was supported by National Sciences Foundation of China (30630045, 30471117), the earmarked fund for Modern Agro-industry Technology Research System of China, National Technology Supporting Program (2006BAD02B02), National 863 program (2006AA10Z157, 2008AA10Z158) and Hebei Natural Sciences Foundation (C2004000697, C2006000725, C2007000972). We thank Dr. Song Ge from the Institute of Botany, Chinese Academy of Sciences, Dr. Andrew Doust from Oklahoma State University, USA, for their critical reading and English modification of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Authors' contributions
XD and MC designed the study, supervised the experiment. HZ and WL collected and planted the samples used and made help in laboratory work. CW, JC, LY, HL and YW designed primers and carried out the laboratory work. JC, BZ, CW and XD performed the statistical analysis. JC prepared the first draft of the manuscript and XD made optimization of the manuscript. All authors discussed the result and conclusion and read and approved the final manuscript.
Chunfang Wang, Jinfeng Chen contributed equally to this work.
Electronic supplementary material
12863_2010_837_MOESM1_ESM.PDF
Additional file 1:Schematic diagrams of nine loci and sequenced regions in this study. Exons, introns and UTRs are indicated by blue boxes, lines and open boxes. The primers that were used to PCR and sequencing are marked with black arrowhead, where F and R stand for forward primer and reverse primer respectively. (PDF 39 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, C., Chen, J., Zhi, H. et al. Population genetics of foxtail millet and its wild ancestor. BMC Genet 11, 90 (2010). https://doi.org/10.1186/1471-2156-11-90
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-11-90