
Abstract
Background
The phylogeny of Cetacea (whales) is not fully resolved with substantial support. The ambiguous and conflicting results of multiple phylogenetic studies may be the result of the use of too little data, phylogenetic methods that do not adequately capture the complex nature of DNA evolution, or both. In addition, there is also evidence that the generic taxonomy of Delphinidae (dolphins) underestimates its diversity. To remedy these problems, we sequenced the complete mitochondrial genomes of seven dolphins and analyzed these data with partitioned Bayesian analyses. Moreover, we incorporate a newly-developed "relaxed" molecular clock to model heterogenous rates of evolution among cetacean lineages.
Results
The "deep" phylogenetic relationships are well supported including the monophyly of Cetacea and Odontoceti. However, there is ambiguity in the phylogenetic affinities of two of the river dolphin clades Platanistidae (Indian River dolphins) and Lipotidae (Yangtze River dolphins). The phylogenetic analyses support a sister relationship between Delphinidae and Monodontidae + Phocoenidae. Additionally, there is statistically significant support for the paraphyly of Tursiops (bottlenose dolphins) and Stenella (spotted dolphins).
Conclusion
Our phylogenetic analysis of complete mitochondrial genomes using recently developed models of rate autocorrelation resolved the phylogenetic relationships of the major Cetacean lineages with a high degree of confidence. Our results indicate that a rapid radiation of lineages explains the lack of support the placement of Platanistidae and Lipotidae. Moreover, our estimation of molecular divergence dates indicates that these radiations occurred in the Middle to Late Oligocene and Middle Miocene, respectively. Furthermore, by collecting and analyzing seven new mitochondrial genomes, we provide strong evidence that the delphinid genera Tursiops and Stenella are not monophyletic, and the current taxonomy masks potentially interesting patterns of morphological, physiological, behavioral, and ecological evolution.
Similar content being viewed by others

Background
Cetaceans (whales, dolphins, and porpoises) have been the subjects of intense phylogenetic inquiry using both morphological (including fossil) and molecular data [1–19]. This attention is not surprising; Cetaceans represent one of the most fascinating evolutionary transitions within vertebrates and a robust phylogenetic framework is the underpinning of any study into morphological, behavioral, and physiological evolution. The results of these phylogenetic inquiries agree on several key relationships including the monophyly of Cetacea and Mysticeti (baleen whales), and the close relationships of the Amazon (Iniidae) and La Plata (Pontoporidae) River dolphins.
However, many of the phylogenetic relationships inferred by the aforementioned studies strongly conflict. Perhaps the most obvious incongruencies are the interrelationships of the major odontocete (toothed whales) clades (Fig. 1). All published phylogenies disagree on the phylogenetic placement of one or more families, and many of these incongruencies are strongly supported with high bootstrap proportions or posterior probabilities (Fig. 1). Thus, we have little confidence in the "deeper" relationships of odontocetes and this situation limits the power of any comparative analyses that incorporate phylogenetic information.

Recent hypotheses of the interrelationships of the major cetacean lineages. Clade support values, when available in the original study, are provided. Decimal numbers represent posterior probabilities and numbers between 50 and 100 represent nonparametric bootstrap proportions.
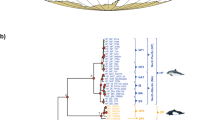
There is also evidence that the traditional taxonomy of dolphins (Delphinidae) does not adequately capture the diversity of the group, especially within the Sousa-Delphinus-Tursiops-Stenella complex (Fig. 2). Bottlenose dolphins (genus Tursiops) have been classified into as few as one and as many as eight different species [20–22], but most recent analyses recognize two distinct species, the common bottlenose dolphin (T. truncatus), and the Indo-Pacific bottlenose dolphin (T. aduncus). The points of contention, however, are the phylogenetic affinities of these two species. Recent molecular evidence lends support to the hypothesis that T. aduncus is not only a distinct species, but is more closely related to the striped dolphin (Stenella coeruleoalba) than to T. truncatus ([9], Fig. 2b). This contrasts with osteological similarities suggesting that the two Tursiops species are sister taxa [23]. In addition, two molecular phylogenetic studies of Delphinidae inferred strong support for the paraphyly of the genus Stenella with respect to Tursiops and Delphinus ([9, 17], Fig. 2). Moreover, the phylogenetic relationships of all of these genera with respect to the genus Sousa (humpback dolphins) is also unresolved (Fig. 2) (see also [19]).

Recent hypotheses of the interrelationships of selected Delphinidae species. The original phylogenies were pruned to include only species used in the current study. Decimal numbers represent posterior probabilities and numbers between 50 and 100 represent nonparametric bootstrap proportions.
Thus, it is clear that the overall state of odontocete phylogenetics, and the evolutionary history of delphinids in particular, remains incompletely resolved. There are myriad possible explanations for ambiguous and conflicting results among these phylogenetic studies. One possiblity is that the phylogenetic methods used in these analyses do not adequately capture the complex nature of DNA evolution, thus allowing systematic error to bias the results. Some studies [8, 9, 12, 14, 24] have used phylogenetic analyses that are known to be misled by the complexity of DNA evolution (e.g., maximum parsimony; [25–29]) or do not implement an optimality criterion to distinguish between competing hypotheses (e.g., neighbor joining [27, 29]). While many studies [5, 11, 15–17, 19, 30] have incorporated phylogenetic methods that explicity incorporate molecular evolution parameters including base frequencies, substitution rates, and rate heterogeneity and an optimality criterion (e.g., maximum likelihood and Bayesian), this does not necessarily mean that they have adequately incorporated all of the critical parameters for modeling DNA evolution. For example, until very recently, there have been insufficient methods to incorporate lineage-specific rates of evolution in phylogenetic analyses. Instead, most phylogenetic methods assume that a phylogeny is unrooted, thereby assuming every lineage evolves at an independent rate (e.g., maximum parsimony, most implementations of maximum likelihood and neighbor-joining) or that all lineages evolve at the same rate (e.g., UPGMA and methods enforcing a molecular clock) [31]. However, the recent development of "relaxed" molecular clocks [31] offers a compromise between the two extremes of a global rate and completely independent species-specific rates. The advantage of incorporating this parameter is the same as incorporating any other parameter that makes a phylogenetic model more "realistic"; the better the model, the more accurate the phylogeny. Moreover, use of relaxed molecular clocks and fossil calibration constraints also permits estimation of divergence times of clades. Therefore, the rich fossil history of cetaceans (see [32]), the development of relaxed molecular clocks, and newly available computer programs to implement this advancement in phyogenetic modeling (e.g., BEAST; [33]) may provide powerful new tools to examine the phylogenetic history of cetaceans.
Furthermore, ambiguous relationships among the delphinid Delphinus-Tursiops-Stenella complex inferred by analyses of mitochondrial DNA may be attributable to the relatively small number of nucleotides used by previous phylogenetic analyses (1140 of cytochrome b [9, 17]). Given the massive number of potentially informative characters (>10,000+ nucleotides) that they contain, complete mitochondrial genomes could potentially resolve the current uncertainty in the phylogeny of these delphinids. While complete genomes exist for all the major families of cetaceans, and have been subsequently used in phylogenetic analyses [5, 6, 30, 34], to date, mitochondrial genomic information is available for only a single delphinid (Lagenorhynchus albirostris).
A reanalysis of cetacean phylogeny and Stenella and Tursiops taxonomy, using recently-developed complex phylogenetic models of DNA evolution and an expanded mitochondrial dataset, is warranted. To this end, we sequenced the complete mitochondrial genomes of seven dolphin species including the common bottlenose dolphin, Indo-Pacific bottlenose dolphin, Long-beaked common dolphin (Delphinus capensis), Pantropical spotted dolphin (Stenella attenuata), striped dolphin (St. coeruleoalba), Risso's dolphin (Grampus griseus), and the Indo-Pacific humpback dolphin (Sousa chinensis). We analyze these data, and existing cetacean mitochondrial genomes, with Bayesian relaxed clock phylogenetic analyses to address two questions in cetacean phylogenetics. First, how do implementing complex phylogenetic models affect our understanding of the relationships of the major odontocete families? Secondly, does mitochondrial genomic data support the hypotheses that the delphinid genera Tursiops and Stenella are polyphyletic? Resolution of both questions may have a profound impact of any future comparative biological analyses of odontocetes, and delphinids in particular.
Results
Characteristics of the mitochondrial genes
In terms of base compositions, gene structure, and gene order, the characteristics of the mitochondrial genomes of the seven newly sequenced delphinids are very similar to the only other existing delphinid mt genome, that of the white-beaked dolphin, L. albirostris [5]. The sizes of the genomes range from 16,384 to 16,393 bp. The base composition of the heavy-strand, excluding the control region, was similar among different mt-genomes, with average values and ranges (in parenthesis) of: A, 31.5% (31.4%–31.7%); C, 28.0% (27.8%–28.3%); G, 11.4% (11.3%–11.6%); and T, 29.0% (28.7%–29.2%). Most genes had standard stop codons, while ND3 and ND4 had incomplete stop codons (TA or T), with the terminal 3' nucleotide being contiguous with the 5' terminal nucleotide of the following tRNA genes. It is assumed that the stop codon is completed with the addition of the poly-A tail [35]. Gene order of all eight available delphinid mt-genomes was consistent and did not deviate from those of other cetaceans or the standard vertebrate gene order.
Phylogenetic results
The two partitioned Bayesian analyses achieved stationarity by 5 million generations, and posterior distributions of each parameter were calculated for the remaining 15 million post burn-in trees. The phylogeny, and 95% credible intervals of divergence times, are provided in Fig. 3. Overall support for the phylogeny is very high with 13 clades strongly (i.e., statistically) supported. The "deeper" relationships of the phylogeny are all well-supported including the monophyly of Cetacea and Odontoceti (toothed whales). The basal divergence within odontocetes is between the physeteroids (sperm whales) and a strongly supported clade (Clade G; PP = 1.0) of remaining odontocete families. The basal relationships within this clade are weakly supported and essentially form a trichotomy including Platanistidae (Indian River dolphins), Ziphiidae (bottlenose whales), and a strongly supported clade (Clade I; PP = 1.0) including other dolphins and porpoises. The relationship between Lipotidae (Yangtze River dolphin) to the two other river dolphin families Pontoporidae (La Plata River dolphins) and Iniidae (Amazon River dolphins) is only weakly supported (PP = 0.61), but support for the sister relationship of the latter two families is significant (PP = 1.0). The remaining marine dolphins and porpoises form a strongly supported clade (Clade L; PP = 1.0), with a basal divergence between a strongly supported Delphinidae (PP = 1.0) and a clade including Phocoenidae (porpoises) and Monodontidae (narwhals and belugas). The interrelationships of G. griseus, L. albirostris, and other delphinid genera are not well supported. Within the strongly supported clade of remaining delphinids (Clade P; PP = 1.0), there is statistically significant support for the paraphyly of Tursiops and Stenella. Stenella attenuata, Sousa chinensis, and T. truncatus form sequentially more exclusive, strongly supported clades (Clades P, Q, and R; PP = 1.0) with the other delphinids. T. aduncus (Indo-Pacific bottlenose dolphin) forms a strongly supported clade (PP = 1.0) with D. capensis and St. coeruleoalba. (but the sister relationship of D. capensis and T. aduncus is highly but not statistically supported [PP = 0.94]).

Chronogram of Cetacea inferred by partitioned Bayesian analyses, enforcing a lognormal relaxed molecular clock, of 12 mitochondrial protein-coding genes. Numbers above the clades represent Bayesian posterior probabilities. Clade letters refer to Table 1. Red boxes indicate nodes for which a prior calibration constraint distribution was used and orange boxes indicate divergence dates estimated without prior calibration constraints for that node. The bounds of the boxes delimit the 95% highest posterior density (HPD) for the clade age. The asterisk indicates that the monophyly of this group was constrained in the phylogenetic analysis.
The divergence dates inferred by the Bayesian relaxed clock analyses suggest that the major odontocete lineages diversified in Mid- to Late Oligocene (Fig. 3; Table 1). The four river dolphin lineages diverged in distinctly different time periods, with the Indian River dolphins (Platanistidae) diverging Late Oligocene, Yangtze River dolphins (Lipotidae) in the very Late Oligocene or Early Miocene, and La Plata River (Pontoporidae) + Amazon River (Iniidae) dolphins in the Mid-Miocene. The divergence of the three extant delphinoid lineages occurred in the Mid-Miocene. The crown Delphinid lineages radiated in the Late Miocene, with the Sousa-Delphinus-Tursiops-Stenella complex diverging recently in the Mid- to Late Pliocene.
Discussion
This study is the first to use partitioned Bayesian analyses with models of rate autocorrelation (i.e., the relaxed molecular clock) and complete mitochondrial genomes to elucidate the phylogenetic relationships of Cetacea and age of each clade. Furthermore, we focused our analysis both at the "deep" phylogenetic level, reconstructing the evolutionary relationships among the major extant odontocete lineages, and the "shallow" level examining interrelationships among delphinid species in the Sousa-Delphinus-Tursiops-Stenella complex. Our results do much to increase our understanding of cetacean phylogenetic relationships, and our estimation of molecular divergence dates allow us to test previous hypotheses of ancestral divergence events.
Odontocete relationships and potential explanations for continuing phylogenetic uncertainty
Although numerous studies have demonstrated that Physeteridae (sperm whales) represent the sister lineage to all other odontocetes [5, 8, 10, 11, 13, 17, 30, 36], there has been significant disagreement about the phylogenetic affinities of the other odontocete families. Although previous studies have supported various hypotheses (Fig. 1), they can essentially be simplified to disagreements about the placement of the river dolphin families Platanistidae (Indian River dolphins) and Lipotidae (Yangtze River dolphins) and relationships among the delphinoids, Delphinidae (marine dolphins), Monodontidae (narwhals and belugas), and Phocoenidae (porpoises).
Numerous studies have refuted the existence of a single river dolphin clade [5, 8, 10, 13, 17, 24, 30, 37–40], and instead have proposed conflicting relationships of the four major river dolphin clades. While most studies strongly support the sister relationship between Iniidae (Amazon River dolphins) and Pontoporidae (La Plata River dolphins), there are numerous strongly supported conflicting hypotheses of evolutionary relationships of Platanistidae (Indian River dolphins) and Lipotidae (Yangtze River dolphins) (Fig. 1).
The results of the current partitioned Bayesian analysis using a relaxed molecular clock (Fig. 3) do not fully resolve the relationships of the river dolphin lineages. Platanistidae is strongly placed in a clade exclusive of physeterids (sperm whales), but its relationship with Ziphiidae (beaked whales) and other odontocetes (Fig. 3, Clade I) is unclear as the branch leading to Ziphiidae + Clade I is poorly supported. The situation with Lipotidae is very similar. Although strongly placed in a clade exclusive of Physeteridae, Ziphiidae, and Platanistidae (Clade I), their relationships with the other river dolphins (Iniidae and Pontoporidae) are only weakly supported.
Given that we were unable to unambiguously resolve all of the odontocete relationships with confidence, how does this study further our understanding of cetacean evolution? First, we note that lack of confidence in phylogenetic reconstructions is preferable to strong support for erroneous reconstructions [28]. Yet, more importantly, because we used fossil calibration constraints and a relaxed molecular clock, the branch lengths of our phylogeny are in units of time. Inspection of the branches critical to understanding the phylogenetic placement of both Platanistidae and Lipotidae and their closely related lineages reveals that they diverged within a very narrow time frame (Fig. 3). Indeed, the branch leading to the clade of non-platanistid river dolphins (branch J) is almost indistinguishable from zero. These results are indicative of a rapid radiation, where lineages diversified so quickly that very few DNA character changes had sufficient time to evolve and become fixed. Rapid radiations are notoriously difficult to reconstruct (e.g., [41–44]), especially those that occurred deeper in time (because synapomorphies are more likely to be lost due to stochastic changes).
If the hypothesis that rapid radiation accounts for the phylogenetic uncertainty in odontocete evolutionary relationships is true, what influenced this radiation? Hamilton et al. (2001) hypothesized that the extreme fluctuations of sea levels in the Middle Miocene promoted diversification in all river dolphin lineages by permitting ecological diversification when seas inundated formerly terrestrial habitat [11]. However, this study also assumed that these lineages diversified ~5 million years more recently than those estimated by our study. Given that these extreme sea level changes occurred throughout the Lower and Middle Miocene [45, 46], this hypothesis may explain the rapid diversification of Lipotidae and Iniidae + Pontoporidae, which diverged some time between ~25 to 17 Mya (Table 1; Fig. 3). However, our results indicate that the lineage leading to extant (clade G) platanistids, ziphiids, and their relatives diverged in the Middle to Late Oligocene, ~32 to 23 Mya (Table 1; Fig. 3) thus precluding Middle Miocene sea level change explanation for this radiation.
The Oligocene is characterized as an "icehouse" period where global environments were significantly cooler than average [47, 48] and was also characterized by extensive expansion of polar ice [49]. Expansion of the polar ice and more extreme ocean thermal gradients both vertically (depth) and horizonally (area) may have resulted in a concomitant expansion of food resources, and have been hypothesized to explain that radiation of cetaceans also seen in the fossil record [50–52]. The relationships of fossil odontocete taxa are the subject of ongoing revision, and it is therefore difficult to determine the phylogenetic relationships of these taxa with any degree of confidence. Nonetheless, there are multiple distinct Oligocene odontocete lineages represented in the fossil record that have subsequently gone extinct (see [32] for review).
Despite partially ambiguous results, our analysis nonetheless provides insight into the evolution of river dolphins. Extant river dolphins are relict lineages whose adaptation to riverine habitats permitted their survival for many millions of years [10]. Additionally, our results reject the hypothesis of a single ecological shift to riverine habitats in the river dolphins [12], and instead support multiple shifts [10, 11, 13, 17, 30]. Based on the geographical distribution of Lipotidae (China) and Pontoporidae + Iniidae (South America), and extinct Parapontoporia [53] (Pacific North America), a sole invasion of the riverine environment by their most recent common ancestor is unlikely [12]. Based on our phylogenetic inference, we predict three older invasions into the riverine habitats by 1) platanistids in the Early to Late Oligocene, 2) lipotids in the Early Miocene, and 3) the most recent common ancestor of Iniidae and Pontoporidae occurring in the Early to Middle Miocene in addition to a relatively recent reinvasion into the marine coastal habitat by the La Plata river dolphin.
The evolutionary history picture is much clearer within the Delphinoidea. Numerous studies have disagreed on the interrelationships of Delphinidae (dolphins), Monodontidae (narwhals and belugas), and Phocoenidae (porpoises), with some studies supporting the sister relationship of Delphinidae and Phocoenidae [5, 12, 30], Monodontidae and Phocoenidae [10, 14, 17, 54] or are unable to resolve the relationships [8, 11, 13, 55]. Our results strongly support the Monodontidae + Phocoenidae hypothesis. Moreover, our divergence date estimates (Table 1; Fig. 3) indicate that the major extant lineages radiated in the Middle Miocene, a time, as noted above, when marine environments underwent cyclical diversification and contraction.
The application of recent advances in modeling DNA evolution and use of a large (over 10 kb) data set could not resolve, with significant support, the closest living relatives of Platanistidae or Lipotidae. However, given our results, we propose a potential explanation of why these relationships are so difficult to reconstruct – these lineages are part of a rapid radiation. Thus, future phylogenetic efforts should focus on multiple, independently evolving loci in the nuclear genome. A larger number of independent loci would provide additional phylogenetically informative characters and permit the use of direct statistical tests of the rapid radiation hypothesis (e.g., [43]) as these tests have little power when used with a single locus (e.g., mitochondrial DNA).
The taxonomy of Delphinidae underestimates its diversity
As with the odontocetes, there has been considerable phylogenetic uncertainty among species within the Delphinidae. Our phylogenetic analyses including seven newly collected complete mitochondrial genomes strongly reject the monophyly of the genera Stenella and Tursiops. Stenella coeruleoalba and T. aduncus form a strongly supported clade with D. capensis (however, the sister relationship between St. coeruleoalba and D. capensis is marginally insignificant [PP = 0.94]) and there is strong support for the exclusion of T. truncatus and St. attenuata from this clade (Fig. 3). These results mirror those of LeDuc et al.'s (1999) study using the cytochrome b gene and all three species of Delphinus [9]; this study also inferred a trichotomy of Delphinus spp., St. coeruleoalba, and T. aduncus. However, unlike LeDuc et al. (1999) [9] (Fig. 2b) and May-Collado and Agnarsson (2006) [17] (Fig. 2a) and Caballero et al. (2008) [19], our results are able to resolve the placement of Sousa chinensis and St. attenuata with very strong support (PP = 1.0).
The monophyly of the genera Tursiops and Stenella has been questioned for more than a century [20, 56, 57]. There exists a complex of cranial characters not shared by all species of the genus Stenella, some of which may actually be more closely related to Tursiops or Delphinus than to their congeners [56]. To add to the confusing phylogenetic signal from morphological data, some osteological studies suggest a greater affinity between the two bottlenose dolphins (T. aduncus and T. truncatus) than between T. aduncus and any Stenella species [23].
Nonetheless, the results of our phylogenetic analysis are clear – Tursiops and Stenella are not monophyletic and the current taxonomy of Delphinidae masks potentially interesting patterns of morphological, physiological, behavioral, and ecological evolution. That these two genera are not monophyletic suggests that the morphological similarity among species in Tursiops and Stenella (as currently defined) could be due to adaptive convergence, primitive retention of ancestral body form, reversal to ancestral body form (in St. coeruleoalba), or a combination of these factors. Unfortunately, the current taxon sampling of Delphinidae prohibits us from testing these two scenarios (and indeed, making taxonomic revisions), but it certainly deserves intense scrutiny in future phylogenetic and evolutionary analyses of Delphinidae.
Another possible explanation for the phylogenetic pattern is mitochondrial introgression. Although no record of the historical geographical distribution of Tursiops or Stenella species is available, the two extant bottlenose dolphins (Tursiops) are sympatric across parts of their range, particularly around the Penghu Archipelago which is situated midway across the Taiwan Strait [58]. Similarly, both St. coeruleoalba and St. attenuata are cosmopolitan in tropical and temperate waters around the world. Considering numerous cases of hybridization in captivity or in the wild among inter-species and inter-genera in the family Delphinidae, it is possible that ancient mitochondrial introgression may confound the true evolutionary relationships [9]. Although answering this question with certainty will require collection of multiple nuclear loci, our results demonstrate that recent introgression, at least, is an unlikely explanation for our results. Although closely related, all of our sampled delphinid species are genetically distinct. In other words, there are genetic changes on the terminal branches leading to each species (i.e., autapomorphies). If there was recent mitochondrial introgression, we would expect the introgressed species to be genetically identical (or nearly so) to another species.
Conclusion
Our phylogenetic analysis of complete mitochondrial genomes using recently developed models of rate autocorrelation resolved the phylogenetic relationships of the major Cetacean lineages with a high degree of confidence. Moreover, our estimation of molecular divergence dates allowed us to construct hypotheses explaining the lack of resolution of the river dolphin lineages. Furthermore, by collecting and analyzing seven new mitochondrial genomes, we provide strong evidence that the generic taxonomy of Delphinidae underestimates the evolutionary history of the group and that the genera Tursiops and Stenella are not monophyletic. This result has important implications for the morphological evolution (and potentially physiological, behavioral, and ecological evolution) within these genera and indicates adaptive convergence, retention of ancestral body form, or both.
Methods
Sample collection and location
We sequenced complete mitochondrial (mt) genomes for seven species representing two species of Stenella and Tursiops as well as potentially closely related genera (Table 2). Complete mt-genome sequences from these specimens are deposited in GenBank under accession numbers EU557091–EU557097. Muscle tissue was preserved in a solution of 0.25 M disodium EDTA, 20% DMSO, and saturated with NaCl [59], and blood was stored in a vacutainer containing 1.5 mg/ml potassium-EDTA. All tissues were subsequently frozen at -20°C. The voucher specimens were preserved in 95% ethanol at Nanjing Normal University. Fourteen additional mt-genomes for other cetartiodactyla (artiodactyls and cetaceans) were obtained from the GenBank and included in our analyses (Table 2).
Laboratory protocols
Total genomic DNA from muscle tissue was extracted with a standard phenol/chloroform procedure followed by ethanol precipitation [60]. For blood, we used the DNeasy Blood Extaction Kit (Qiagen) in a separate laboratory facility. In order to avoid amplifying nuclear pseudogene copies, approximately 7 kb and 9 kb fragments were amplified using the same long-range PCR protocol as Sasaki et al. [34]. These large mtDNA products were subsequently used as template for short-range PCR of 0.5–1.5 kb using conserved primers (see Additional file 1). PCR was set up with approximately 100 ng of template DNA, 10 × PCR buffer, 2–3 mM MgCl2, 1.5 uM each primer, 1 mM dNTPs, 2 U of rTaq (Takara, Japan) and increased to a reaction volume of 50 ul with ultrapure water. Amplification conditions involved an initial denaturing step at 95°C for 5 min, followed by 35 cycles of 95°C for 40 s, 50–55°C for 40 s and 72°C for 60 s, with a final extension step at 72°C for 10 min. Negative controls were run for all amplifications to check for possible contamination. Amplified products were isolated by gel electrophoresis, then excised and purified on Wizard® SV Gel and PCR Clean-Up System (Promega, USA). Sequencing reactions were performed according to the manufacturer's protocols, run on a 3100 or 3700 ABI DNA sequencer, and sequenced in both forward and reverse directions.
Phylogenetic analyses
Overlapping contigs were compiled using SeqMan II (DNASTAR, Inc. Madison, WI) to generate continuous sequences. To minimize alignment error inherent in phylogenetic analysis of RNA data, we analyzed only protein-coding genes encoded by the mitochondrial heavy-strand, for a total of 10,803 nucleotides from 12 genes (ND6 was excluded to make the data set comparable to existing mt-genome studies).
We employed Bayesian phylogenetic analysis to infer the phylogeny of the sampled cetacean mt genomes. Bayesian analysis incorporates the likelihood equation and models of nucleotide evolution, and has been repeatedly shown to outperform other phylogenetic methods (e.g., [25, 26, 28]). However, unlike maximum likelihood, model parameters (e.g., tree topology, branch lengths, substitution rates, base frequencies, site rate heterogeneity) in Bayesian analysis are sampled according to their posterior probability [61–64]. Thus, Bayesian analysis inherits many of the attractive qualities of maximum likelihood, but with the added benefit of simultaneously inferring the posterior probabilities of model parameters. In the case of the tree topology, these posterior probabilities can be interpreted as the probability that the clade is correct, given the model [65]. Bayesian analysis is also useful because the use of posterior probabilities allows for very simple hypothesis testing (e.g., the posterior probability of alternative hypotheses can be calculated). Furthermore, the recent development of "relaxed" molecular clocks permits the modeling of autocorrelated rates of evolution [31], thereby relaxing the assumption that every lineage evolves at the same rate and permitting the simultaneous estimated of divergence between lineages.
All phylogenetic analyses were conducted using BEAST v1.4.8 [33] incorporating a lognormal relaxed molecular clock to model rate autocorrelation. Incorporating autocorrelation requires some a priori estimate of molecular rates. Because locus-specific rates of evolution are rarely known, one can estimate these rates (i.e., "calibrate" the rate) for one or more lineages using fossil constraints. Modern Bayesian methods allow for the incorporation of a prior distribution of ages ("age constraints"), and thus uncertainty, into these fossil calibrations and ultimately, autocorrelation estimates. Fortunately, Cetacea has an extensive fossil record (see [32]) permitting the use of multiple calibration constraints. All fossil calibrations were lognormal distributions of ages as they best represent the information that fossils provide about the age of a clade (i.e., a lineage most likely diverged around the age of the fossil lineage, but may have diverged earlier [66]). Note that, unlike calibration points, these are distributions of ages that permit the explicit incorporation of error in the fossil age and times that lineages actually diverged.
We chose prior age distributions so that the youngest age of the distribution corresponded with the youngest possible age of the fossil (i.e., the youngest possible age that lineage existed). We chose a standard deviation of the distribution so that 95% of the distribution fell within the geological time period of the fossil (i.e., 5% of the tail extended into older ages). For example, if a fossil was from Late Miocene deposits (11.2 – 6.5 million years ago [Mya]), we would choose a distribution so that the youngest age of the distribution was 6.5 and 95% of the distribution fell within 11.2 to 6.5 Mya. We used distributions of fossil ages that spanned the entire geological age of the strata in which the fossil was discovered, unless the author provided a more precise fossil age. We used the following fossils as calibration age constraints:
1. The age of the divergence between Hippopotomidae and Cetacea was calibrated using the Ypresian (Eocene: 55.8 – 48.6 Mya) fossil Pakicetus [67, 68]. We chose a lognormal distribution so that the earliest possible sampled age corresponds to 48.6 Mya and the older 95% credible interval (CI) encompasses the beginning of the Ypresian (55.8 Mya) (standard deviation = 1.2). When estimating divergence dates, calibration ages of divergences close to the root are extremely important (e.g., [66, 69]). We therefore enforced the monophyly of this clade in accordance with numerous phylogenetic analyses that have inferred this relationship [4, 5, 17, 70–78].
2. The divergence between Mysticeti (baleen whales) and Odontoceti (toothed whales) was calibrated using the earliest record of mysticetes from the Eocene/Oligocene boundary (see [32] for a review). The Eocene/Oligocene boundary was a critical period in the evolution of both plants and animals (see [79]) and there has been debate about when this transition occurred (38 – 33.5 Mya; [80]). We chose a lognormal distribution so that the earliest possible sampled age corresponds to 33.5 Mya and the older 95% credible interval (CI) encompasses the beginning of the Late Eocene (40 Mya) (standard deviation = 1.138).
3. The age of the root of crown Odontoceti was calibrated using the earliest record of a physeterid (sperm whale) the Late Oligocene Ferecetotherium ([81]). We chose a lognormal distribution so that the earliest possible sampled age corresponds to 23.7 Mya and the older 95% credible interval (CI) encompasses the beginning of the Late Oligocene (30 Mya) (standard deviation = 1.119).
4. The divergence between Iniidae (the Amazon River dolphin) and Pontoporidae (La Plata River dolphin) was calibrated using the earliest record of a pontoporid, the Middle Miocene Brachydelphis [37, 82]. We chose a lognormal distribution so that the earliest possible sampled age corresponds to 11.2 Mya and the older 95% CI encompasses the beginning of the Middle Miocene (16.6 Mya) (standard deviation = 1.025).
5. The divergence between Phocoenidae (porpoises) and Monodontidae (narwhals) was calibrated using the earliest record of a phocoenid, the Late Miocene Salumiphocaena [83]. This fossil is approximately 10–11 million years old [83], and we chose a lognormal distribution so that the earliest possible sampled age corresponds to 10 Mya and the older 95% CI encompasses the beginning of the Late Miocene (11.2 Mya) (standard deviation = 1.138).
Each BEAST analysis consisted of 2 × 107 generations with a random starting tree, birth-death default priors (with the exception that we used a uniform [0, 100] prior distribution for the GTR substitution rates), sampled every 1000 generations. Previous studies have demonstrated that applying separate models of nucleotide evolution to specific subsets of nucleotide data (i.e., "partitioned" or "mixed-model" analyses) improves phylogenetic reconstruction [84–86]. We therefore partitioned the data a priori by codon position for the combined protein coding gene data set (three total partitions). To determine convergence, we constructed cumulative posterior probability plots for each clade using the cumulative function in AWTY [87]. Stationarity was assumed when the cumulative posterior probabilities of all clades stabilized. These plots indicated that excluding the first two million generations as burn-in was sufficient, and the frequency of inferred relationships in the remaining trees represented estimated posterior probabilities. To decrease the chance of reaching apparent stationarity on local optima, two separate analyses were performed. Posterior probability estimates for each clade were then compared between the two analyses using a scatter-plot created by the compare command in AWTY. If posterior probability estimates for clades were similar in both analyses, the results of the analyses were combined. Posterior probabilities ≥ 0.95 are considered statistically significant (i.e., "strong") clade support [65].
References
Arnason U, Best PB: Phylogenetic relationships within the Mysticeti (whalebone whales) based upon studies of highly repetitive DNA in all extant species. Hereditas. 1991, 114: 263-269. 10.1111/j.1601-5223.1991.tb00333.x.
Arnason U, Gullberg A: Comparison between the complete mtDNA sequences of the blue and the fin whale, two species that can hybridize in nature. J Mol Evol. 1993, 37: 312-322.
Arnason U, Gullberg A: Relationship of baleen whales established by cytochrome b gene comparison. Nature. 1994, 367: 726-728. 10.1038/367726a0.
Arnason U, Gullberg A: Cytochrome b nucleotide sequences and the identification of five primary lineages of extant cetaceans. Mol Biol Evol. 1996, 13: 407-417.
Arnason U, Gullberg A, Janke A: Mitogenomic analyses provide new insights into cetacean origin and evolution. Gene. 2004, 333: 27-34. 10.1016/j.gene.2004.02.010.
Arnason U, Gullberg A, Gretarsdottir S, Ursing B, Janke A: The mitochondrial genome of the sperm whale and a new molecular reference for estimating eutherian divergence dates. J Mol Evol. 2000, 50: 569-578.
Milinkovitch MC: Molecular phylogeny of cetaceans prompts revision of morphological transformations. Trends Ecol Evol. 1995, 10: 328-334. 10.1016/S0169-5347(00)89120-X.
Messenger SL, McGuire JA: Morphology, molecules, and the phylogenetics of Cetaceans. Syst Biol. 1998, 47: 90-124. 10.1080/106351598261058.
LeDuc RG, Perrin WF, Dizon AE: Phylogenetic relationships among the delphinid cetaceans based on full cytochrome b sequences. Mar Mamm Sci. 1999, 15: 619-648. 10.1111/j.1748-7692.1999.tb00833.x.
Cassens I, Vicario S, Wadde VG, Balchowsky H, Balle DV, Wang D, Chen F, Mohan RSL, Simoes-Lopes PC, Bastida R, Meyer A, Stanhope MJ, Millinkovitch MC: Independent adaptation to riverine habitats allowed survival of ancient cetacean lineages. Proc Natl Acad Sci USA. 2000, 97: 11343-11347. 10.1073/pnas.97.21.11343.
Hamilton H, Caballero S, Collins AG, Browbnell RL: Evolution of river dolphins. Proc R Soc Lond B. 2001, 268: 549-558. 10.1098/rspb.2000.1385.
Geisler JH, Sanders A: Morphological evidence for the phylogeny of Cetacea. J Mamm Evol. 2003, 10: 23-129. 10.1023/A:1025552007291.
Nikaido M, Matsuno F, Hamilton H, Brownell JRL, Cao Y, Wang D, Zhu ZY, Shedlock AM, Fordyce RE, Hasegawa M, Okada N: Retroposon analysis of major cetacean lineages: the monophyly of the toothed whales and the paraphyly of river dolphins. Proc Natl Acad Sci. 2001, 98: 7384-7389. 10.1073/pnas.121139198.
Nishida S, Pastene LA, Goto M, Koike H: SRY gene structure and phylogeny in the cetacean species. Mamm Stud. 2003, 28: 57-66. 10.3106/mammalstudy.28.57.
Rychel AL, Reeder TW, Berta A: Phylogeny of mysticete whales based on mitochondrial and nuclear data. Mol Phylogenet Evol. 2004, 32: 892-901. 10.1016/j.ympev.2004.02.020.
Harlin-Cognato AD, Honeycutt RL: Multi-locus phylogeny of dolphins in the subfamily Lissodelphininae: Character synergy improves phylogenetic resolution. BMC Evol Biol. 2006, 6: 87-92. 10.1186/1471-2148-6-87.
May-Collado L, Agnarsson I: Cytochrome b and Bayesian inference of whale phylogeny. Mol Phylogenet Evol. 2006, 38: 344-354. 10.1016/j.ympev.2005.09.019.
Nishida S, Goto M, Pastene LA, Kanda N, Koike H: Phylogenetic relationships among cetaceans revealed by Y-chromosome sequences. Zool Sci. 2007, 7: 723-732. 10.2108/zsj.24.723.
Caballero S, Jackson J, Mignucci-Giannoni AA, Barrios-Garrido H, Beltran-Pedreros S, Montiel-Villalobos MG, Robertson KM, Baker CS: Molecular systematics of South American dolphins Sotalia: Sister taxa determination and phylogenetic relationships, with insights into a multi-locus phylogeny of the Delphinidae. Mol Phylogenet. 2008, 46: 252-268. 10.1016/j.ympev.2007.10.015.
Hershkovitz P: Catalog of living whales. US Natl Mus Bull. 1966, 246: 1-259.
Rice DW: Marine mammals of the world: Systematics and distribution. The Society for Marine Mammalogy. Special Publication No. 4. Edited by: Lawrence KS. 1998, 1-231.
Ross GJB, Cockcroft VG: Comments on Australian bottlenose dolphins and the taxonomic status of Tursiops aduncus (Ehrenberg, 1832). The bottle dolphin. Edited by: Leatherwood S, Reeves RR. 1990, San Diego: Academic Press, 101-128.
Wang JY, Chou LS, White BN: Osteological differences between two sympatric forms of bottlenose dolphins (genus Tursiops) in Chinese waters. J Zool Lond. 2000, 252: 147-162. 10.1111/j.1469-7998.2000.tb00611.x.
Verma SK, Sinha RK, Singh L: Phylogenetic position of Platanista gangetica: insights from the mitochondrial cytochrome b gene and nuclear interphotoreceptor retinoid-binding protein gene sequences. Mol Phylogenet Evol. 2004, 33: 280-288. 10.1016/j.ympev.2004.06.018.
Huelsenbeck JP: The performance of phylogenetic methods in simulation. Syst Biol. 1995, 44: 17-48. 10.2307/2413481.
Huelsenbeck JP: The robustness of two phylogenetic methods: Four-taxon simulations reveal a slight superiority of maximum likelihood over neighbor joining. Mol Biol Evol. 1995, 12: 843-849.
Swofford DL, Olsen GJ, Waddell PJ, Hillis DM: Phylogenetic inference. Molecular Systematics. Edited by: Hillis DM, Moritz C, Mable B. 1996, Sunderland: Sinauer Associates, 407-514. 2
Swofford DL, Waddell PJ, Huelsenbeck JP, Foster PG, Lewis PO, Rogers JS: Bias in phylogenetic estimation and its relevance to the choice between parsimony and likelihood methods. Syst Biol. 2001, 50: 525-539. 10.1080/106351501750435086.
Felsenstein J: Inferring Phylogenies. 2004, Sunderland: Sinauer Associates
Yan J, Zhou K, Yang G: Molecular phylogenetics of 'river dolphins' and the baiji mitochondrial genome. Mol Phylogenet Evol. 2005, 37: 743-750. 10.1016/j.ympev.2005.06.009.
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A: Relaxed phylogenetics and dating with confidence. PLOS Biology. 2006, 4: e88-10.1371/journal.pbio.0040088.
Fordyce E, de Muizon C: Evolutionary history of cetaceans: A review. Secondary Adaptation of Tetrapods to Life in Water. Edited by: Mazin JM, de Buffrenil V. 2001, Munchen: Verlag, F. Pfeil, 169-233.
Drummond AJ, Rambaut A: BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007, 7: 214-10.1186/1471-2148-7-214.
Sasaki T, Nikaido M, Hamilton H, Goto M, Kato H, Kanda N, Pastene LA, Cao Y, Fordyce RE, Hasegawa M: Mitochondrial phylogenetics and evolution of Mysticete whales. Syst Biol. 2005, 54: 77-90. 10.1080/10635150590905939.
Ojala D, Crews S, Montoya J, Gelfand R, Attardi G: A small polyadenylated RNA (7S RNA), containing a putative ribosome attachment site, maps near the origin of human mitochondrial DNA replication. J Mol Biol. 1981, 150: 303-314. 10.1016/0022-2836(81)90454-X.
Nikaido M, Piskurek O, Okada N: Toothed whale monophyly reassessed by SINE insertion analysis: The absence of lineage sorting effects suggests a small population of a common ancestral species. Mol Phylogenet Evol. 2007, 43: 216-224. 10.1016/j.ympev.2006.08.005.
deMuizon C: Les vertébrés fossiles de la Formation Pisco (Pérou). III. Les odontocètes du Miocène. Editions Recherche sur les Civilisations Mémoire. 1988, 78: 1-244.
deMuizon C: A new Ziphiidae from the Early Miocene of Washington state and a phylogenetic analysis of the major group of odontocetes. Bull Mus Natl Hist Nat Paris. 1991, 3–4: 279-326.
Heyning JE: Comparative facial anatomy of beaked whales (Ziphiidae) and a systematic revision among the families of extant Odontoceti. Contrib Sci Nat Hist Mus Los Angeles. 1989, 405: 1-64.
Yang G, Zhou K: A study on the molecular phylogeny of river dolphins. Acta Theriol Sin. 1999, 19: 1-9.
Jackman TR, Larson A, de Queiroz K, Losos JB: Phylogenetic relationships and tempo of early diversification in Anolis lizards. Syst Biol. 1999, 48: 254-285. 10.1080/106351599260283.
McGuire JA, Kiew BH: Phylogenetic systematics of Southeast Asian flying lizards (Iguania: Agamidae: Draco) as inferred from mitochondrial DNA sequence data. Biol J Linn Soc. 2001, 73: 203-229.
Poe S, Chubb AL: Birds in a bush: five genes indicate explosive evolution of avian orders. Evolution. 2004, 58: 404-415.
Schulte JA, Valladares JP, Larson A: Phylogenetic relationships within Iguanidae using molecular and morphological data and a phylogenetic taxonomy of iguanian lizards. Herpetologica. 2003, 59: 399-419. 10.1655/02-48.
Haq BH, Hardenbol J, Vail PR: Chronology of fluctuating sea levels since the Triassic. Science. 1987, 235: 1156-1162. 10.1126/science.235.4793.1156.
Wright JD, Miller KG, Fairbanks RG: Early and Middle Miocene stable isotopes: implications for deepwater circulation and climate. Paleoceanography. 1992, 7: 357-389. 10.1029/92PA00760.
Miller KG, Fairbanks RG, Mountain GS: Tertiary oxygen isotope synthesis, sea level history, and continental margin erosion. Paleoceanography. 1987, 2: 1-19. 10.1029/PA002i001p00001.
Zanazzi Z, Kohn1 MJ, MacFadden BJ, Terry DO: Large temperature drop across the Eocene-Oligocene transition in central North America. Nature. 2006, 445: 639-642. 10.1038/nature05551.
Merico A, Tyrrell T, Wilson PA: Letters to Nature: Eocene/Oligocene ocean de-acidification linked to Antarctic glaciation by sea-level fall. Nature. 2008, 452: 979-982. 10.1038/nature06853.
Fordyce RE: Whale evolution and Oligocene Southern Ocean environments. Palaeogeogr Palaeoclimatol Palaeoecol. 1980, 31: 319-336. 10.1016/0031-0182(80)90024-3.
Fordyce RE: Origins and evolution of Antarctic marine mammals. Geological Society, London, special publications. 1989, 47: 269-281. 10.1144/GSL.SP.1989.047.01.20.
Fordyce RE: etacean evolution and Eocene/Oligocene environments. Eocene-Oligocene Climatic and Biotic Evolution. Edited by: Prothero DR, Berggren WA. 1992, New Jersey: Princeton University Press, 368-381.
Barnes LG: Fossil pontoporiid dolphins (Mammalia: Cetacea) from the Pacific coast of North America. Contrib Sci Nat Hist Mus Los Angeles. 1985, 363: 1-34.
Waddell VG, Milinkovitch MC, Berube M, Stanhope MJ: Molecular phylogenetic examination of the Delphinoidea trichotomy: Congruent evidence from three nuclear loci indicates that porpoises (Phocoenidae) share a more recent common ancestry with white whales (Monodontidae) than they do with true dolphins (Delphinidae). Mol Phylogenet Evol. 2000, 15: 314-318. 10.1006/mpev.1999.0751.
Heyning JE: Sperm whale phylogeny revisited: Analysis of the morphological evidence. Marine Mammal Science. 1997, 13: 596-613. 10.1111/j.1748-7692.1997.tb00086.x.
Perrin WF, Mitchell ED, Mead JG, Caldwell DK, van Bree PJH: Stenella clymene, a rediscovered tropical dolphin of the Atlantic. J Mammal. 1981, 62: 583-598. 10.2307/1380405.
True FW: Contributions to the natural history of the cetaceans, a review of the family Delphinidae. US Natl Mus Bull. 1889, 36: l-191.
Wang JY, Chou LS, White BN: Mitochondrial DNA analysis of sympatric morphotypes of bottlenose dolphins (genus Tursiops) in Chinese waters. Mol Ecol. 1999, 8: 1603-1612. 10.1046/j.1365-294x.1999.00741.x.
Seutin G, White BN, Boag PT: Preservation of avian blood and tissue samples for DNA analysis. Can J Zool. 1991, 69: 82-90. 10.1139/z91-013.
Sambrook J, Russell DW: Molecular cloning: a laboratory manual. 2001, New York: Cold Spring Harbor Laboratory Press, 3
Huelsenbeck JP, Larget B, Miller RE, Ronquist F: Potential applications and pitfalls of Bayesian inference of phylogeny. Syst Biol. 2002, 51: 673-688. 10.1080/10635150290102366.
Larget B, Simon DL: Markov chain Monte Carlo algorithms for the Bayesian analysis of phylogenetic trees. Mol Biol Evol. 1999, 16: 750-759.
Rannala B, Yang Z: Probability distribution of molecular evolutionary trees: a new method of phylogenetic inference. J Mol Evol. 1996, 43: 304-311. 10.1007/BF02338839.
Yang Z, Rannala B: Bayesian phylogenetic inference using DNA sequences: a Markov chain Monte Carlo method. Mol Biol Evol. 1997, 14: 717-724.
Huelsenbeck JP, Rannala B: Frequentist properties of Bayesian posterior probabilities of phylogenetic trees under simple and complex substitution models. Syst Biol. 2004, 53: 904-913. 10.1080/10635150490522629.
Ho SYW: Calibrating molecular estimates of substitution rates and divergence times in birds. J Avian Biol. 2007, 38: 409-414.
Gingerich PD, Russel DE: Pakicetus inachus, a new archaeocete (Mammalia, Cetacea) from the Early-Middle Eocene Kuldana Formation of Kohat (Pakistan). Contributions from the Museum of Paleontology, University of Michigan. 1981, 28: 1-20.
Thewissen JGM, Williams EM, Roe LJ, Hussain ST: Skeletons of terrestrial cetaceans and the relationship of whales to artiodactyls. Nature. 2001, 413: 277-281. 10.1038/35095005.
Ho SYW, Phillips MJ, Cooper A, Drummond AJ: Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol Biol Evol. 2005, 22: 1561-1568. 10.1093/molbev/msi145.
Gatesy J, Hayashi C, Cronin M, Arctander P: Evidence from milk casein genes that cetaceans are close relatives of hippopotamid artiodactyls. Mol Biol Evol. 1996, 13: 954-963.
Gatesy J: More support for a Cetacea/Hippopotamidae clade: The blood clotting protein gene g-fibrinogen. Mol Biol Evol. 1997, 14: 537-543.
Ursing BM, Arnason U: Analyses of mitochondrial genomes strongly support a hippopotamus-whale clade. Proc Biol Sci. 1998, 265 (1412): 2251-2255. 10.1098/rspb.1998.0567.
Gatesy J, Milinkovitch M, Waddell V, Stanhope M: Stability of cladistic relationships between Cetacea and higher-level artiodactyl taxa. Syst Biol. 1999, 48: 6-20. 10.1080/106351599260409.
Nikaido M, Rooney AP, Okada N: Phylogenetic relationships among cetartiodactyls based on insertions of short and long interpersed elements: Hippopotamuses are the closest extant relatives of whales. Proc Natl Acad Sci. 1999, 96: 10261-10266. 10.1073/pnas.96.18.10261.
Lum JK, Nikaido M, Shimamura M, Shimodaira H, Shedlock AM, Okada N, Hasegawa M: Consistency of SINE insertion topology and flanking sequence tree: quantifying relationships among cetartiodactyls. Mol Biol Evol. 2000, 17: 1417-1424.
Mathee CA, Burzlaff JD, Taylor JF, Davis S: Mining the mammalian genome for artiodactyl systematics. Syst Biol. 2001, 50: 367-390. 10.1080/106351501300317987.
Boissere JR, Lihoreau F, Brunet M: The position of Hippopotamidae within Cetartiodactyla. Proc Natl Acad Sci. 2005, 102: 1537-1541. 10.1073/pnas.0409518102.
O'Leary MA, Gatesy J: Impact of increased character sampling on the phylogeny of Cetartiodactyla (Mammalia): combined analysis including fossils. Palaeontology. 2008, 24: 397-442.
Prothero DR, Berggren WA: Eocene-Oligocene climatic and biotic evolution. 1992, New Jersey: Princeton University Press
Berggren WA, Kent DV, Obradovich JD, Swisher CC: Toward a revised Paleogene geochronology. Eocene-Oligocene Climatic and Biotic Evolution. Edited by: Prothero DR, Berggren WA. 1992, New Jersey: Princeton University Press, 29-45.
Mchedlidze GA: Nekotorye obshie cherty istorii kitoobraznykh. (Some general characteristics of the evolution of cetaceans). 1970, Metsniereba Press, Institut Paleobiologii Akademia Nauk, Gruzinskoi SSR, Tbilisi, Georgia, 1-112.
deMuizon C: Pliopontos littoralis, un noveau Platanistidae Cetacea du Pliocène de la côte pèruvienne. Comptes Rendus de L'Academie des Sciences. 1983, 296: 625-628.
Barnes LG: Evolution, taxonomy, and antitropical distributions of the porpoises (Phocoenidae, Mammalia). Mar Mammal Sci. 1985, 1: 149-165. 10.1111/j.1748-7692.1985.tb00003.x.
Brandley MC, Schmitz A, Reeder TW: Partitioned bayesian analyses, partition choice, and the phylogenetic relationships of scincid lizards. Syst Biol. 2005, 54: 373-390. 10.1080/10635150590946808.
Castoe TA, Doan TM, Parkinson CL: Data partitions and complex models in Bayesian analysis: the phylogeny of gymnophthalmid lizards. Syst Biol. 2004, 53: 448-469. 10.1080/10635150490445797.
Nylander JAA, Ronquist F, Huelsenbeck JP, Nieves-Aldrey JL: Bayesian phylogenetic analysis of combined data. Syst Biol. 2004, 53: 47-67. 10.1080/10635150490264699.
Nylander JAA, Wilgenbusch JC, Warren DL, Swofford DL: AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics. 2008, 24: 581-583. 10.1093/bioinformatics/btm388.
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (NSFC) key project grant number 30830016 and NSFC grant numbers 30670294 and 30470253 to GY, the Program for New Century Excellent Talents in University (NCET-07-0445), the Ministry of Education of China to GY, the Specialized Research Fund for the Doctoral Program of Higher Education (SRFDP 20060319002), the Ministry of Education of China to GY, and the major project of the Natural Science Foundation of the Jiangsu Higher Education Insitutions of China (07KJA18016) to GY. We are grateful to Mr Anli Gao, Xinrong Xu, Hua Chen, and Qing Chang for their assistance and contribution to collecting samples for many years. The Penglai Oceanic Polar World, Penglai, China, kindheartedly provided us with fresh blood samples of the common bottlenose dolphin. We especially thank Jonathan J. Fong, Adam D. Leaché, and Ainsley E. Seago for their critical reading on this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
YX participated in the design of the study and performed most of the laboratory work. MCB and YX conducted all phylogenetic analyses and prepared the manuscript. SX helped to sequence the mitochondrial genome of the Indo-Pacific humpbacked dolphin. KZ helped to improve the manuscript. GY made the overall design of the study, and helped to draft and improve the manuscript. All authors read and approved the last version of the manuscript.
Electronic supplementary material
12862_2008_934_MOESM1_ESM.xls
Additional file 1: Primers used for amplifying and sequence the Delphinidae mtDNA in this study. This table provides names and relevant sequence information of the primers used for amplifying and sequencing the complete mtDNA genome of seven delphinids. (XLS 17 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Xiong, Y., Brandley, M.C., Xu, S. et al. Seven new dolphin mitochondrial genomes and a time-calibrated phylogeny of whales. BMC Evol Biol 9, 20 (2009). https://doi.org/10.1186/1471-2148-9-20
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-9-20