
Abstract
Background
Populations of the teleost fish Fundulus heteroclitus appear to flourish in heavily polluted and geographically separated Superfund sites. Populations from three Superfund sites (New Bedford Harbor, MA, Newark Bay, NJ, and Elizabeth River, VA) have independently evolved adaptive resistance to chemical pollutants. In these polluted populations, natural selection likely has altered allele frequencies of loci that affect fitness or that are linked to these loci. The aim of this study was to identify loci that exhibit non-neutral behavior in the F. heteroclitus genome in polluted populations versus clean reference populations.
Results
To detect signatures of natural selection and thus identify genetic bases for adaptation to anthropogenic stressors, we examined allele frequencies for many hundreds of amplified fragment length polymorphism markers among populations of F. heteroclitus. Specifically, we contrasted populations from three Superfund sites (New Bedford Harbor, MA, Newark Bay, NJ, and Elizabeth River, VA) to clean reference populations flanking the polluted sites. When empirical FST values were compared to a simulated distribution of FST values, 24 distinct outlier loci were identified among pairwise comparisons of pollutant impacted F. heteroclitus populations and both surrounding reference populations. Upon removal of all outlier loci, there was a strong correlation (R2 = 0.79, p < 0.0001) between genetic and geographical distance. This apparently neutral evolutionary pattern was not evident when outlier loci were included (R2 = 0.092, p = 0.0721). Two outlier loci were shared between New Bedford Harbor and Elizabeth River populations, and two different loci were shared between Newark Bay and Elizabeth River populations.
Conclusion
In total, 1% to 6% of loci are implicated as being under selection or linked to areas of the genome under selection in three F. heteroclitus populations that reside in polluted estuaries. Shared loci among polluted sites indicate that selection may be acting on multiple loci involved in adaptation, and loci shared between polluted sites potentially are involved in a generalized adaptive response.
Similar content being viewed by others
Background
The genetic basis of adaptation is a fundamental issue in evolutionary biology. Much of the research in this field has been focused on the classic model systems of Drosophila [1–13] and Arabidopsis [14–18]. Recently, insight into adaptation in non-model species has become possible due to advances in molecular biology and statistics [19–31]. This recent expansion into studies of non-model systems allows further development of evolutionary inferences [32], such as the role that selection, mutation, gene flow, and drift play in adaptation [33]. A powerful approach to understand genome-wide adaptation is to investigate independent natural populations that inhabit environments with strong selective pressures.
One species that has adapted to a wide range of estuarine environments is the teleost fish, Fundulus heteroclitus [34]. F. heteroclitus is widely distributed along the United States' eastern seaboard from the Gulf of St. Lawrence to northeastern Florida [35]. Subpopulations of F. heteroclitus inhabit clean estuaries as well as those heavily impacted by chemical pollutants (reviewed in [36]). Three well-known polluted sites where F. heteroclitus reside are New Bedford Harbor (Massachusetts), Newark Bay (New Jersey), and Elizabeth River (Norfolk, VA). All three sites have been identified by the Environmental Protection Agency (EPA) as Superfund sites (part of the federal government's program to clean up the nation's uncontrolled hazardous waste sites) and contain high levels of a variety of lipophilic, persistent and toxic contaminants worthy of remediation using Federal funds. All three Superfund sites are highly contaminated with chemical pollutants that are broadly classified as aromatics. New Bedford Harbor is polluted with extremely high levels of polychlorinated biphenyls [37] as well as polychlorinated dibenzo-p-dioxins (PCDD), polychlorinated dibenzofurans (PCD), polycyclic aromatic hydrocarbons (PAH), and several trace metals [37, 38]. Newark Bay is most notorious for containing 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) as well as other dioxins [39, 40] and also is contaminated with heavy metals, pesticides, PCBs and PAHs [41]. The Elizabeth River is predominantly contaminated with creosote, comprised of a complex mixture of PAHs [42–44].
F. heteroclitus from these chronically polluted areas are resistant to the aromatic hydrocarbons in their environment as compared to nearby fish from relatively clean environments [45–52]. Resistance in first and second generation embryos from New Bedford Harbor and Elizabeth River and first generation embryos from depurated Newark Bay fish suggests that differential survival is due to genetic adaptation rather than physiological induction. Investigating and comparing F. heteroclitus from these three sites provides the opportunity to study similarities and differences in adaptation to differing chemical pollutant and resistance to general stress conditions among populations.
Previous work to elucidate mechanisms of resistance and the underlying genetic basis in F. heteroclitus from these three sites has investigated the refractory phenotype of the xenobiotic metabolizing enzyme cytochrome P4501A (CYP1A) in polluted populations [47, 48, 53–55], epigenetic silencing through CpG methylation of promoter regions of the CYP1A 5' promoter region [56], and elimination of contaminants through the induction of other phase I, II, and III enzymes [55, 57–59], many by way of the aryl hydrocarbon receptor (AHR) pathway (reviewed in [60]). Yet, none of these research efforts has completely accounted for the differences in the resistance phenotypes between polluted and reference site fish in New Bedford Harbor, Newark Bay and Elizabeth River, nor has the genetic basis for resistance been elucidated.
In contrast to a candidate gene approach, our strategy to begin to understand the genetic mechanisms that enable F. heteroclitus populations to inhabit these highly polluted sites was to screen the genome for selectively important loci. The premise is that loci under selection will have patterns of variation statistically different from the majority of neutral loci [61]. Loci that have a large difference in allele frequencies between populations with respect to what would be expected under the neutral expectation are outliers. The identification of these outliers provides evidence for which and how many loci may be involved in the evolutionary adaptation to anthropogenic pollution.
Loci can have significantly different frequencies relative to other neutral loci for many reasons. To obviate the detection of outliers due to genetic drift rather than selection, our sampling scheme contrasted each polluted population with two reference populations that were geographically more distant from each other than either was to the polluted population. This provides a control for each Superfund site by identifying which loci are significant outliers relative to two reference sites that are demographically distant from each other. To provide extensive coverage of the genome, we used approximately 300 amplified fragment length polymorphisms (AFLP) [62] to genotype 288 individuals from nine F. heteroclitus populations and used a modeling approach to reveal significant outliers. Furthermore, we investigated whether outlier loci were shared among polluted populations, suggesting similar patterns of selection on the genome despite differences in pollutant compositions and local conditions.
Methods
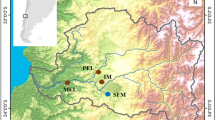
F. heteroclitus were collected using minnow traps during the spring of 2005. Fin clip samples from 32 individuals were sampled from each of the nine collection sites along the east coast of the United States (Fig. 1; Table 1). Three of the collection sites were Superfund sites: New Bedford (EPA ID: MAD980731335), Newark (EPA ID: NJD980528996), and Elizabeth River (EPA ID: VAD990710410). Two non-polluted reference sites flanked each Superfund site, approximately equidistant on either side of each polluted site (Fig. 1; Table 1).

Sample locations. Sampling locations for Fundulus heteroclitus populations. Circles are reference sites and stars are Superfund sites.
Genomic DNA was extracted from fin clips using a modified version of Aljanabi and Martinez [63]. Fin clips were incubated at 55°C for two hours in 300 μL of 75 mM NaCl, 25 mM EDTA, and 1% SDS with Proteinase K (3 μL of 20 mg/mL). Following incubation, 0.5 volumes of 7.5 M ammonium acetate were added and DNA was precipitated on ice with the addition of 0.7 volumes of isopropanol. Subsequently, DNA was pelleted through centrifugation and washed with 70% ethanol. DNA was resuspended overnight at 4°C in 0.1× TE.
The AFLP analysis was performed in replicate following the ligation of the DNA for each individual using a modified version of Vos et al. [62] to generate approximately 300 loci. Genomic DNA (500 ng) was digested with 5 U EcoRI (New England Biolabs, MA) and 5 U MseI (New England Biolabs, MA) overnight at 37°C in a total volume of 45 μL containing 1× T4 DNA ligase buffer (Epicentre) supplemented with 100 μg/mL BSA. Following incubation, 50 pmol adaptor oligonucleotides (Applied Biosystems) and 1 U T4 DNA ligase (Epicentre) were added and incubated overnight at 16°C. Preselective PCRs were performed in a 15 μL volume using 5 μL of diluted (1:10) ligation product with EcoRI + (C/A) primer (Integrated DNA Technologies; 10 pmol), MseI + (C/A) primer (Integrated DNA Technologies; 10 pmol) and 1 U Taq. PCR conditions were 20 cycles of 94°C for 10 sec, 56° for 30 sec, and 72°C for 2 min. Selective Eco + 3NT primers (Integrated DNA Technologies; 10 pmol) labeled with FAM dye at the 5' end and MseI + 3NT primers (Integrated DNA Technologies; 10 pmol) were added to diluted (1:10) pre-selective PCR product in a 15 μL volume. PCR conditions in the first cycle were 94°C for 10 sec, 65°C for 30 sec, and 72°C for 2 min with the annealing temperature reduced by 0.4°C for 12 cycles, then 30 cycles of 94°C for 10 sec, 56°C for 30 sec, and 72°C for 2 min. Semi-automated analysis of the selective PCR products was performed on MegaBACE 1000 DNA sequencing system (GE Healthcare). Peak patterns were calculated using MegaBACE Geneprofiler software v. 1.0 (GE Healthcare). The criteria for distinct peaks were a size between 50 and 400 base pairs and an absolute intensity greater than or equal to 1000. Replicated fragments were obtained from all samples (the same template was used for independent PCRs) and replicate fragments were scored as being present or absent using Peakmatcher software [64]. Peakmatcher software automatically creates marker categories and generates a binary table for the presence and absence of markers based on the minimum 75 percent repeatability of markers across replicates.
Statistical Analysis
The frequency of band presence allele was calculated using the formula P = 1 - ((N - C)/N)0.5 where N equals the sample size and C is the number of individuals with the band [65]. This formula assumes Hardy-Weinberg equilibrium. However, because AFLPs are dominant markers and heterozygotes are not observed, Hardy-Weinberg equilibrium cannot be directly tested. Due to strong selection or increased mutational rates, some of the loci may not be in Hardy-Weinberg equilibrium. Though not directly comparable, microsatellites are in Hardy-Weinberg equilibrium in these F. heteroclitus populations [66]. This calculation also assumes that shared band presence or absence between two individuals is due to common evolutionary origin and not homoplasy. Pairwise FST values between populations were calculated for each locus by the method of Nei [67] with the correction of Nei and Chesser [68] for finite sample sizes, and a null distribution of FST values versus allele frequency was simulated using the Winkles program ([69], Fig. 2).

F ST versus allele frequency values. FST values estimated from approximately 300 variable AFLP loci plotted against mean allele frequency. The solid line represents the 0.99 quantile estimated from a simulation model for each comparison. Loci shared among the same Superfund site are labeled with their primer set (letter) and number. Loci shared between Superfund sites are starred. §Shared loci included in these points are: A2, A19, A34, A56, D87, E118, E127, E137, E150, E156, C186, C194, C205, and C252. E118 also is shared between New Bedford Harbor and Elizabeth River populations.
Winkles is based on the model described in Beaumont and Nichols [20] which employs coalescent simulations using the Island model and an infinite alleles mutational model. Samples of the same size and number as the data are simulated, where each sample is taken from a different island. This simulation uses two populations of size N diploid individuals, with a set mutation rate, μ, and a migration rate, m, per generation. Parameters for the simulation are estimated through the calculation FST = 1/(1 + 16Nm + 16Nμ). The FST value is found by calculating the mean FST from any given pairwise comparison and adjusting that value by -0.0093 to account for the upward bias in the model reported by Wilding et al. [69]; this bias is consistent with previous simulations using Nei's methods to calculate pairwise FST values [70]. The Nm factor is calculated by solving for that parameter in the above equation. Each simulation used 103 and 10-4 as estimates of N and μ, respectively. Simulated FST values are relatively unaffected by changing either the sample size of the simulated population or the mutation rate [20]. Five simulations were run on each pairwise comparison to generate an expected null distribution of 25,000 values. Each simulation started with 500 simulation bi-allelic loci in each of the two populations with uniform random distribution and was allowed to drift for 10N generations. The 99th percentile of FST values within each of the 40 binned mean allele frequency values (each bin representing a set of 0.025 frequency values from 0 to 1) was calculated after removing monomorphic loci because FST is strongly dependent on allele frequencies [20].
The model we used [20] is robust to a wide range of alternative models such as colonization and stepping-stone [5]. It is likely to detect outliers with unusually high FST values and will identify adaptive selection at one or many loci through pairwise comparisons of populations [5, 11]. This model is not able to identify loci under balancing selection and tends to generate discrepancies when numbers of immigrants per generation are unequal, the true population history consists of repeated branching events, or the connectivity of populations is uneven [5]. Isolation, population bottlenecks, and populations which are heterogeneous with respect to their demographic parameters further bias to the model [20]. There is no evidence for isolation and bottleneck history [66] or reduced genetic diversity [71] in our populations. However, if non-homogenous demographic parameters exist (e.g., skewed age structure or sex ratios), this model may be biased. Given the relative robustness of the model to identify loci under adaptive selection, we used theoretical versus experimentally derived allele frequencies for loci to determine significant deviations from the neutral expectation.
Results
Total number of loci among populations
Five different primer combinations (Table 2) were used to amplify approximately 300 loci from 288 individuals from nine different F. heteroclitus populations. Among New Bedford Harbor and its reference sites, Sandwich and Point Judith, a total of 296 loci were scored. Of those 296 loci, 11 bands were found to be monomorphic (3.7%). Newark and its two reference sites, Tuckerton and Clinton, had a total of 336 loci, of which 7 loci were monomorphic (2.1%). Elizabeth River and its two reference sites, Magotha and Manteo, had a total of 299 loci, with 4 loci found to be monomorphic (1.3%). Among all populations, 450 distinct loci were scored.
Outlier loci among populations
In comparisons of the three Superfund sites and their clean reference sites, twenty-four loci show patterns indicative of selection. The criteria for identifying these selective loci are that they were identified as outliers in pairwise comparisons of each Superfund site population relative to its two reference site populations (polluted versus both references, analyzed separately, i.e. the union of polluted versus reference 1 and polluted versus reference 2) but not in comparisons between the reference site populations. Eighteen of these twenty-four loci were found in the New Bedford Harbor comparisons, four were found in the Newark Bay comparisons, and six were found in the Elizabeth River comparisons (Fig. 3). Four of these loci were shared between two Superfund site populations suggesting conserved mechanisms of adaptation (Fig. 4).

Venn diagrams of shared outlier loci in each Superfund comparison. Outlier loci in comparisons of each Superfund populations to both its clean reference sites; numbers in the unions of circles represent outlier loci shared among populations. A) New Bedford Harbor, MA Sandwich, MA and Pt. Judith, RI comparison. B) Newark Bay, NJ, Clinton, CT, and Tuckerton, NJ comparison. C) Elizabeth River, VA, Magotha, VA and Manteo, NC comparison.

Venn diagram of shared outlier loci among Superfund populations. Shared outlier loci among Superfund population comparisons to both clean reference sites; numbers in the unions of circles represent outlier loci shared between two Superfund populations.
In the northern most Superfund site, New Bedford Harbor, 42 loci representing 14% of total analyzed loci were located above the simulated 0.99 quantile in the polluted versus one of the references' comparisons. That is, these 42 loci have FST values that lie outside the expected neutral distribution of 99% of all loci. This is more than 10 fold greater than the 3 that are expected by chance from the approximately 300 amplified loci. These 42 loci are outliers in the New Bedford Harbor comparison to the Point Judith, RI reference population (36 loci), the Sandwich, MA reference population (23 loci) or relative to both reference sites (18 loci). The 18 outlier loci found in the comparisons of New Bedford Harbor to both of its reference populations were amplified from three different primer combinations, spanning a 100 base pair range (Fig. 3A). The joint probability (<0.01 squared or <0.0001) indicates that less than one locus should be different in both clean sites versus the Superfund site. These 18 loci are thus implicated as separate loci under selection or linked to areas of the genome under selection. There are 16 loci that are outliers when comparing the two reference populations to each other. Only one of these 16 outlier loci is specific to the clean reference sites; the other 15 are also found in the comparison to the New Bedford Harbor Superfund site to one of these reference sites. No locus was an outlier in all pairwise comparisons.
Newark Bay, NJ is close to the phylogeographic boundary that separates northern and southern populations of F. heteroclitus [66, 72]. The Clinton reference population is on the northern side and the Tuckerton reference population is on the southern side. The Newark Bay Superfund site has 26 outlier loci (8% versus 1% expected) relative to these two reference sites: 18 (5%) in the comparison with the Clinton reference population and 13 (4%) in the comparison with the Tuckerton reference site population. Four outlier loci are found in both comparisons between the Newark Bay Superfund site and its two clean reference sites (Fig. 3B) and not among clean sites. These four loci are greater than that predicted from the joint probability of differences in both clean sites versus the Superfund site. In pairwise comparisons of the two clean reference sites, 18 loci are outliers. Ten of these 18 loci are common outliers between a northern and two different southern populations i.e., Clinton and Newark Bay populations and Clinton and Tuckerton populations.
Elizabeth River is the most southern Superfund site. The Elizabeth River population, in comparisons to its two reference site populations, had 9 outlier loci (3%). The Elizabeth River and Magotha reference site comparison had 8 outlier loci (2.7% of the total loci) whereas the Elizabeth River and Manteo reference site comparison had 7 (2.4% of the total loci). Six outlier loci were found in both comparisons (Fig. 3C) and not found in the comparison among clean sites. Among the two reference sites (Magotha and Manteo) only three loci were outliers and none of these were unique to the reference-reference comparison. Two loci were in common with outliers from the Elizabeth River-Magotha comparison and one locus was in common with the Elizabeth River-Manteo comparison.
Among the twenty-three loci that were outliers in comparisons only among Superfund sites and both reference sites, four loci are outliers in two of the three Superfund sites (Fig. 4; Table 3). Two of these four outlier loci are shared between New Bedford Harbor and Elizabeth River populations, and two are shared between Newark Bay and Elizabeth River. None is shared between New Bedford and Newark Bay, nor are any shared among all three Superfund site populations.
FST values were calculated for comparisons between all sites with and without outlier loci (Table 4). As would be expected, average FST values were higher in all comparisons before the removal of the outliers. The average FST value (with outliers) between New Bedford Harbor and its reference sites is 0.038, between Newark and its reference sites it is 0.039, and between Elizabeth River and its reference sites it is 0.018. Upon removal of the outliers, average FST values fall to 0.010, 0.016, and 0.011 for New Bedford Harbor, Newark Bay, and Elizabeth River, respectively. These values were plotted against log-ten of geographic distance between sites versus genetic distance [FST/(1 - FST), [73]]. There is no apparent pattern in the distribution of pairwise comparisons corresponding to reference-reference, polluted-reference, or polluted-polluted sites. When outliers were included in the calculation of average FST and plotted against distance, there was no significant linear relationship (R2 = 0.092, p = .0721). Upon removal of the outliers, there was a significant and strong linear relationship (R2 = 0.79, p < 0.0001) between geographic and genetic distance (Fig. 5). Mantel tests that account for multiple comparisons confirmed the significance of both relationships (data not shown). This relationship indicates that 79% of the variability in the neutral genetic distance (without outlier loci) between sites can be explained by geographic distance.

Geographic versus genetic distance. Relationship between genetic distance and geographic distance. Genetic distance was calculated from the mean FST for each pair of populations with (A) and without (B) outlier loci. Circles represent a pairwise comparison of a Superfund versus a reference site, squares represent a Superfund versus a Superfund site comparison, and crosses represent a reference versus a reference site comparison.
Discussion
Multiple F. heteroclitus populations have independently evolved adaptive resistance to complex suites of pollutants [45–52, 74, 75]. These different populations provide independent contrasts for identifying loci involved in adaptation. We identified loci suggestive of adaptation for each polluted population by identifying outlier loci in the polluted population relative to two nearby reference populations. These loci are outliers because they are statistically different from the neutral distribution among populations. Only loci exhibiting a non-neutral distribution in comparisons of the polluted population versus both a north and south reference population were considered to be adaptive. Through this comparison, we are more likely to identify loci whose non-neutral distribution is due to pollution rather than geography. Similarly, while the model used to identify outlier loci has a false positive rate of approximately 7% [11], it is unlikely that the same loci will be falsely identified in multiple comparisons (i.e., in the polluted population versus both a north and south reference populations). In each of the Superfund sites, 1% to 6% (four to 18 loci out of approximately 300) of amplified fragments were identified as being loci under selection or linked to areas of the genome under selection. Four of these loci were outliers in two separate Superfund population comparisons.
We only consider loci exhibiting a non-neutral distribution in comparisons of the polluted population versus both a north and south reference population to be adaptive. These populations make up a geographic triangle formed among the northern and southern clean reference populations and a latitudinally intermediate polluted population (Fig. 1). This double comparison ensures that we are not identifying loci that differ simply due to genetic drift or clinal variation common to this species. This contrast, in addition to the joint comparison among populations, address most of the possible neutral or demographic models. Population isolation can alter allele frequencies among populations. One would expect that a single population that suffered from unique isolation would have significantly greater FST values among many loci in comparison to similarly geographically distance populations that were not uniquely isolated. This demographic explanation does not fit the data for two reasons: 1) it is the statistically different FST value for a few loci in comparison to all other loci that we define as being important, and 2) all non-outlier loci follow the more common demographic trend of isolation by distance (Fig. 5). However, differences in FST values also can result if loci under functional constraints evolve more slowly than loci without functional constraints. Thus, loci with large FST values would have few, if any constraints, relative to the hundreds of other AFLP loci. However, our comparisons were based on both a significant FST between both reference sites versus a polluted site and insignificant differences among reference sites (as well as a difference from the permutation model, see methods). Because we are using three criteria (significant difference versus the joint distribution in two reference sites, lack of a difference among reference sites, and a statistical difference from a neutral permutation model), it seems most parsimonious to suggest that these outlier loci are due to natural selection. However, lack of Hardy-Weinberg equilibrium or recent mutations also might cause loci to be outliers. We suggest that the most obvious cause for this evolved difference is chronic exposure to the aromatic hydrocarbons and other anthropogenic pollutants; yet, we cannot explicitly control every variable in natural environments. Other selective forces also could be different between the three sites. For instance, site complexity differs among the nine sites with the three polluted sites tending to be less complex (have less edges) than the reference sites. Thus, predation or food availability might differ among sites. Similarly, salinity might affect food availability or absorption, and although all populations inhabit brackish waters, the Elizabeth River population is less coastal than the reference populations to which it is compared. Under controlled laboratory conditions, survival differs among fish from clean populations exposed to polluted sediments and fish from polluted populations exposed to clean sediments. This phenomenon points towards adaptation to anthropogenic contaminants rather than differing local conditions for the differences seen between polluted and reference populations. Thus we postulate that outlier loci are due to pollution, especially those loci shared among separate Superfund populations.
Most of the outlier loci are unique to a single polluted population rather than shared across polluted populations (Fig. 4). One explanation for the lack of shared loci is that different loci are involved in the adaptation to a particular pollutant or stress. Alternatively, some of these outliers might be linked to the same locus in the different populations and only appear to be different because the locus under selection dragged different polymorphisms to fixation. This could occur because different polymorphisms existed in the different ancestral populations.
Resistance to pollution is a modern phenotype in F. heteroclitus due to recent exposure (approximately within the last 60 years), suggesting that F. heteroclitus have rapid evolutionary responses with respect to their environment. Our data and other data on survival and development indicate that populations of Fundulus have adapted to local pollutants and thus selection has favored a few alleles. Resistance phenotypes resulting from rapid evolution have been well documented in plants [76] and benthic invertebrates [77] in response to metals as well as in insects in response to pesticides [78] and depend both on population dynamics as well as the strength of selection. F. heteroclitus populations residing in chronically polluted areas provide an advantageous situation whereby strong selective pressures and rapid evolution can be studied. F. heteroclitus have high standing genetic variation [79], high reproductive potential [80], limited home ranges [81] and large population sizes exceeding 10,000 in a single tidal creek [66]. These attributes can and have resulted in locally adapted F. heteroclitus populations. Adaptation due to positive selection often reduces genetic variation among natural populations because of selective sweeps. For example, reduced genetic variation has occurred in brown rats resistant to the rodenticide, warfarin [25, 82, 83], tobacco budworm exposed to the pyrethroid insecticide [84], and the human malarial parasite, Plasmodium falciparum, exposed to antimalarial agents [85]. However, genetic diversity is not reduced in the polluted F. heteroclitus populations compared to the reference site populations for either neutral markers [71, 86, 87] or gene expression [88]. Maintenance of genetic diversity in these populations subjected to significant selection most likely represents steady influx of alternative alleles by migration. If migration and resulting gene flow is strong enough to prevent the reduction of genetic diversity at non-selected loci, it suggests that selection at adaptively important loci is equally strong. Importantly, with constant influx of allelic variation at loci without adaptive value, there should be fewer spurious allelic differences among populations. Thus, shared loci between Superfund populations are likely to be affected by selection and therefore biologically important.
Among three F. heteroclitus populations inhabiting highly polluted Superfund sites and flanking reference populations, 63 different loci (14% of the collective 450 loci) have FST values outside the 99% quantile. Using all loci (i.e., including outliers) our FST values based on AFLP (0.038, 0.039, and 0.018 for New Bedford Harbor, Newark Bay and Elizabeth River, respectively) are approximately one-half of those found for microsatellites (0.077, 0.068, and 0.043, respectively [66]) although these genetic measures are difficult to compare due to differences in genomic coverage and mutation rates [89]. Using AFLPs, McMillan et al. [71] found similar FST values for the New Bedford Harbor population (0.056). For the Elizabeth River population, Mulvey et al. [86] also found similar FST values (0.014) using allozymes. Notice that these calculated FST values use all loci and do not distinguish between neutral and non-neutral loci. If selection affects the frequency of alleles among these molecular markers, the perceived genetic distance (FST) will be exaggerated.
The neutral hypothesis is a powerful tool to explore differences among populations [90]. However, in order to test evolutionary hypotheses, one needs to distinguish between neutral and non-neutral loci. Among populations for each Superfund site, the genetic distances among local populations are affected by the outlier loci. New Bedford Harbor and Newark Bay populations are more differentiated in comparison to their reference site populations than the Elizabeth River populations (FST values of 0.038 and 0.039 versus 0.018) because the Elizabeth River population has the fewest outlier loci (2.4% – 2.7%) in comparison to neutral loci. These differences among Superfund sites do not exist upon removal of outliers: FST values among loci without outlier values are similar for New Bedford Harbor, Newark Bay and Elizabeth River (0.01, 0.016, and 0.011, respectively). With outliers, there is no relationship between FST values and geographic distance. However, upon removal of outlier loci, there is a strong relationship between genetic and geographical distance indicating an equilibrium model of isolation-by-distance. Similar findings have been shown in other F. heteroclitus studies [66, 87], with the intertidal snail [69], and sea trout [91]. Not surprisingly, these data indicate that loci with unusually large FST values have a large and potentially misleading effect on the perceived genetic distance among populations. The 63 outliers exhibit this effect; once removed from the data set, the neutral expectation of increasing genetic distance with geographic distance holds true. For twenty-four of these outlier loci, this non-neutral distribution is most likely caused by evolution by natural selection due to pollution or another strong selective force unique to the polluted sites since the geographical effect was taken into account through the comparison of the polluted sites with both a north and south reference population. Ten other loci have a larger than expected distance at the north-south phylogenetic boundary and likely reflect the historic split among northern and southern F. heteroclitus populations [92–94]. Outlier loci in reference-reference pairwise comparisons likely reflect genetic drift although some may be due to selection. While we can only speculate why these and the remaining 29 loci affect the relationship between genetic and geographic distance, this illustrates the need to distinguish among potentially selected and neutral loci to determine expected differences and posit hypotheses.
Conclusion
Contrasting populations that experience different selective pressures provides insight into evolution by natural selection. Our goal is to understand the genetic basis of adaptive resistance to pollution in chronically contaminated natural populations. Future analyses will address whether polymorphisms between populations are functional and potentially responsible for conferring resistance in populations adapted to chronic exposure to chemical pollutants in the different Superfund sites. We have shown that between 1 to 6% of loci are implicated as being under selection or linked to areas of the genome under selection in three distinct F. heteroclitus populations that reside in polluted Superfund estuaries. Shared loci affected by natural selection among polluted sites indicate that there may be a similar mechanism of resistance in these different populations. This study suggests that multiple loci may be involved in adaptation and a few of these loci have a generalized adaptive response.
References
Posthuma L, Vanstraalen NM: Heavy-Metal Adaptation in Terrestrial Invertebrates – a Review of Occurrence, Genetics, Physiology and Ecological Consequences. Comparative Biochemistry and Physiology C-Pharmacology Toxicology & Endocrinology. 1993, 106 (1): 11-38. 10.1016/0742-8413(93)90251-F.
Beaumont MA, Nichols RA: Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society of London Series B-Biological Sciences. 1996, 263 (1377): 1619-1626. 10.1098/rspb.1996.0237.
Feder ME: Engineering candidate genes in studies of adaptation: The heat-shock protein Hsp70 in Drosophila melanogaster. American Naturalist. 1999, 154: S55-S66. 10.1086/303283.
Kopp A, Duncan I, Godt D, Carroll SB: Genetic control and evolution of sexually dimorphic characters in Drosophila. Nature. 2000, 408 (6812): 553-559. 10.1038/35046017.
Vitalis R, Dawson K, Boursot P: Interpretation of variation across marker loci as evidence of selection. Genetics. 2001, 158 (4): 1811-1823.
Daborn PJ, Yen JL, Bogwitz MR, Le Goff G, Feil E, Jeffers S, Tijet N, Perry T, Heckel D, Batterham P, et al: A single P450 allele associated with insecticide resistance in Drosophila. Science (New York, NY). 2002, 297 (5590): 2253-2256.
Kauer M, Zangerl B, Dieringer D, Schlotterer C: Chromosomal patterns of microsatellite variability contrast sharply in African and non-African populations of Drosophila melanogaster. Genetics. 2002, 160 (1): 247-256.
Hoffmann AA, Sorensen JG, Loeschcke V: Adaptation of Drosophila to temperature extremes: bringing together quantitative and molecular approaches. Journal of Thermal Biology. 2003, 28 (3): 175-216. 10.1016/S0306-4565(02)00057-8.
Presgraves DC, Balagopalan L, Abmayr SM, Orr HA: Adaptive evolution drives divergence of a hybrid inviability gene between two species of Drosophila. Nature. 2003, 423 (6941): 715-719. 10.1038/nature01679.
Riley RM, Jin W, Gibson G: Contrasting selection pressures on components of the Ras-mediated signal transduction pathway in Drosophila. Molecular ecology. 2003, 12 (5): 1315-1323. 10.1046/j.1365-294X.2003.01741.x.
Beaumont MA, Balding DJ: Identifying adaptive genetic divergence among populations from genome scans. Molecular ecology. 2004, 13 (4): 969-980. 10.1111/j.1365-294X.2004.02125.x.
Nuzhdin SV, Wayne ML, Harmon KL, McIntyre LM: Common pattern of evolution of gene expression level and protein sequence in Drosophila. Molecular biology and evolution. 2004, 21 (7): 1308-1317. 10.1093/molbev/msh128.
Wiehe T, Nolte V, Zivkovic D, Schlotterer C: Identification of selective sweeps using a dynamically adjusted number of linked microsatellites. Genetics. 2007, 175 (1): 207-218. 10.1534/genetics.106.063677.
Maloof JN, Borevitz JO, Dabi T, Lutes J, Nehring RB, Redfern JL, Trainer GT, Wilson JM, Asami T, Berry CC, et al: Natural variation in light sensitivity of Arabidopsis. Nature genetics. 2001, 29 (4): 441-446. 10.1038/ng777.
Fowler S, Thomashow MF: Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. The Plant cell. 2002, 14 (8): 1675-1690. 10.1105/tpc.003483.
Weinig C, Ungerer MC, Dorn LA, Kane NC, Toyonaga Y, Halldorsdottir SS, Mackay TF, Purugganan MD, Schmitt J: Novel loci control variation in reproductive timing in Arabidopsis thaliana in natural environments. Genetics. 2002, 162 (4): 1875-1884.
Pigliucci M, Pollard H, Cruzan MB: Comparative studies of evolutionary responses to light environments in Arabidopsis. The American naturalist. 2003, 161 (1): 68-82. 10.1086/345460.
Tian D, Traw MB, Chen JQ, Kreitman M, Bergelson J: Fitness costs of R-gene-mediated resistance in Arabidopsis thaliana. Nature. 2003, 423 (6935): 74-77. 10.1038/nature01588.
Pogson GH, Mesa KA, Boutilier RG: Genetic population structure and gene flow in the Atlantic cod Gadus morhua: a comparison of allozyme and nuclear RFLP loci. Genetics. 1995, 139 (1): 375-385.
Beaumont MA, Nichols RA: Evaluating loci for use in the genetic analysis of population structure. Proc R Soc B. 1996, 263: 1619-1626. 10.1098/rspb.1996.0237.
Yan G, Romero-Severson J, Walton M, Chadee DD, Severson DW: Population genetics of the yellow fever mosquito in Trinidad: comparisons of amplified fragment length polymorphism (AFLP) and restriction fragment length polymorphism (RFLP) markers. Molecular ecology. 1999, 8 (6): 951-963. 10.1046/j.1365-294x.1999.00647.x.
Allendorf FW, Seeb LW: Concordance of genetic divergence among sockeye salmon populations at allozyme, nuclear DNA, and mitochondrial DNA markers. Evolution; international journal of organic evolution. 2000, 54 (2): 640-651.
Bradshaw HD, Ceulemans R, Davis J, Stettler R: Emerging model systems in plant biology: Poplar (Populus) as a model forest tree. Journal of Plant Growth Regulation. 2000, 19 (3): 306-313. 10.1007/s003440000030.
Frary A, Nesbitt TC, Grandillo S, Knaap E, Cong B, Liu J, Meller J, Elber R, Alpert KB, Tanksley SD: fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science (New York, NY). 2000, 289 (5476): 85-88.
Kohn MH, Pelz HJ, Wayne RK: Locus-specific genetic differentiation at Rw among warfarin-resistant rat (Rattus norvegicus) populations. Genetics. 2003, 164 (3): 1055-1070.
Mock KE, Theimer TC, Rhodes OE, Greenberg DL, Keim P: Genetic variation across the historical range of the wild turkey (Meleagris gallopavo). Molecular ecology. 2002, 11 (4): 643-657. 10.1046/j.1365-294X.2002.01467.x.
Parsons YM, Shaw KL: Species boundaries and genetic diversity among Hawaiian crickets of the genus Laupala identified using amplified fragment length polymorphism. Molecular ecology. 2001, 10 (7): 1765-1772. 10.1046/j.1365-294X.2001.01318.x.
Peichel CL, Nereng KS, Ohgi KA, Cole BL, Colosimo PF, Buerkle CA, Schluter D, Kingsley DM: The genetic architecture of divergence between threespine stickleback species. Nature. 2001, 414 (6866): 901-905. 10.1038/414901a.
Storz JF, Dubach JM: Natural selection drives altitudinal divergence at the albumin locus in deer mice, Peromyscus maniculatus. Evolution; international journal of organic evolution. 2004, 58 (6): 1342-1352.
Storz JF, Nachman MW: Natural selection on protein polymorphism in the rodent genus Peromyscus: evidence from interlocus contrasts. Evolution; international journal of organic evolution. 2003, 57 (11): 2628-2635.
Whitehead A, Anderson SL, Kuivila KM, Roach JL, May B: Genetic variation among interconnected populations of Catostomus occidentalis: implications for distinguishing impacts of contaminants from biogeographical structuring. Molecular ecology. 2003, 12 (10): 2817-2833. 10.1046/j.1365-294X.2003.01933.x.
Luikart G, England PR, Tallmon D, Jordan S, Taberlet P: The power and promise of population genomics: from genotyping to genome typing. Nature reviews. 2003, 4 (12): 981-994.
Wang Z, Baker AJ, Hill GE, Edwards SV: Reconciling actual and inferred population histories in the house finch (Carpodacus mexicanus) by AFLP analysis. Evolution; international journal of organic evolution. 2003, 57 (12): 2852-2864.
Griffith RW: Environmental and salinity tolerance in the genus Fundulus. Copeia. 1974, 2: 319-331. 10.2307/1442526.
Duvernell DD, Lindmeier JB, Faust KE, Whitehead A: Relative influences of historical and contemporary forces shaping the distribution of genetic variation in the Atlantic killifish, Fundulus heteroclitus. Molecular ecology. 2008, 17 (5): 1344-1360. 10.1111/j.1365-294X.2007.03648.x.
Wirgin I, Waldman JR: Resistance to contaminants in North American fish populations. Mutation research. 2004, 552 (1–2): 73-100.
Pruell R, Norwood C, Bowen R, Boothman W, Rogerson P, Hackett M, Butterworth B: Geochemical study of sediment contamination in New Bedford Harbor, Massachusetts. Marine Environmental Research. 1990, 29: 77-101. 10.1016/0141-1136(90)90030-R.
Bergen BJ, Rahn K, Nelson WG: Remediation at a Marine Superfund Site: Surficial Sediment PCB Congener Concentration, Composition and Redistribution. Environmental Science and Technology. 1998, 32: 3496-3501. 10.1021/es980413o.
Prince R, Cooper KR: Comparisons of the the effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on chemically impacted and nonimpacted subpopulations of Fundulus heteroclitus: I. TCDD Toxicity. Environ Toxicol Chem. 1995, 14 (4): 579-587. 10.1897/1552-8618(1995)14[579:COTEOD]2.0.CO;2.
Weis J: Tolerance to environmental contaminants in the mummichug, Fundulus heteroclitus. Human and Ecological Risk Assessment. 2002, 8: 933-953. 10.1080/1080-700291905756.
Iannuzzi TJ, Armstrong TN, Thelen JB, Ludwig DF, Firstenberg CE: Characterization of chemical contamination in shallow-water estuarine habitats of an industrialized river. Part 1: Organic compounds. Soil & Sediment Contamination. 2005, 14 (1): 13-33. 10.1080/15320380590891790.
Bieri R, Hein C, Huggett R, Shou P, Slone H: Polycyclic Aromatic Hydrocarbons in Surface Sediments from the Elizabeth River Subestuary. International Journal of Environmental Analytical Chemistry. 1986, 26: 97-113. 10.1080/03067318608077108.
Huggett R, Van Veld P, Smith C, Hargis W, Vogelbein W: The Effects of Contaminated Sediments in the Elizabeth River. 1992, Boca Raton: Lewis Publishers
Padma T, Hale R, Roberts M: Toxicity of water-soluble fractions derived from whole creosote and creosote-contaminated sediments. Environmental Toxicology and Chemistry. 1998, 17: 1606-1610. 10.1897/1551-5028(1998)017<1606:TOWSFD>2.3.CO;2.
Vogelbein WK, Fournie JW, Vanveld PA, Huggett RJ: Hepatic Neoplasms in the Mummichog Fundulus-Heteroclitus from a Creosote-Contaminated Site. Cancer Research. 1990, 50 (18): 5978-5986.
Black DE, Gutjahr-Gobell R, Pruell RJ, Bergen B, Mills L, McElroy AE: Reproduction and polychlorinated biphenyls in Fundulus heteroclitus (Linnaeus) from New Bedford Harbor, Massachusetts, USA. Environmental Toxicology and Chemistry. 1998, 17 (7): 1405-1414. 10.1897/1551-5028(1998)017<1405:RAPBIF>2.3.CO;2.
Elskus AA, Monosson E, McElroy AE, Stegeman JJ, Woltering DS: Altered CYP1A expression in Fundulus heteroclitus adults and larvae: a sign of pollutant resistance?. Aquatic Toxicology. 1999, 45 (2–3): 99-113. 10.1016/S0166-445X(98)00102-7.
Nacci D, Coiro L, Champlin D, Jayaraman S, McKinney R, Gleason TR, Munns WR, Specker JL, Cooper KR: Adaptations of wild populations of the estuarine fish Fundulus heteroclitus to persistent environmental contaminants. Marine Biology. 1999, 134 (1): 9-17. 10.1007/s002270050520.
Meyer JN, Nacci DE, Di Giulio RT: Cytochrome P4501A (CYP1A) in killifish (Fundulus heteroclitus): Heritability of altered expression and relationship to survival in contaminated sediments. Toxicological Sciences. 2002, 68 (1): 69-81. 10.1093/toxsci/68.1.69.
Nacci DE, Champlin D, Coiro L, McKinney R, Jayaraman S: Predicting the occurrence of genetic adaptation to dioxinlike compounds in populations of the estuarine fish Fundulus heteroclitus. Environmental toxicology and chemistry/SETAC. 2002, 21 (7): 1525-1532. 10.1897/1551-5028(2002)021<1525:PTOOGA>2.0.CO;2.
Ownby DR, Newman MC, Mulvey M, Vogelbein WK, Unger MA, Arzayus LF: Fish (Fundulus heteroclitus) populations with different exposure histories differ in tolerance of creosote-contaminated sediments. Environmental toxicology and chemistry/SETAC. 2002, 21 (9): 1897-1902. 10.1897/1551-5028(2002)021<1897:FFHPWD>2.0.CO;2.
Meyer JN, Di Giuliuo RT: Heritable adaptation and fitness costs in killifish (Fundulus beteroclitus) inhabiting a polluted estuary. Ecological Applications. 2003, 13 (2): 490-503. 10.1890/1051-0761(2003)013[0490:HAAFCI]2.0.CO;2.
Prince R, Cooper KR: Comparisons of the Effects of 2,3,7,8-Tetrachlorodibenzo-P-Dioxin on Chemically Impacted and Nonimpacted Subpopulations of Fundulus-Heteroclitus .1. Tcdd Toxicity. Environmental Toxicology and Chemistry. 1995, 14 (4): 579-587. 10.1897/1552-8618(1995)14[579:COTEOD]2.0.CO;2.
Van Veld PA, Westbrook DJ: Evidence for depression of cytochrome P4501A in a population of chemically resistant mummichog (Fundulus heteroclitus). Environ Sci. 1995, 3: 221-234.
Bello SM, Franks DG, Stegeman JJ, Hahn ME: Acquired resistance to Ah receptor agonists in a population of Atlantic killifish (Fundulus heteroclitus) inhabiting a marine superfund site: In vivo and in vitro studies on the inducibility of xenobiotic metabolizing enzymes. Toxicological Science. 2001, 60: 77-91. 10.1093/toxsci/60.1.77.
Timme-Laragy AR, Meyer JN, Waterland RA, Di Giulio RT: Analysis of CpG methylation in the killifish CYP1A promoter. Comp Biochem Physiol C Toxicol Pharmacol. 2005, 141 (4): 406-411.
Bard SM, Bello SM, Hahn ME, Stegeman JJ: Expression of P-glycoprotein in killifish (Fundulus heteroclitus) exposed to environmental xenobiotics. Aquatic Toxicology. 2002, 59 (3–4): 237-251. 10.1016/S0166-445X(01)00256-9.
Cooper PS, Vogelbein WK, Van Veld PA: Altered expression of the xenobiotic transporter P-glycoprotein in liver and liver tumours of mummichog (Fundulus heteroclitus) from a creosote-contaminated environment. Biomarkers. 1999, 4 (1): 48-58. 10.1080/135475099230994.
Armknecht SL, Kaattari SL, Van Veld PA: An elevated glutathione S-transferase in creosote-resistant mummichog (Fundulus heteroclitus). Aquatic Toxicology. 1998, 41 (1–2): 1-16. 10.1016/S0166-445X(97)00074-X.
Hahn ME: Mechanisms of innate and acquired resistance to dioxin-like compounds. Rev Toxicol. 1998, 2: 395-443.
Lewontin RC, Krakauer J: Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics. 1973, 74 (1): 175-195.
Vos P, Hogers R, Bleeker M, Reijans M, Lee van de T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, et al: AFLP: a new technique for DNA fingerprinting. Nucleic acids research. 1995, 23 (21): 4407-4414. 10.1093/nar/23.21.4407.
Aljanabi SM, Martinez I: Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic acids research. 1997, 25 (22): 4692-4693. 10.1093/nar/25.22.4692.
DeHaan LR, Belina RAK, Ehlke NJ: Peakmatcher: Software for semi-automated fluorescence-based AFLP. Crop Science. 2002, 42 (4): 1361-1364.
Wilding C, Butlin R, Grahame J: Differential gene exchange between parapatric morphs of Littorina saxatilis detected using AFLP markers. Journal of Evolutionary Biology. 2001, 14: 611-619. 10.1046/j.1420-9101.2001.00304.x.
Adams SM, Lindmeier JB, Duvernell DD: Microsatellite analysis of the phylogeography, Pleistocene history and secondary contact hypotheses for the killifish, Fundulus heteroclitus. Molecular ecology. 2006, 15 (4): 1109-1123. 10.1111/j.1365-294X.2006.02859.x.
Nei M: F-statistics and analysis of gene diversity in subdivided populations. Annals of human genetics. 1977, 41 (2): 225-233. 10.1111/j.1469-1809.1977.tb01918.x.
Nei M, Chesser RK: Estimation of fixation indices and gene diversities. Annals of human genetics. 1983, 47 (Pt 3): 253-259. 10.1111/j.1469-1809.1983.tb00993.x.
Wilding CS, Butlin RK, Grahame J: Differential gene exchange between parapatric morphs of Littorina saxatilis detected using AFLP markers. Journal of Evolutionary Biology. 2001, 14 (4): 611-619. 10.1046/j.1420-9101.2001.00304.x.
Slatkin M, Barton NH: A Comparison of 3 Indirect Methods for Estimating Average Levels of Gene Flow. Evolution. 1989, 43 (7): 1349-1368. 10.2307/2409452.
McMillan AM, Bagley MJ, Jackson SA, Nacci DE: Genetic diversity and structure of an estuarine fish (Fundulus heteroclitus) indigenous to sites associated with a highly contaminated urban harbor. Ecotoxicology (London, England). 2006, 15 (6): 539-548.
Smith MW, Chapman RW, Powers DA: Mitochondrial DNA analysis of Atlantic Coast, Chesapeake Bay, and Delaware Bay populations of the teleost Fundulus heteroclitus indicates temporally unstable distributions over geologic time. Molecular Marine Biology and Biotechnology. 1998, 7 (2): 79-87.
Rousset F: Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics. 1997, 145 (4): 1219-1228.
Gutjahr-Gobell RE, Black DE, Mills LJ, Pruell RJ, Taplin BK, Jayaraman S: Feeding the mummichog (Fundulus heteroclitus) a diet spiked with non-ortho- and mono-ortho-substituted polychlorinated biphenyls: Accumulation and effects. Environmental Toxicology and Chemistry. 1999, 18 (4): 699-707. 10.1897/1551-5028(1999)018<0699:FTMFHA>2.3.CO;2.
Powell WH, Bright R, Bello SM, Hahn ME: Developmental and tissue-specific expression of AHR1, AHR2, and ARNT2 in dioxin-sensitive and -resistant populations of the marine fish Fundulus heteroclitus. Toxicol Sci. 2000, 57 (2): 229-239. 10.1093/toxsci/57.2.229.
Forbes VE: Genetics and ecotoxicology. 1999, Philadelphia, PA: Taylor & Francis
Klerks PL, Levinton JS: Rapid Evolution of Metal Resistance in a Benthic Oligochaete Inhabiting a Metal-Polluted Site. Biological Bulletin. 1989, 176 (2): 135-141. 10.2307/1541580.
McKenzie JA: Ecological and evolutionary aspects of insecticide resistance. 1996, Austin, Tex.: R.G. Landes
Mitton JB, Koehn RK: Genetic organization and adaptive response of allozymes to ecological variables in Fundulus heteroclitus. Genetics. 1975, 79 (1): 97-111.
Weis JS: Tolerance to environmental contaminants in the mummichog, Fundulus heteroclitus. Human and Ecological Risk Assessment. 2002, 8 (5): 933-953. 10.1080/1080-700291905756.
Lotrich VA: Summer Home Range and Movements of Fundulus-Heteroclitus (Pisces-Cyprinodontidae) in a Tidal Creek. Ecology. 1975, 56 (1): 191-198. 10.2307/1935311.
Kohn MH, Pelz HJ: Genomic assignment of the warfarin resistance locus, Rw, in the rat. Mamm Genome. 1999, 10 (7): 696-698. 10.1007/s003359901073.
Kohn MH, Pelz HJ, Wayne RK: Natural selection mapping of the warfarin-resistance gene. Proceedings of the National Academy of Sciences of the United States of America. 2000, 97 (14): 7911-7915. 10.1073/pnas.97.14.7911.
Taylor MFJ, Shen Y, Kreitman ME: A Population Genetic Test of Selection at the Molecular-Level. Science (New York, NY). 1995, 270 (5241): 1497-1499.
Wootton JC, Feng XR, Ferdig MT, Cooper RA, Mu JB, Baruch DI, Magill AJ, Su XZ: Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature. 2002, 418 (6895): 320-323. 10.1038/nature00813.
Mulvey M, Newman MC, Vogelbein WK, Unger MA, Ownby DR: Genetic structure and mtDNA diversity of Fundulus heteroclitus populations from polycyclic aromatic hydrocarbon-contaminated sites. Environmental toxicology and chemistry/SETAC. 2003, 22 (3): 671-677. 10.1897/1551-5028(2003)022<0671:GSAMDO>2.0.CO;2.
Roark SA, Nacci D, Coiro L, Champlin D, Guttman SI: Population genetic structure of a nonmigratory estuarine fish (Fundulus heteroclitus) across a strong gradient of polychlorinated biphenyl contamination. Environmental toxicology and chemistry/SETAC. 2005, 24 (3): 717-725. 10.1897/03-687.1.
Fisher MA, Oleksiak MF: Convergence and divergence in gene expression among natural populations exposed to pollution. BMC genomics. 2007, 8: 108-10.1186/1471-2164-8-108.
Gaudeul M, Till-Bottraud I, Barjon F, Manel S: Genetic diversity and differentiation in Eryngium alpinum L. (Apiaceae): comparison of AFLP and microsatellite markers. Heredity. 2004, 92 (6): 508-518. 10.1038/sj.hdy.6800443.
Kreitman M: The neutral theory is dead. Long live the neutral theory. Bioessays. 1996, 18 (8): 678-683. 10.1002/bies.950180812. discussion 683
Hansen MM, Mensberg KLD: Genetic differentiation and relationship between genetic and geographical distance in Danish sea trout (Salmo trutta L.) populations. Heredity. 1998, 81: 493-504. 10.1046/j.1365-2540.1998.00408.x.
Cashon RE, Vanbeneden RJ, Powers DA: Biochemical Genetics of Fundulus-Heteroclitus (L) .4. Spatial Variation in Gene-Frequencies of Idh-a, Idh-B, 6-Pgdh-a, and Est-S. Biochem Genet. 1981, 19 (7–8): 715-728. 10.1007/BF00484004.
Gonzalezvillasenor LI, Powers DA: Mitochondrial-DNA Restriction-Site Polymorphisms in the Teleost Fundulus-Heteroclitus Support Secondary Intergradation. Evolution. 1990, 44 (1): 27-37. 10.2307/2409522.
Ropson IJ, Brown DC, Powers DA: Biochemical Genetics of Fundulus-Heteroclitus (L) .6. Geographical Variation in the Gene-Frequencies of 15 Loci. Evolution. 1990, 44 (1): 16-26. 10.2307/2409521.
Acknowledgements
The Authors thank G. Bozinovic for assistance in field collections and D. Crawford for valuable comments on an earlier version of this manuscript. Funding was partially provided by NIEHS Training Grant ES525163 award from the Department of Environmental and Molecular Toxicology at North Carolina State University to LMW and NIH 5 RO1 ES011588 to MFO.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
LMW designed experiments, carried out laboratory and statistical analysis, and drafted the manuscript. MFO designed experiments, assisted on statistical analysis, and helped draft the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Williams, L.M., Oleksiak, M.F. Signatures of selection in natural populations adapted to chronic pollution. BMC Evol Biol 8, 282 (2008). https://doi.org/10.1186/1471-2148-8-282
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-8-282