
Abstract
Background
The genus Oryza is composed of 10 distinct genome types, 6 diploid and 4 polyploid, and includes the world's most important food crop – rice (Oryza sativa [AA]). Genome size variation in the Oryza is more than 3-fold and ranges from 357 Mbp in Oryza glaberrima [AA] to 1283 Mbp in the polyploid Oryza ridleyi [HHJJ]. Because repetitive elements are known to play a significant role in genome size variation, we constructed random sheared small insert genomic libraries from 12 representative Oryza species and conducted a comprehensive study of the repetitive element composition, distribution and phylogeny in this genus. Particular attention was paid to the role played by the most important classes of transposable elements (Long Terminal Repeats Retrotransposons, Long interspersed Nuclear Elements, helitrons, DNA transposable elements) in shaping these genomes and in their contributing to genome size variation.
Results
We identified the elements primarily responsible for the most strikingly genome size variation in Oryza. We demonstrated how Long Terminal Repeat retrotransposons belonging to the same families have proliferated to very different extents in various species. We also showed that the pool of Long Terminal Repeat Retrotransposons is substantially conserved and ubiquitous throughout the Oryza and so its origin is ancient and its existence predates the speciation events that originated the genus. Finally we described the peculiar behavior of repeats in the species Oryza coarctata [HHKK] whose placement in the Oryza genus is controversial.
Conclusion
Long Terminal Repeat retrotransposons are the major component of the Oryza genomes analyzed and, along with polyploidization, are the most important contributors to the genome size variation across the Oryza genus. Two families of Ty3-gypsy elements (RIRE2 and Atlantys) account for a significant portion of the genome size variations present in the Oryza genus.
Similar content being viewed by others


Background
The C-value paradox, the phenomenon describing the lack of correlation between biological complexity of an organism and its genome size [1], is probably best exemplified in plants were genome sizes span several orders of magnitude ranging from the 98 Mbp Fragaria viridis genome [2] to more than 110,000 Mbp genome of the lily Fritillaria assiriaca [3]. Significant genome size variations are also not uncommon even within single genera such as rice, cotton and sorghum were 3.6, 3 and 8.1 fold genome size variation have been reported, respectively [4–6]. The primary mechanisms that contribute to this variation in plants are polyploidization [7, 8] and transposable element (TE) proliferation [9–11] and elimination [12–17]. TEs are classified according to the different molecules used as intermediates in the replicative mechanism of transposition as class 1(or RNA elements) and class 2 (or DNA elements) [18]. Class 1 elements transpose via an RNA intermediate and include long terminal repeat (LTR) retrotransposons (LTR-RTs), long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs) [19]. Class 2 elements transpose via a DNA intermediate and have been classified into superfamilies (hAT, CACTA and Mutator-like elements) according to the similarity of transposases, the element-encoded protein that catalyzes transposition and integration [20]. Other classes of DNA transposable elements are represented by helitrons [21] and polintons [22].
The genus Oryza is an ideal model system to study the role of TEs on genome size variation. The genus is composed of 23 species: two cultivated (Oryza sativa and Oryza glaberrima) and 21 wild [23, 24]. Based on evidence derived from interspecific crossing, cytogenetics and genomic DNA hybridization, ten Oryza genome types have been recognized including 6 diploid (2n = 24) and 4 tetraploid (2n = 48). The relevance of rice as staple food resource for the world, its compact 389 Mbp genome [25] and its role as a "model" species for genomic studies of cereals [26, 27] have driven massive research efforts that include the production of the first finished genome sequence of any crop plant [25]. Additionally, our laboratory has developed a comprehensive set of BAC libraries, BAC end sequences and integrated physical maps representing the 10 Oryza genome types [28, 29].
To better understand the role transposable elements have played in genome size variation in Oryza we generated and sequenced a set of random sheared genomic libraries from 12 species representative of the 10 genome types of the Oryza. Random sheared libraries represent an unbiased sampling of genome content and enable the characterization of their most relevant features without undertaking a massive sequencing effort. The sequences obtained were analyzed in order to describe, classify and compare the repetitive fraction of the genus Oryza. Particular attention was paid to the role played by the most important classes of TEs (LTR RTs, LINEs, helitrons and other DNA transposable elements) in shaping these genomes and in contributing to genome size variation. In the case of four groups of TEs (the class 1 LTR-RTs and LINEs and the class 2 CACTA and Mutator-like elements) phylogenetic analyses were carried out.
Results
To better understand the repeat content of the genus Oryza, 12 random sheared libraries were constructed and sequenced from 12 vouchered accessions that represent the 10 genome types of Oryza (Figure 1). The total number of clones produced was 42,432, which roughly corresponded to 0.015× coverage of each genome. Mean insert size ranged from 2.5 kbp for O. australiensis [genome type:EE; genome size:965 Mbp] to 3.9 kbp for O. rufipogon [AA; 439 Mbp]. Cloned inserts were bi-directionally sequenced and after removing low quality data (sequences shorter than 50 bases and all those similar to plastid genomes), 72,245 high quality sequence reads were obtained for a total of 51.76 Mb of sequence (0.56% of the total size of all the genomes studied (Table 1)).

Phylogeny of genus Oryza. Phylogenetic tree is a modified version of that proposed by Ge et al [53]. Figures between parentheses indicate the genome size in Million of nucleotides.
Repeats abundance and distribution
Repetitive sequences were identified by similarity searches against databases of previously characterized repetitive elements, isolated from O. sativa and other Oryza species, including centromeric repeats, helitrons, SINEs, LINEs, MITEs, LTR-RTs, DNA transposable elements (DNA-TEs), ribosomal sequences and telomeric repeats (see Methods). All the major classes of repeats were represented throughout the genus (Table 2). The overall amount of repeats was quite variable in different species and ranged from the 25.04% of reads in O. coarctata [HHKK] to 66.42% of reads in O. officinalis [CC; 651 Mbp].
In all the genomes studied the largest class of repeats consisted of LTR-RTs with amounts ranging from the 55% of reads in O. granulata [GG; 882 Mbp] to 12.7% of reads in O. brachyantha [FF; 362 Mbp]. DNA transposons, excluding MITEs and helitrons, appeared to be more abundant in O. ridleyi [HHJJ; 1283 Mbp] (11.93% of reads) and were depleted in O. coarctata [HHKK] (3.44% of reads).
The MITE sequences used as queries in similarity searches were isolated in O. sativa and not surprisingly they tag the highest value of hits in the three AA genome species: O. glaberrima (17% of reads), O. nivara (11.73% of reads) and O. rufipogon (14.36% of reads). The quantity of MITEs identified in the remaining genome types was less than 6% of reads except for the notable exception of the distantly related species O. brachyantha [FF; 362 Mbp] with 15.28% of reads. The lower than expected MITE content in the non-AA genomes species is likely due to the fact that MITEs are highly species specific and the repeat database used to identify MITEs was a curated data set from the O. sativa species only.
Differences in TE size, host genome size and degrees of interspecific conservation make it particularly difficult to perform a detailed comparison based solely on the percentage of significantly similar sequences out of the total. A more rigorous approach was recently proposed by Hawkins et al. [9] where an equation was derived (see Methods) that takes into account TE mean size, host genome size, the minimum length needed in order to have a significant BLASTN hit, and the number of matches out of total reads available to estimate the absolute number of repeats in a given genome. We used this equation for all classes of repeats for which reliable estimates of the average length were available and then, in order to have comparable results, normalized the data to the single Mbp (Figure 2 and Additional file 1). Here we considered only the most represented and conserved families of elements: LTR-RTs, LINEs, helitrons, and the DNA-TEs – CACTA and Mutator like. It should however be noted how the use of average lengths for entire classes of elements could introduce a certain degree of uncertainty and so the following values should be considered as an approximation. Not surprisingly the LTR-RTs remained the major contributor to the repeat pool of each species. Species with the highest occurrence of LTR-RTs per Mbp were O. granulata [GG; 882 Mbp] (49.5) and O. australiensis [EE; 965 Mbp] (50.9) whereas the most depleted was O. brachyanta [FF; 362 Mbp] (12.9). In almost all cases, the Ty3-gypsy elements outnumbered Ty1-copia elements. This disproportion was particularly striking in the case of O. nivara [AA; 448 Mbp], O. rufipogon [AA; 439 Mbp] and O. granulata [GG; 882 Mbp] were the ratios Ty3-gypsy to Ty1-copia elements were 4.80, 4.91 and 4.6 respectively. The only exception to the overwhelming presence of Ty3-gypsy elements in comparison to Ty1-copia was found in O. coarctata [HHKK] where twice as many Ty1-copia elements (ratio of Ty3-gypsy/Ty1-copia = 0.5) were found.

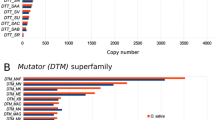
Occurrence per single Mbp for the major classes of repeats in different Oryza genomes. Y-axis: the estimated number of occurrences calculated using the equation from Hawkins et al (2006) [9] and normalized to 1 Mbp. The confidence intervals, displayed as error bars, were calculated assuming a Poisson distribution of repeats in the genome. The mean length used in calculation for different repeats were as follows: Ty3-gypsy elements: 12 Kbp; Ty1-copia elements: 5.5 Kbp; others LTR-RTs not classified (LTR-retrotransposons): 8.75 Kbp; LINEs: 3.5 kbp; helitrons (complete autonomous): 12.8 Kbp; CACTA: 15.2 Kbp; hAT: 3.6 Kbp *In this case all the calculations are based on a rough estimate of the genome size of this species: the real value is unknown, we therefore used the value estimated for O. ridleyi [HHJJ; 1283 Mbp], which is also an allotetraploid species and shares the HH genome type with O. coarctata.
Among other repeats, it is worth to note that CACTA elements seemed to be significantly more frequent in the BB (O. punctata; 3.3 elements per Mbp), CC (O. officinalis; 4.0 elements per Mbp), BBCC (O. minuta; 3.4. hits per Mbp), CCDD (O. alta; 2.7 hits per Mbp), EE (O. australiensis; 3.2 hits per Mbp) and HHJJ (O. ridleyi; 2.5 hits per Mbp) genomes as compared with all 3 AA genomes (O. glaberrima, O. rufipogon, O. nivara) where the number of hits was less than 2 per Mbp. In almost all the genomes the frequency of CACTA elements was greater than the hAT elements, with the exception of O. brachyantha [FF; 362 Mbp] (CACTA = 0.5 hits/Mbp; hAT 0.6 hits/Mbp) and O. granulata [GG; 882 Mbp] in which hAT elements outnumber the CACTA elements 6 to 1 (CACTA = 0.3 hits/Mb; hAT = 1.9/Mbp). The distribution of LINEs was higher in the AA genomes where it was greater than 2.5 hits per Mbp and very low in O. granulata [GG; 882 Mbp] with only 0.7 hits per Mbp.
The significant variance of LTR-RT representation presented above prompted us to perform a more in depth analysis of the distribution of the different families in each genome. To identify the primary LTR-RT families responsible for this variance and possibly link them to the genome size variation in each host species, all sequences previously identified as being similar to LTR-RTs were screened a second time against a reduced database containing representatives of twenty-six of the most abundant LTR-RT families isolated in Oryza genus (11 Ty1-copia and 15 Ty3-gypsy) (Table 3). In all species the overall majority of sequences already annotated as LTR-RTs could be easily classified according to these parameters (Table 3). Elements related to the most abundant families of LTR-RTs isolated in O. sativa [AA; 389 Mbp] were identified in all the Oryza genome types considered with very few exceptions. This indicated that the complement of LTR-RTs previously identified in rice is shared throughout the Oryza genus. However, as shown in Table 3, different LTR-RTs had different fates in different species.
A paradigmatic case was that of two Ty3-gypsy families Atlantys and RIRE2 (that is related to the element Wallabi [10]). Atlantys was the most abundant element in 7 of the 10 Oyrza genome types (O. punctata [BB; 425 Mbp], 6.36% of total reads; O. officinalis [CC; 651 Mbp], 10.15% of total reads; O. minuta [BBCC; 1124 Mbp], 8.70% of total reads; O. alta [CCDD; 1008 Mbp], 7.32% of total reads; O. australiensis [EE; 965 Mbp], 10.98% of total reads; O. coarctata [HHKK], 4.08% of total reads and O. ridleyi [HHJJ; 1283 Mbp], 7.62% of total reads). It was also the second most abundant element in O. granulata [GG; 882 Mbp], 7.28% of total reads. However in the AA and FF genomes, it did not expand significantly in comparison to the other elements. RIRE2 related elements (i.e. Wallabi [10] in O. australiensis) expanded significantly in O. australiensis [EE; 965 Mbp], 8.56% of the total reads and in O. granulata [GG; 882 Mbp], 21.5% of total reads.
It is also worth to note that the RIRE8 Ty3-gypsy element is more widespread in the AA genomes (5.90% and 5.21% of total reads in O. nivara and O. rufipogon, respectively) than in the remaining genome types.
The O. minuta [BBCC; 1124 Mbp] genome represents the only polyploid genome for which a comparison with both the original genome counterparts BB (O. punctata) and CC (O. officinalis) is possible. All LTR-RT families identified in O. minuta [BBCC: 1124 Mbp] were found in at least one of the diploid counterparts and there were only few cases where an element was significantly more represented in the polyploid genome than in the two diploid counterparts. The most convincing example was that of the Ty1-copia element RIRE1 (0.16%, 0.1% and 0.6% of total reads in O. punctata, O. officinalis and O. minuta respectively).
For all the genome types it was possible to identify a fraction of LTR-RT related sequences that did not show any significant similarity with the major families isolated from O. sativa. The amount of this fraction expressed as percentage out of the total number of sequence reads ranged from 0.71% in O. brachyantha [FF; 362 Mbp] to 5.6% in O. ridleyi [HHJJ; 1283 Mbp] and represented the most species specific sets of these elements (or the most diverged from the O. sativa retroelement pool).
Our strategy to rely on similarity searches in order to identify the repetitive fraction of a genome has the obvious pitfall that, if a repeat is not present in the repeat database it will not be detected. To check for this possibility we adopted a different approach to screen all sequence reads that did not show any significant hits with already characterized repeats. Sequences were clustered using the program Blastclust and the results are shown in Table 4. Only a few clusters containing more than 3 sequences were isolated in all the species with the exception of the AA and BB genomes in which no clusters were identified. The largest cluster identified was found in O. ridleyi [HHJJ; 1283 Mbp] and contained 69 sequences out of 11,091 (0.62%). The sequences present in these clusters very likely represent previously uncharacterized repeats. However as the figures for O. ridleyi [HHJJ; 1283 Mbp] demonstrate, they do not constitute a significant portion of the genomes in which they were isolated.
Phylogenetic analysis of repeats
To establish the phylogeneic relationships among the most prominent superfamilies of TEs in Oryza, we used amino acid sequences of the most conserved domain tracts (see Methods) of Ty1-copia and Ty3-gypsy LTR-RTs, LINEs and CACTA and Mutator-like DNA-TEs to build neighbor-joining trees.
For Ty1-copia elements (Figure 3) it was possible to retrieve 269 reverse transcriptase (RT) sequences out of the 72,245 available sequences. These sequences were resolved into at least eight lineages as representatives of the major Ty1-copia retroelements isolated. The amount of sequences in each lineage reflected the abundance of that family in the genome. Furthermore the most abundant lineages contained representatives of all the different species and there was not a single species-specific lineage with the partial exception of RIRE1 in O. australiensis [EE; 965 Mbp]. This lineage, also including a few sequences from O. minuta [BBCC; 1124 Mbp], was separated from the others and it was possible to see how the branch lengths are short suggesting that amplification of this element took place after speciation (with the possible exception represented by the few O. minuta RIRE1 related sequences).

Phylogenetic analysis of Ty1- copia retroelements. A) 269 sequences similar to the Ty1-copia reverse transcriptase domain were used to build a phylogenetic tree using the neighbor-joining method. Bootstrap values were calculated for 1000 replicates; only those with values greater than 50 are proposed B) distribution of the domains isolated in different species. Bar colors are the same of those used in the circles marking, on the neighbor-joining tree sequences from different species.
RT-like sequences from Ty3-gypsy elements (508 elements) were isolated and used to build a phylogenetic tree (Figure 4). The complete set is less heterogeneous than the Ty1-copia set, however all the major families are represented. In the case of RIRE2 and Atlantys, the two most abundant Ty3-gypsy families throughout the genus, all the species were included in the correspondent lineages indicating that their presence in the genus predates its speciation. However, considering the different species in which the major amplification events of elements belonging to these two families took place, it was possible to note how the elements isolated in different species usually do not mix together in the internal lineages suggesting that their amplification has happened after speciation.

Phylogenetic analysis of Ty3- gypsy retroelements. A) 508 sequences similar to the Ty3-gypsy reverse transcriptase domain were used to build a phylogenetic tree using the neighbor-joining method. Bootstrap values were calculated for 1000 replicates; only those with values greater than 50 are proposed B) distribution of the domains isolated in different species. Bar colors are the same of those used in the circles marking, on the neighbor-joining tree sequences from different species.
LINEs are usually not very abundant in plants as compared to LTR-RTs with some exceptions such as the del2 element in Lilium speciosum [30]. The genus Oryza in this sense appears to follow this general rule. This trend is reflected in the number of LINE reverse transcriptase like sequences [38] isolated from our set. Twenty-two of them cluster in 4 major bootstrap supported lineages and many are characterized by very long branches suggestive of an ancient origin of these retroelements in the genus (Figure 5). These findings suggest that even in the case of the Oryza genus, LINEs exhibit, as in other plants, high sequence divergence and extreme heterogeneity [31–33].

Phylogenetic analysis of LINE retroelements. A) 38 sequences similar to the LINE reverse transcriptase domain were used to build a phylogenetic tree using the neighbor-joining method. Bootstrap values were calculated for 1000 replicates; only those with values greater than 50 are proposed B) distribution of the domains isolated in different species. Bar colors are the same of those used in the circles marking, on the neighbor-joining tree sequences from different species.
The two major groups of DNA TEs, CACTA and Mutator-like were phylogenetically investigated using tracts of their transposase coding domains. In the case of CACTA elements, we identified 125 transposase-like sequences, whereas 55 were isolated for Mutator-like elements. For both classes of TE elements, it was not possible to identify species specific lineages in the corresponding Neighbor Joining trees (Figures 6 and 7). The only exception was represented by a large lineage including 14 of the 18 CACTA transposase domains isolated in O. alta [CCDD; 1008 Mbp]. However this lineage lacks consistent bootstrap support.

Phylogenetic analysis of CACTA transposable elements. A) 125 sequences similar to the CACTA transposase domain were used to build a phylogenetic tree using the neighbor-joining method. Bootstrap values were calculated for 1000 replicates; only those with values greater than 50 are proposed B) distribution of the domains isolated in different species. Bar colors are the same of those used in the circles marking, on the neighbor-joining tree sequences from different species.

Phylogenetic analysis of Mutator -like transposable elements. A) 55 sequences similar to the Mutator-like transposase domain were used to build a phylogenetic tree using the neighbor-joining method. Bootstrap values were calculated for 1000 replicates; only those with values greater than 50 are proposed B) distribution of the domains isolated in different species. Bar colors are the same of those used in the circles marking, on the neighbor-joining tree sequences from different species.
Discussion
Genome survey sequences from 12 random sheared genomic libraries representative of the 10 genome types of the genus Oryza were generated and analyzed to obtain an unbiased sampling of the repeat content of the genus as a whole. Our results showed that the genomic composition of the Oryza species is not exceptional when compared to other plant species in that the repetitive fraction consistently makes up a significant portion of each of the 10 genome types.
Among TEs, the major class of repeats in Oryza was represented by LTR-RTs whose absolute occurrence was estimated to be in the order of thousands of copies for each species. Detailed analysis of the major O. sativa LTR-RT families enabled us to identify the elements responsible for the major differences in genome size variation across the genus Oryza. For example, LTR-RTs related to RIRE2 represent more than 8.5% and 21% of all the sequences obtained for O. australiensis [EE; 965 Mbp] and O. granulata [GG; 882 Mbp] respectively. This is consistent with previous studies which showed that the proliferation of 3 family members (RIRE1, the RIRE2 related element Wallabi and Kangaroo) in EE genomes [10] and a single family (Gran3) in the GG genome [Jetty et al., 2007 accepted with revisions] accounts for nearly 50% and 25% increases in their genome sizes respectively. Another family that has played a significantly similar role in genome size variation is the Ty3-gypsy element Atlantys. The Atlantys family has been surprisingly overlooked by previous studies and with its amplification has impacted not only the O. granulata and O. australiensis genomes but also the BB, CC, BBCC and CCDD genomes. The events leading to the explosive proliferation of both the RIRE2 and Atlantys families appear to have taken place after each speciation event.
The consequences of TE proliferation subsequent to speciation on genome size variation is reminiscent of Zea mays in which five LTR-RT families represent nearly 25% of the genome [34] and of the species Gossypium herabaceum and G. exiguum where a group of Ty3-gypsy elements, named GORGE-3, has extensively proliferated in comparison to other elements and constitutes a significant portion of each genome [9]. In contrast, no single LTR-RT family was found to constitute significant portions of the AA genome Oryza species with the only exception being represented by the Ty3-gypsy element RIRE8. The finding that, apart from the polyploids, the largest genomes in the Oryza (O. australiensis and O. granulata) are those that have seen the "explosive" proliferation of one or two LTR-RT families, once again identifies the pivotal role of this class of TEs in the dynamics of genome size variation.
The differential proliferation of LTR-RTs in the Oryza genus could be explained by taking into account the effects of varying genomic backgrounds on element life cycles, different rates of LTR-RT removal by unequal recombination and/or illegitimate recombination [8, 15, 16] and inherently different retrotranspositional potentials of various element families [35]. However no single convincing explanation of this phenomenon has so far been advanced for other plant species in which similar events have been described [9, 10].
Here we have demonstrated that the pool of LTR-RTs in the Oryza is substantially conserved throughout the genus because in almost all cases relatives of the LTR-RT families identified in O. sativa [AA] could also be found in the other Oryza species. We therefore conclude that its existence predates the speciation events that originated the genus as we know. This finding is well supported by our phylogenetic analyses and it is in accordance with the results of previous studies analyzing the Ty1-copia elements phylogeny and distribution across different genera of the Gramineae family [36, 37].
Besides the identification of different amounts of elements from the same LTR-RT families, all the genomes studied, except O. coarctata, showed the same quantitative relationship between Ty3-gypsy and Ty1-copia elements where the Ty3-gypsy elements constantly outnumber Ty1-copia elements, even if the ratio between the two elements varied (from more than 4.6 in O. nivara [AA; 448 Mbp], O. rufipogon [AA; 439 Mbp] and O. granulata [GG; 882 Mbp] to 1.52 in O. brachyantha [FF; 362 Mbp]). This data is completely consistent with previous analysis of the cultivated japonica and indica genomes [25, 38–41] and can now be extended to the genus Oryza, with the notable exception of O. coarctata [HHKK] where we found a ratio close to 0.5 between the Ty3-gypsy and Ty1-copia elements.
An even more striking difference between the O. coarctata [HHKK] genome and the other Oryza spp genomes is that it was found to contain the least amount repetitive elements despite it being polyploid and its predicted large genome size. It should be noted that we have not been able to obtain live plants or tissues in the U.S. in order to determine an accurate genome size measurement by flow cytometry due to quarantine restrictions. To rule out the possibility that our similarity searches could not detect highly diverged elements we analyzed all the sequences to try to identify repeats de novo through a clustering approach. In this case no significant de novo clusters were obtained in O. coarctata. These findings rule out the existence of a single recently originated family of repeats, highly species specific and constituting a significant portion of the genome. However, the results do not discount the possibility that repeats missed by previous searches in O. coarctata, if present, belong to several different families, possibly diverged (and so ancient). All evidence combined supports the old taxonomic view that places O. coarctata in the genus Porteresia [42] and not Oryza. However more in depth and accurate analyses are needed in order to ascertain the dynamics acting towards this apparently repeat depletion in O. coarctata.
The evolutionary events leading to the present day Oryza genus spanned at least 9 million years from O. sativa [AA; 389 Mbp] to the the most basal species O. granulata [GG; 882 Mbp] [43]. Similar to what was observed in O. coarctata [HHKK], the use of repeat collections mainly based on elements isolated in O. sativa [AA;389 Mbp] to screen other species of the genus raises some concerns about how nucleotide divergence can impact estimates of LTR-RTs abundance. With the exceptions of O. coarctata (discussed above) and O. brachyantha [FF; 362 Mbp] (in which the depletion of repeats can be easily explained taking into account the small size of this genome), our work demonstrated that the amount of LTR-RTs isolated does not decrease substantially when our analysis moved from the AA genome types to the most distant ones. The evidence has been indirectly confirmed by the clustering analysis that did not revealed the existence any significantly repeat family previously missed. This, in turn, demonstrates how similarity searches of LTR-RT families have a good level of "portability" throughout the genus giving substantially reliable and reasonably exhaustive results, if the proper settings are used.
A completely different scenario could be described for TEs lacking conserved domains and having higher species sequence specificity such as MITEs and SINEs. In this case the evolutionary distance from AA genomes consistently results in a steady drop in the amount TEs identified (with the significant exception of MITEs in O. brachyantha) prompting the need of de novo and ad hoc identification tools based on structural features beside the nucleotidic sequence.
Conclusion
Here we report the results of a comprehensive analysis of the abundance and relative distribution of major TE classes across twelve species of the genus Oryza.
We have demonstrated how the LTR-RT complement in of these species is ancient and conserved throughout the genus, and has attained different retrotranpositional success in different species.
We also identified two LTR-RT families (RIRE2 and Atlantys) that are responsible for a significant portion of genome size variation in the genus and we demonstrated how their massive increase in copy number is recent.
Methods
Plant material (put in genome designations)
Total genomic DNA was isolated from young leaf tissue from the following Oryza species: O. brachyantha [FF; 362 Mbp] (Acc# 101232), O. alta [CCDD;1008 Mbp] (Acc# 105143), O. officinalis [CC; 651 Mbp] (Acc# 100896), O. ridleyi [HHJJ; 1283 mbp](Acc# 100821), O. punctata [BB; 425 Mbp](Acc# 105690), O. coarctata [HHKK](Acc# 104502), O. minuta [BBCC; 1124 mbp](Acc# 101141), O. granulata [GG; 882 Mbp](Acc# 102118), O. glaberrima [AA; 357 Mbp](Acc# 96717), O. rufipogon [AA; 439 mbp] (Acc# 105491), O. nivara [AA; 448 Mbp] (Acc# 100897) and O. australiensis [EE; 965 Mbp] (Acc# 100882). Plant materials were grown at the International Rice Research Institute, Philippines.
Library construction and DNA sequencing
Genomic DNA was extracted using CTAB [44]. DNA was sheared using the Hydroshear (GeneMachines) to produce 2–5 kb fragments. DNA fragments were end-repaired using Epicientre's End-it Repair kit and size selected on 0.6% agarose gels. DNA fragments in the size range of 2–5 kb were cut out of the gel, eluted using a QIAEX II Gel extraction kit and ligated to linearized pBluescriptII KS+ at 16°C overnight. Ligation products (1 uL) were transformed into DH10B electrocompetent cells (Invitrogen). Shotgun plasmids were sequenced bi-directionally on ABI 3730XL DNA sequencers (ABI) using standard protocols. Sequence data was extracted using ABI sequence analysis software and base called using Phred [45]. Vector screening and low quality sequence removal was done using the program Lucy [46].
Similarity searches
Sequences were used as queries in similarity searches against different repeat databases: TIGR rice repeats version 3.1 [47], Repbase version 11_09 [48] and a proprietary collection of retroelements isolated in Oryza sativa and other Oryza species. Searches were carried out using the BLASTN algorithm [49] run under relaxed settings (-q -2 -r 3) in order to accommodate the divergence between species searched from the major source of repeats in our database (O. sativa). Only hits having an E value equal or lower to 1e-10 were used. For all sequences that did not result in any significant hits, a second round of searches was carried out using the algorithm BLASTX against the non-redundant division of GenBank. Under these conditions only hits having an E value equal or lower to 1e-5 were parsed. To estimate the number of significantly similar hits present in the entire genome of each of the 12 Oryza species, we used this equation: n = (Xobs/N)*(G/(L-2m+e) that is a slight modification of that proposed by Hawkins et al. [9], where "Xobs" is the observed number of copies, "N" is the total number of sequence reads, "n" is number of targets in the genome, "L" is length of target sequence, "m" is estimated minimum length required to identify a sequence in a BLAST search (we used the extremely conservative value of 100 bp), "e" is number of bp sequenced from each insert and "G" is genome size. Published sequences for various repetitive elements were used to estimate "L".
A third round of similarity searches was performed using only representative elements from the LTR-RT families listed in Table 3.
All sequences that were not classified as repeats were used in a cluster analysis using "Blastclust" [50]. The program was used with the following settings, L 0.51 S 80, meaning that all sequences sharing at least 80% similarity over at least 51% of their length were included in the same cluster. Only clusters containing 3 or more hits were considered in analyzed further.
Phylogenetic analysis
Sequences homologous to the Reverse Transcriptase domains of Ty1-copia ([GenBank: CAD40165.2], 92 AA from residues 781 to 872), Ty3-gypsy ([GenBank: CAH66235.1],162 AA from residues 471 to 632) and LINEs (122 residues from RT domain of element OSLINE1_5 in Repbase [48]) and sequences homologous to transposase domains of CACTA ([GenBank: DAA02108.1], 100 AA from residue 1 to 100) and Mutator-like elements ([GenBank: AAM94534.1], 238 AA from residues 361 to 598), were identified in the random sheared libraries through tBLASTN searches [49]. The corresponding amino acid domains were retrieved and aligned using Muscle [51]. Neighbor-joining trees were produced and edited using the program MEGA version 3 [52].
Accession numbers
Sequences for this paper were submitted to the GSS division of GenBank under the following accessions numbers: [GenBank: EI028463–EI035999] (O. alta), [GenBank: EI36000–EI043358] (O. australiensis), [GenBank: EI043359–EI046735] (O. brachyantha), [GenBank: EI46736–EI56259] (O. coarctata), [GenBank: EI056260–EI59753] (O. glaberrima), [GenBank: EI059754–EI066441] (O. granulata), [GenBank: EI066442–EI075900] (O. minuta), [GenBank: EI-075901–EI079003] (O. nivara), [GenBank: EI079004–EI083851] (O. officinalis), [GenBank: EI083852–EI86995] (O. punctata), [GenBank: EI086996–EI098086] (O. ridleyi), [GenBank: EI098087–EI100641] (O. rufipogon).
References
Thomas CA: The genetic organization of chromosomes. Annu Rev Genet. 1971, 5: 237-256. 10.1146/annurev.ge.05.120171.001321.
Antonius K, Ahokas H: Flow cytometric determination of polyploidy level in spontaneous clones of strawberries. Hereditas. 1996, 124: 285-10.1111/j.1601-5223.1996.t01-1-00285.x.
Arumuganathan K, Earle ED: Nuclear DNA Content of Some Important Plant Species. Plant Molecular Biology Reporter. 1991, 9: 211-215.
Ammiraju JS, Luo M, Goicoechea JL, Wang W, Kudrna D, Mueller C, Talag J, Kim H, Sisneros NB, Blackmon B, Fang E, Tomkins JB, Brar D, MacKill D, McCouch S, Kurata N, Lambert G, Galbraith DW, Arumuganathan K, Rao K, Walling JG, Gill N, Yu Y, SanMiguel P, Soderlund C, Jackson S, Wing RA: The Oryza bacterial artificial chromosome library resource: construction and analysis of 12 deep-coverage large-insert BAC libraries that represent the 10 genome types of the genus Oryza. Genome Res. 2006, 16: 140-147. 10.1101/gr.3766306.
Hendrix B, Stewart JM: Estimation of the nuclear DNA content of Gossypium species. Ann Bot (Lond). 2005, 95: 789-797. 10.1093/aob/mci078.
Price HJ, Dillon SL, Hodnett G, Rooney WL, Ross L, Johnston JS: Genome evolution in the genus Sorghum (Poaceae). Ann Bot (Lond). 2005, 95: 219-227. 10.1093/aob/mci015.
Adams KL, Wendel JF: Polyploidy and genome evolution in plants. Curr Opin Plant Biol. 2005, 8: 135-141. 10.1016/j.pbi.2005.01.001.
Bennetzen JL, Ma J, Devos KM: Mechanisms of recent genome size variation in flowering plants. Ann Bot (Lond). 2005, 95: 127-132. 10.1093/aob/mci008.
Hawkins JS, Kim H, Nason JD, Wing RA, Wendel JF: Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006, 16: 1252-1261. 10.1101/gr.5282906.
Piegu B, Guyot R, Picault N, Roulin A, Saniyal A, Kim H, Collura K, Brar DS, Jackson S, Wing RA, Panaud O: Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006, 16: 1262-1269. 10.1101/gr.5290206.
Neumann P, Koblizkova A, Navratilova A, Macas J: Significant expansion of Vicia pannonica genome size mediated by amplification of a single type of giant retroelement. Genetics. 2006, 173: 1047-1056. 10.1534/genetics.106.056259.
Rabinowicz PD: Are obese plant genomes on a diet?. Genome Res. 2000, 10: 893-894. 10.1101/gr.10.7.893.
Vicient CM, Suoniemi A, Anamthawat-Jonsson K, Tanskanen J, Beharav A, Nevo E, Schulman AH: Retrotransposon BARE-1 and Its Role in Genome Evolution in the Genus Hordeum. Plant Cell. 1999, 11: 1769-1784. 10.1105/tpc.11.9.1769.
Vitte C, Panaud O: Formation of solo-LTRs through unequal homologous recombination counterbalances amplifications of LTR retrotransposons in rice Oryza sativa L. Mol Biol Evol. 2003, 20: 528-540. 10.1093/molbev/msg055.
Devos KM, Brown JK, Bennetzen JL: Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 2002, 12: 1075-1079. 10.1101/gr.132102.
Ma J, Devos KM, Bennetzen JL: Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 2004, 14: 860-869. 10.1101/gr.1466204.
Vitte C, Panaud O: LTR retrotransposons and flowering plant genome size: emergence of the increase/decrease model. Cytogenet Genome Res. 2005, 110: 91-107. 10.1159/000084941.
Capy P, Bazin C, Langin T, Higuet D: Dynamics and Evolution of Transposable Elements. 1997, Kluwer Academic Publishers, 197-
Xiong Y, Eickbush TH: Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 1990, 9: 3353-3362.
Flavell AJ: Retroelements, reverse transcriptase and evolution. Comp Biochem Physiol B Biochem Mol Biol. 1995, 110: 3-15. 10.1016/0305-0491(94)00122-B.
Kapitonov VV, Jurka J: Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci USA. 2001, 98: 8714-8719. 10.1073/pnas.151269298.
Kapitonov VV, Jurka J: Self-synthesizing DNA transposons in eukaryotes. Proc Natl Acad Sci USA. 2006, 103: 4540-4545. 10.1073/pnas.0600833103.
Khush GS: Origin, dispersal, cultivation and variation of rice. Plant Mol Biol. 1997, 35: 25-34. 10.1023/A:1005810616885.
Vaughan DA, Morishima H, Kadowaki K: Diversity in the Oryza genus. Curr Opin Plant Biol. 2003, 6: 139-146. 10.1016/S1369-5266(03)00009-8.
International Rice Genome Sequencing Project: The map-based sequence of the rice genome. Nature. 2005, 436: 793-800. 10.1038/nature03895.
Goff SA: Rice as a model for cereal genomics. Curr Opin Plant Biol. 1999, 2: 86-89. 10.1016/S1369-5266(99)80018-1.
Bennetzen JL: The rice genome. Opening the door to comparative plant biology. Science. 2002, 296: 60-63. 10.1126/science.1071402.
The oryza map alignment project. [http://www.omap.org]
Wing RA, Ammiraju JS, Luo M, Kim H, Yu Y, Kudrna D, Goicoechea JL, Wang W, Nelson W, Rao K, Brar D, Mackill DJ, Han B, Soderlund C, Stein L, SanMiguel P, Jackson S: The oryza map alignment project: the golden path to unlocking the genetic potential of wild rice species. Plant Mol Biol. 2005, 59: 53-62. 10.1007/s11103-004-6237-x.
Leeton PR, Smyth DR: An abundant LINE-like element amplified in the genome of Lilium speciosum. Mol Gen Genet. 1993, 237: 97-104. 10.1007/BF00282789.
Schmidt T: LINEs, SINEs and repetitive DNA: non-LTR retrotransposons in plant genomes. Plant Mol Biol. 1999, 40: 903-910. 10.1023/A:1006212929794.
Wright DA, Ke N, Smalle J, Hauge BM, Goodman HM, Voytas DF: Multiple non-LTR retrotransposons in the genome of Arabidopsis thaliana. Genetics. 1996, 142: 569-78.
Hill P, Burford D, Martin DM, Flavell AJ: Retrotransposon populations of Vicia species with varying genome size. Mol Genet Genomics. 2005, 273: 371-81. 10.1007/s00438-005-1141-x.
SanMiguel P, Tikhonov A, Jin YK, Motchoulskaia N, Zakharov D, Melake-Berhan A, Springer PS, Edwards KJ, Lee M, Avramova Z, Bennetzen JL: Nested retrotransposons in the intergenic regions of the maize genome. Science. 1996, 274: 765-768. 10.1126/science.274.5288.765.
Vitte C, Bennetzen JL: Analysis of retrotransposon structural diversity uncovers properties and propensities in angiosperm genome evolution. Proc Natl Acad Sci USA. 2006, 103: 17638-1764. 10.1073/pnas.0605618103.
Matsuoka Y, Tsunewaki K: Presence of wheat retrotransposons in Gramineae species and the origin of wheat retrotransposon families. Genes Genet Syst. 1997, 72: 335-43. 10.1266/ggs.72.335.
Gribbon BM, Pearce SR, Kalendar R, Schulman AH, Paulin L, Jack P, Kumar A, Flavell AJ: Phylogeny and transpositional activity of Ty1-copia group retrotransposons in cereal genomes. Mol Gen Genet. 1999, 261: 883-91. 10.1007/PL00008635.
Mao L, Wood TC, Yu Y, Budiman MA, Tomkins J, Woo S, Sasinowski M, Presting G, Frisch D, Goff S, Dean RA, Wing RA: Rice transposable elements: a survey of 73,000 sequence-tagged-connectors. Genome Res. 2000, 10: 982-90. 10.1101/gr.10.7.982.
McCarthy EM, Liu J, Lizhi G, McDonald JF: Long terminal repeat retrotransposons of Oryza sativa. Genome Biol. 2002, 3: RESEARCH0053-10.1186/gb-2002-3-10-research0053.
Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchison D, Martin C, Katagiri F, Lange BM, Moughamer T, Xia Y, Budworth P, Zhong J, Miguel T, Paszkowski U, Zhang S, Colbert M, Sun WL, Chen L, Cooper B, Park S, Wood TC, Mao L, Quail P, Wing R, Dean R, Yu Y, Zharkikh A, Shen R, Sahasrabudhe S, Thomas A, Cannings R, Gutin A, Pruss D, Reid J, Tavtigian S, Mitchell J, Eldredge G, Scholl T, Miller RM, Bhatnagar S, Adey N, Rubano T, Tusneem N, Robinson R, Feldhaus J, Macalma T, Oliphant A, Briggs S: A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science. 2002, 296: 92-100. 10.1126/science.1068275.
Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, Cao M, Liu J, Sun J, Tang J, Chen Y, Huang X, Lin W, Ye C, Tong W, Cong L, Geng J, Han Y, Li L, Li W, Hu G, Huang X, Li W, Li J, Liu Z, Li L, Liu J, Qi Q, Liu J, Li L, Li T, Wang X, Lu H, Wu T, Zhu M, Ni P, Han H, Dong W, Ren X, Feng X, Cui P, Li X, Wang H, Xu X, Zhai W, Xu Z, Zhang J, He S, Zhang J, Xu J, Zhang K, Zheng X, Dong J, Zeng W, Tao L, Ye J, Tan J, Ren X, Chen X, He J, Liu D, Tian W, Tian C, Xia H, Bao Q, Li G, Gao H, Cao T, Wang J, Zhao W, Li P, Chen W, Wang X, Zhang Y, Hu J, Wang J, Liu S, Yang J, Zhang G, Xiong Y, Li Z, Mao L, Zhou C, Zhu Z, Chen R, Hao B, Zheng W, Chen S, Guo W, Li G, Liu S, Tao M, Wang J, Zhu L, Yuan L, Yang H: A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science. 2002, 296: 79-92. 10.1126/science.1068037.
Tateoka T: Porteresia a new genus of Graminae. Bullettin of National Science Museum Rokyo. 1965, 8: 405-406.
Guo Y, Ge S: Molecular phylogeny of Oryzeae (Poaceae) based on DNA sequences from chloroplast, mitochondrial, and nuclear genomes. American Journal of Botany. 2005, 92: 1548-1558.
Doyle JJ, Doyle JL: A rapid DNA isolation procedure from small quantities of fresh leaf tissues. Phytochem Bull. 1987, 19: 11-15.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequences traces using phred. I. Accuracy assessment. Genome Res. 1998, 8: 175-185.
Chou HH, Holmes MH: DNA sequence quality trimming and vector removal. Bioinformatics. 2001, 17: 1093-1104. 10.1093/bioinformatics/17.12.1093.
J. Craig Venter Institute. [http://www.tigr.org]
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J: Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005, 110: 462-467. 10.1159/000084979.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Blastclust. [ftp://ftp.ncbi.nih.gov/blast/documents/blastclust.html]
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004, 32: 1792-1797. 10.1093/nar/gkh340.
Kumar S, Tamura K, Nei M: MEGA3: Integrated Software for Molecular Evolutionary Genetics Analysis and Sequence Alignment. Briefings in Bioinformatics. 2004, 5: 150-163. 10.1093/bib/5.2.150.
Ge S, Sang T, Lu BR, Hong DY: Phylogeny of rice genomes with emphasis on origins of allotetraploid species. Proc Natl Acad Sci USA. 1999, 96: 14400-14405. 10.1073/pnas.96.25.14400.
Martinez CP, Arunuganathan K, Kikuchi H, Earle ED: Nuclear DNA content of ten rice species as determined by flow cytometry. Jpn J Genet. 1994, 69: 513-523. 10.1266/jjg.69.513.
Acknowledgements
This work was funded by the NSF Plant Genome Program award #DBI-0321678 and the Bud Antle Endowed Chair to RAW. We also thank the AGI technical staff that supported this project, especially members of the BAC/EST Resource, Sequencing, and Bioinformatics Centers.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AZ carried out the analysis, wrote the manuscript and participated in the design of this study. AS: contributed to the phylogenetic analysis and to the data parsing. JT: constructed the genomic libraries. YY, HK, KC and DK participated in the library construction and the production of sequencing data. RAW contributed to the design of the study, supervised the experimental steps and contributed to the preparation and editing of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12862_2007_443_MOESM1_ESM.doc
Additional file 1: Occurrences per single Mbp for the major classes of repeats in different Oryza genomes. A) The absolute number of significant hits to the Oryza repeat data bases. C: Estimated number of occurrences calculated using the equation from Hawkins et al (2006) [9]. E: The number normalized to 1 Mbp. Columns B, D and F are the confidence intervals calculated assuming a Poisson distribution of repeats in the genome, calculated for values in columns A, C and E respectively. The mean length used for different repeats were as follows: Ty3-gypsy elements: 12 Kbp; Ty1-copia elements: 5.5 Kbp; others LTR-RTs not classified: 8.75 Kbp; LINEs: 3.5 kbp; Helitrons (complete autonomous): 12.8 Kbp; CACTA: 15.2 Kbp; hAT: 3.6 Kbp. *In this case all the calculations are based on a rough estimate of the genome size of this species: the real value is unknown; we therefore used the value estimated for O. ridleyi [HHJJ; 1283 Mbp], which is also an allotetraploid species and shares the HH genome type with O. coarctata. (DOC 264 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zuccolo, A., Sebastian, A., Talag, J. et al. Transposable element distribution, abundance and role in genome size variation in the genus Oryza. BMC Evol Biol 7, 152 (2007). https://doi.org/10.1186/1471-2148-7-152
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-7-152